Helicene-Based Chiral Auxiliaries and Chirogenesis


Department of Chemistry and Biotechnology, Tallinn University of Technology, Akadeemia tee 15, 12618 Tallinn, Estonia
*
Author to whom correspondence should be addressed.
Symmetry 2018, 10(1), 10; https://0-doi-org.brum.beds.ac.uk/10.3390/sym10010010
Submission received: 7 December 2017
/
Revised: 25 December 2017
/
Accepted: 26 December 2017
/
Published: 29 December 2017
(This article belongs to the Special Issue Chiral Auxiliaries and Chirogenesis)
Abstract
:Helicenes are unique helical chromophores possessing advanced and well-controlled spectral and chemical properties owing to their diverse functionalization and defined structures. Specific modification of these molecules by introducing aromatic rings of differing nature and different functional groups results in special chiroptical properties, making them effective chiral auxiliaries and supramolecular chirogenic hosts. This review aims to highlight these distinct structural features of helicenes; the different synthetic and supramolecular approaches responsible for their efficient chirality control; and their employment in the chirogenic systems, which are still not fully explored. It further covers the limitation, scope, and future prospects of helicene chromophores in chiral chemistry.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
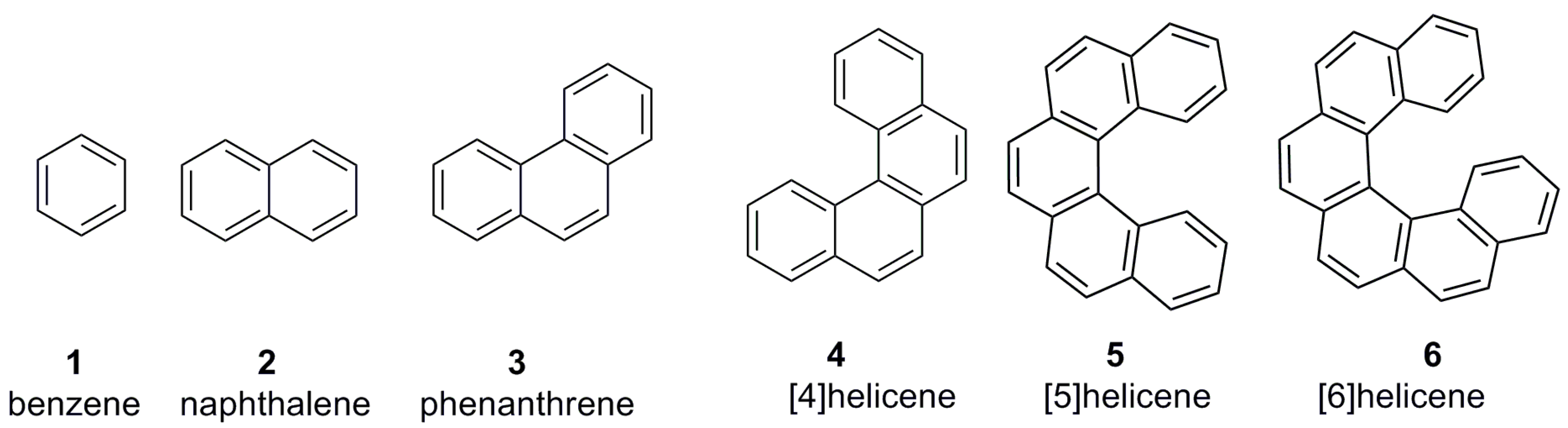
Aromatic characteristics in organic compounds are very interesting due to the resonance electronic features, stability, and inter- and intramolecular π-π interactions along with the specific geometric properties and reactivity, making it an important topic for researchers. Among the diverse aromatic compounds, there is a class of the molecules carrying ortho-annulated polyaromatic structures, classified as helicenes. In order to understand their unique structural features, helicenes should be compared with the conventional aromatic molecules (Figure 1).
Benzene 1, naphthalene 2, and phenanthrene 3 are spatially flat aromatic structures (Figure 1); hence, they are not helicenes. As the number of ortho-fused aromatic rings is increased beyond three units, such as in 4–6 (Figure 1), the space constraint in the molecule is also enhanced, forcing it to adopt a nonplanar helical arrangement. These types of aromatic structures are called helicenes. As a consequence, the electron resonance ability is restricted throughout the molecule. Similar to conventional aromatics, helicenes can be functionalized by various synthetic protocols to further enhance their spectral and chemical properties. However, the diversity of helical aromatic structures makes helicenes into unique and advanced chromophoric systems with tunable electronic and steric properties for corresponding applications in all types of stereochemical processes, including chiral auxiliaries and supramolecular chirogenesis. It is of note that, whilst there is a great variety of aromatic structures used in different chiral fields [1], helicenes still lag behind, despite their attractive properties discussed below.
2. Overview of Latest Reviews
Firstly, the existing reviews on helicenes from within the last five years should be briefly analyzed. Indeed, the chemistry of helicenes has existed for more than a century. However, this topic has been reviewed only recently to a rather limited extent [2,3,4,5,6,7,8,9,10,11]. Furthermore, these reviews mainly focused on the synthesis of helicenes and much less on their applications. One of the most comprehensive reviews was published in 2012 by Chen et al., describing helicene synthesis and applications [2]. In 2013, Gingras accumulated three sets of reviews targeting only carbohelicenes and covering the topics of its non-stereoselective [3] and stereoselective synthesis, chiral separation [4], and applications, along with the corresponding properties [5]. Yet, in 2014, three small review articles appeared on the topics of photochemical methods of helicene synthesis [6], helicene-based transition metal complexes [7], and helicene-like chiral auxiliaries [8]. Further, in 2015, Virieux [9] summarized the synthesis and application of helical phosphorus derivatives, where the helicene-based phosphorus ligands were also described. Thus, it is clear that—despite several consecutive reviews and monographs—this field is not fully covered, especially from the point of view of chiral applications. Further, the helicenes, being polyaromatic compounds, have been discussed in several chapters of a few books devoted to polyaromatic compounds and material chemistry [10]. Very recently, in 2017, a book on helicene chemistry [11] was published, covering most of the topic as it had appeared in previous reviews and presenting the progress up until the period of 2015.
Despite these recent efforts described above, a general tutorial and critical summary devoted to the design and tuning of helicene structures for suitable general and specific applications in the areas of chiral auxiliaries and chirogenesis is not yet available. Hence, the present review aims to fill in this gap, with a specific focus on the potential applicability and usefulness of helicene chemistry in these chirality fields—including the latest updates.
3. Classification and Nomenclature
3.1. Structural Diversity
In order to understand the structure and type of helicene molecules, a brief introduction into the classification and nomenclature should be made. As a part of classical organic chemistry, helicenes can be structurally divided into two major groups: carbohelicenes and heterohelicenes.
3.1.1. Carbohelicenes
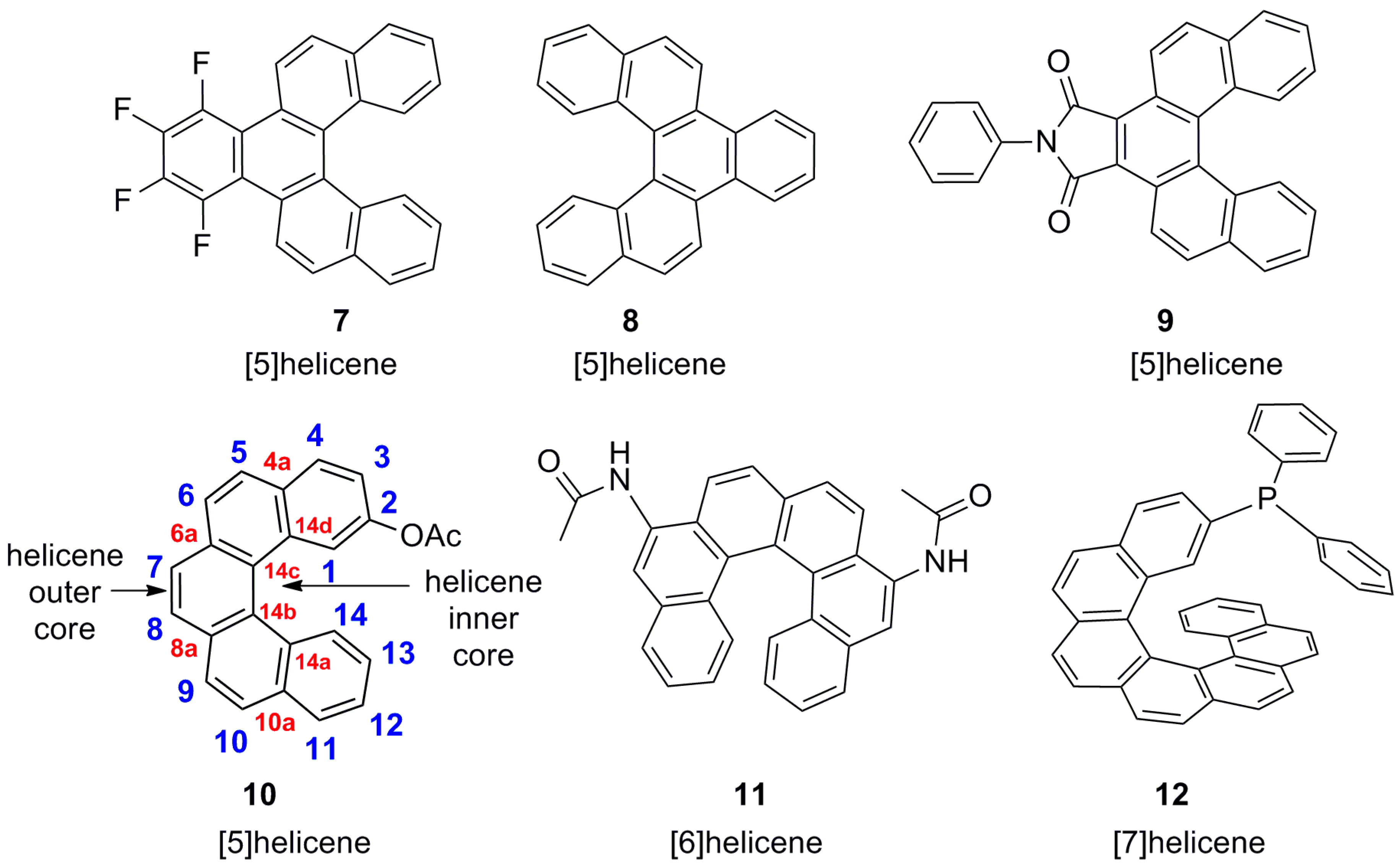
Carbohelicenes are all benzenoid (aromatic) ortho-fused systems and named as [n]helicenes, where n represents the number of rings forming a helix in the ortho-fused fashion. Thus, the structures 4 [12], 5 [13,14], and 6 [15,16], are [4]helicene, [5]helicene, and [6]helicene, respectively (Figure 1). However, any fused ring which is not present in the ortho-fused manner is not included in the n counting. For example, structures 7 [17], 8 [18], and 9 [19] are all [5]helicenes, but not [6]helicenes, despite the presence of six aromatic rings in total (Figure 2). The functionalized helicenes such as 10 [20], 11 [21], and 12 [22] (Figure 2) which have a heteroatom as a substituent on the aromatic ring, as in the case of amino, hydroxyl (phenolic or alcoholic), thiol, or phosphorus-containing groups, are still carbohelicenes but not heterohelicenes. In order to specify the position of functional group(s), a numbering system is used, as shown in the structure 10. The numbering starts from the innermost position at one end and goes to another end via the outer helical core in such a way that the functional group has the minimal number. Secondly, only unfused carbons (with hydrogen or other groups as substituents) are counted sequentially (shown in blue), whereas fused carbon atoms (of the helicene core) have the previous number with the corresponding a, b, c suffixes based on the sequence order of fused 1, 2, 3 atoms, and so on (shown in red). Thus, the structure 10 can be named as 2-acetoxy[5]helicene.
In the case of nonsymmetric heterohelicene, the numbering should be started from the peripheral ring which will give the least number to the heteroatomic ring core. In the case of more than one substituent, a general nomenclature rule of numbering should be applied. Yet, in both the cases mentioned, one should always follow the sequential numbering of the helicene core (starting from one innermost atom to another innermost atom of the two peripheral rings).
3.1.2. Heterohelicenes
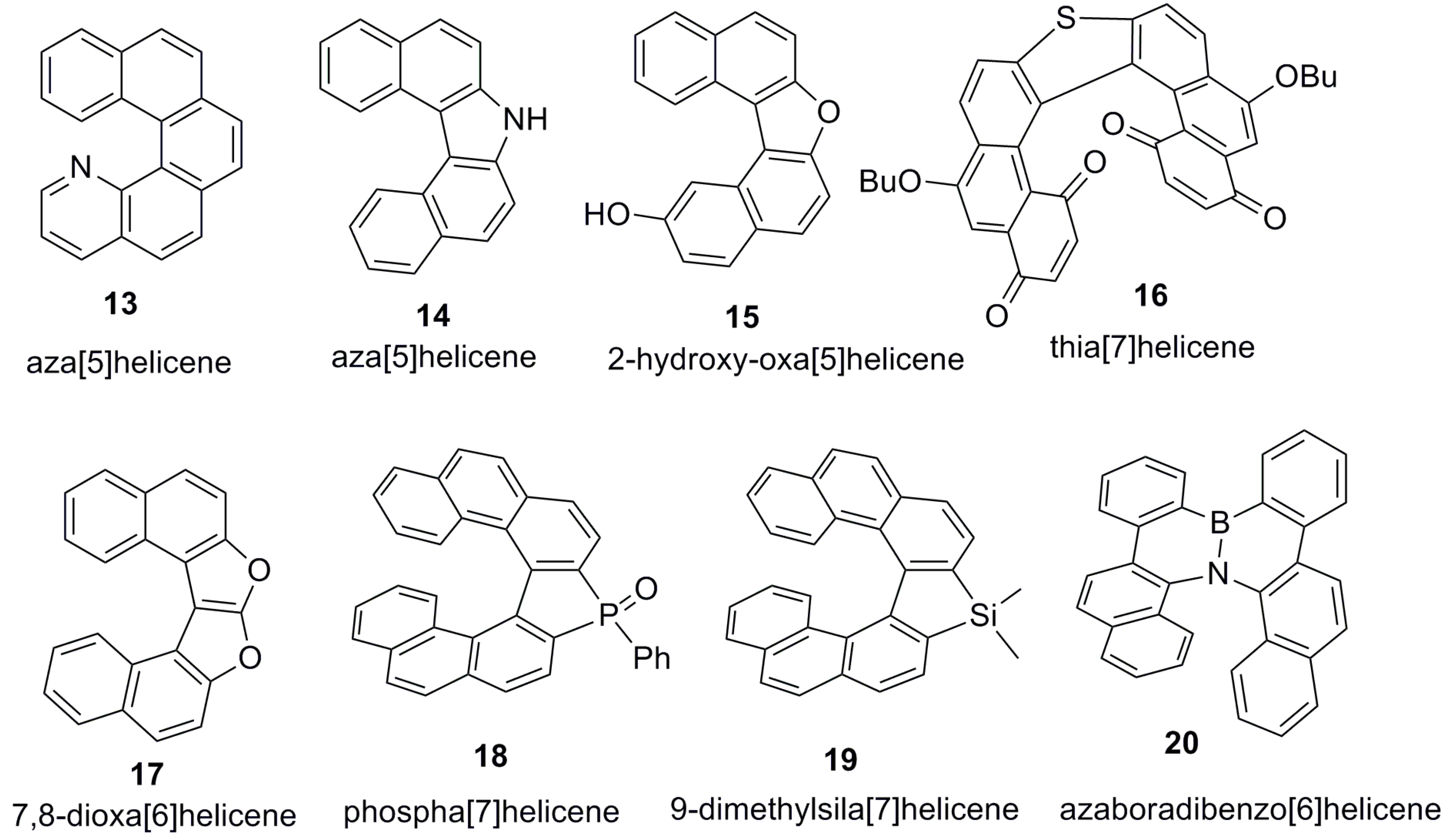
Another class of helicenes is the heterohelicenes, in which one or more benzenoid ring is substituted with a heteroatomic ring (Figure 3). Heterohelicenes are generally based on five-membered rings such as thiophene, pyrrole, furan, etc., and six-membered rings mostly containing pyridine. Additionally, they can be fused and functionalized.
The corresponding heteroatom must be a part of the ring present in the fused ortho fashion but not as the substituent either in a functional group or in a heteroatomic ring. In the latter, whilst the overall molecule is heterocyclic, the helicene core is not. Further, depending on the heteroatom nature, the helicene structures are named aza[5]helicenes (13 [23], 14 [24]), oxa[5]helicenes (15 [25]), and thia[7]helicenes (16 [26]). If there is more than one heteroaromatic ring or heteroatom, the helicene names are simplified as hetero[n]helicenes, [n]heterohelicenes, or simply [n]helicenes. For two or three similar heteroatomic rings, the prefixes “di”, “tri”, etc., are added with or without indicating the exact position. For example, the structure 17 [27] is called 7,8-dioxa[6]helicene or simply dioxa[6]helicene. Heterohelicenes containing phosphorus 18 [28], silicon 19 [29], and boron 20 [30] atoms are relatively rare and their chiral properties have not been well studied so far; therefore, they are excluded from this review.
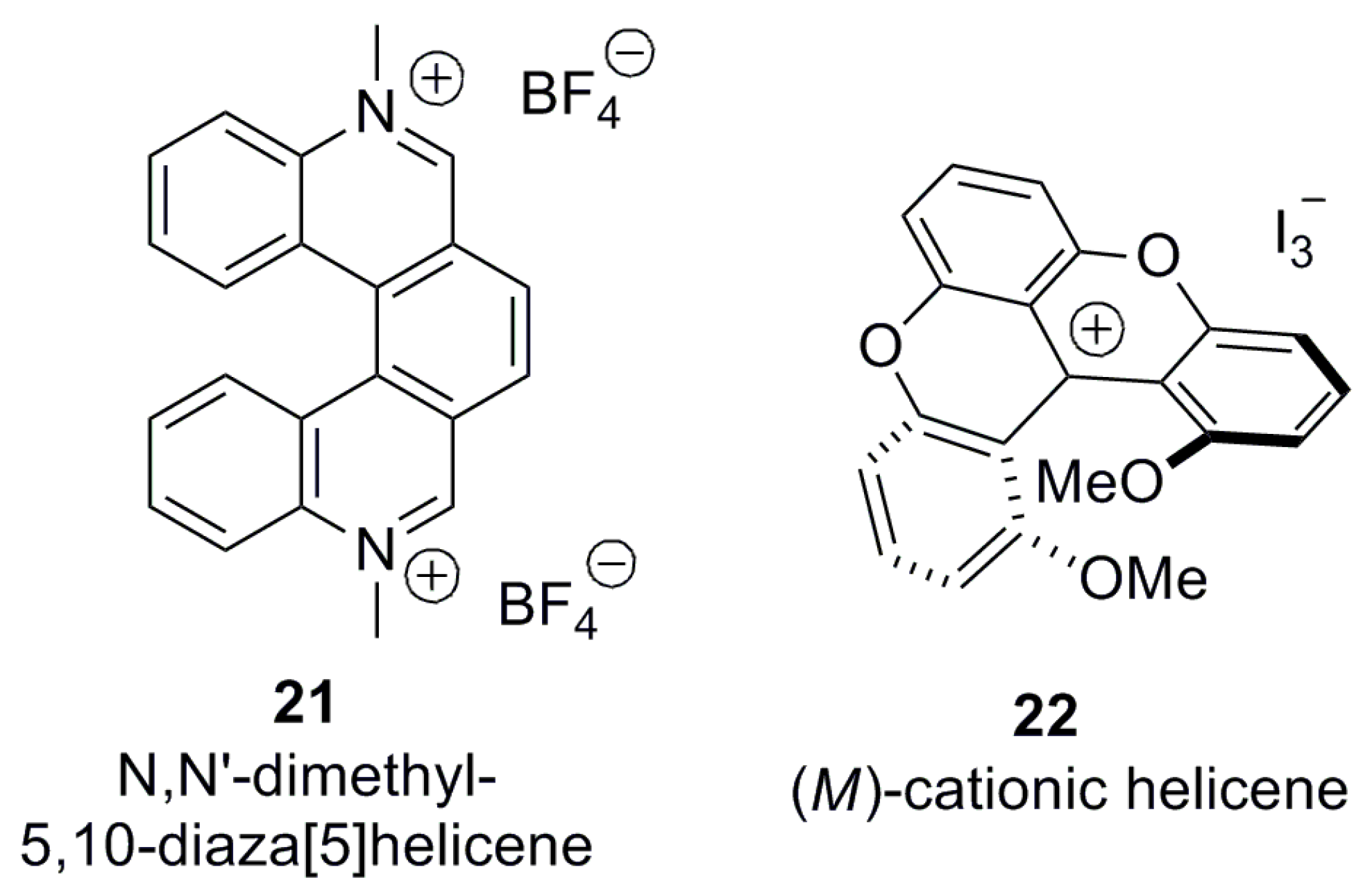
3.1.3. Charged Helicenes
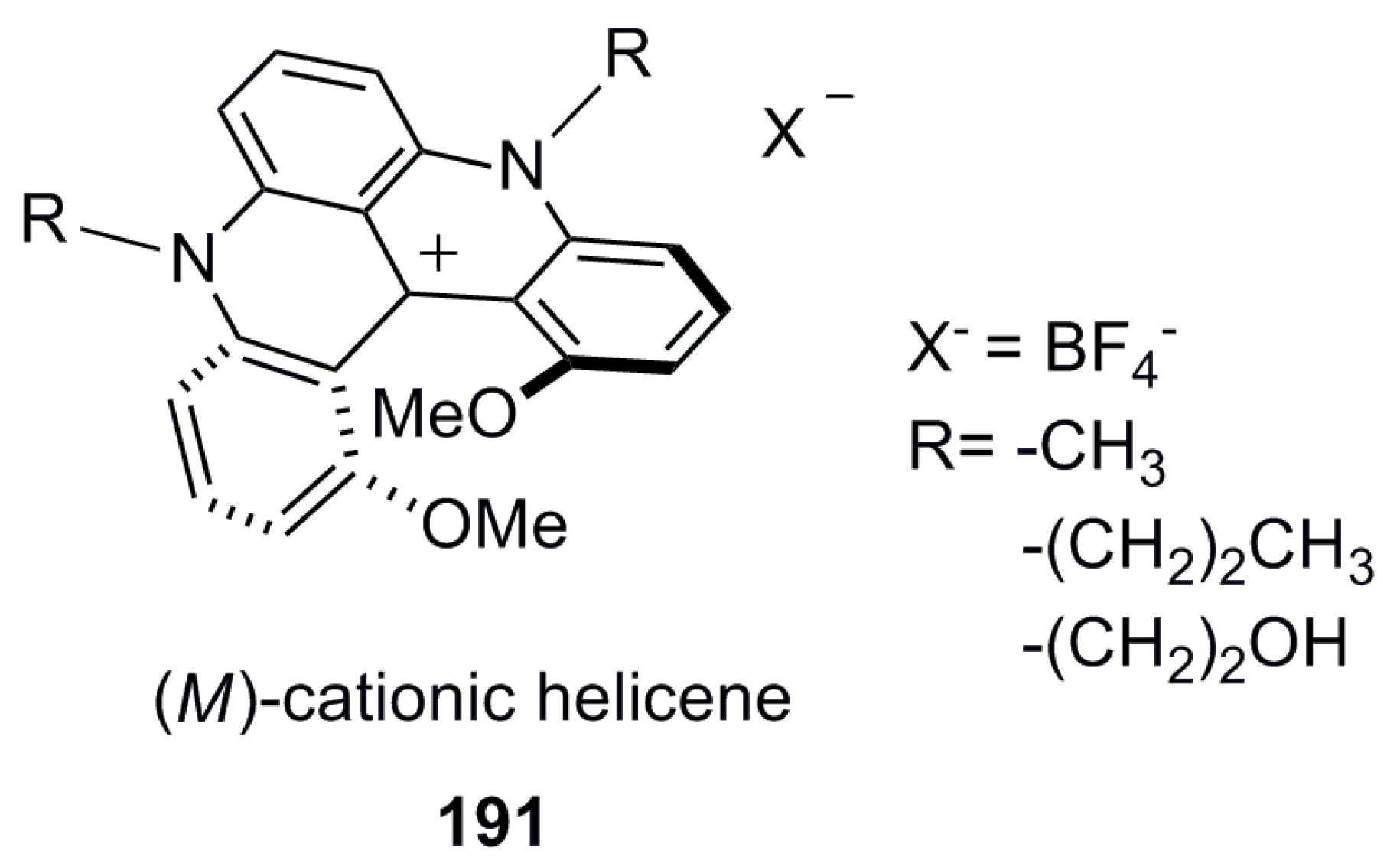
Helicenes can also be categorized into neutral and charged helicene molecules. Most of the above examples are neutral helicene molecules, where the charge of the aromatic moiety is zero. However, if there is a charge on the helicene structure, a counter ion should be presented to balance the overall charge, hence resulting in different properties and applications. In such helicenes, exchanging the counter ion can easily enhance a number of the corresponding derivatives, controlling the solubility and pH effect. This counter ion exchange can sometimes offer a new approach towards optical resolution by changing an achiral counter ion to a chiral one in the case of thermodynamically stable structures. For example, quarternized azahelicene 21 [31] and cationic helicene 22 [32] stabilized by electron resonance/conjugation represent typical charged helicenes (Figure 4).
3.1.4. Special Structural Type of Helicene-Like Molecules
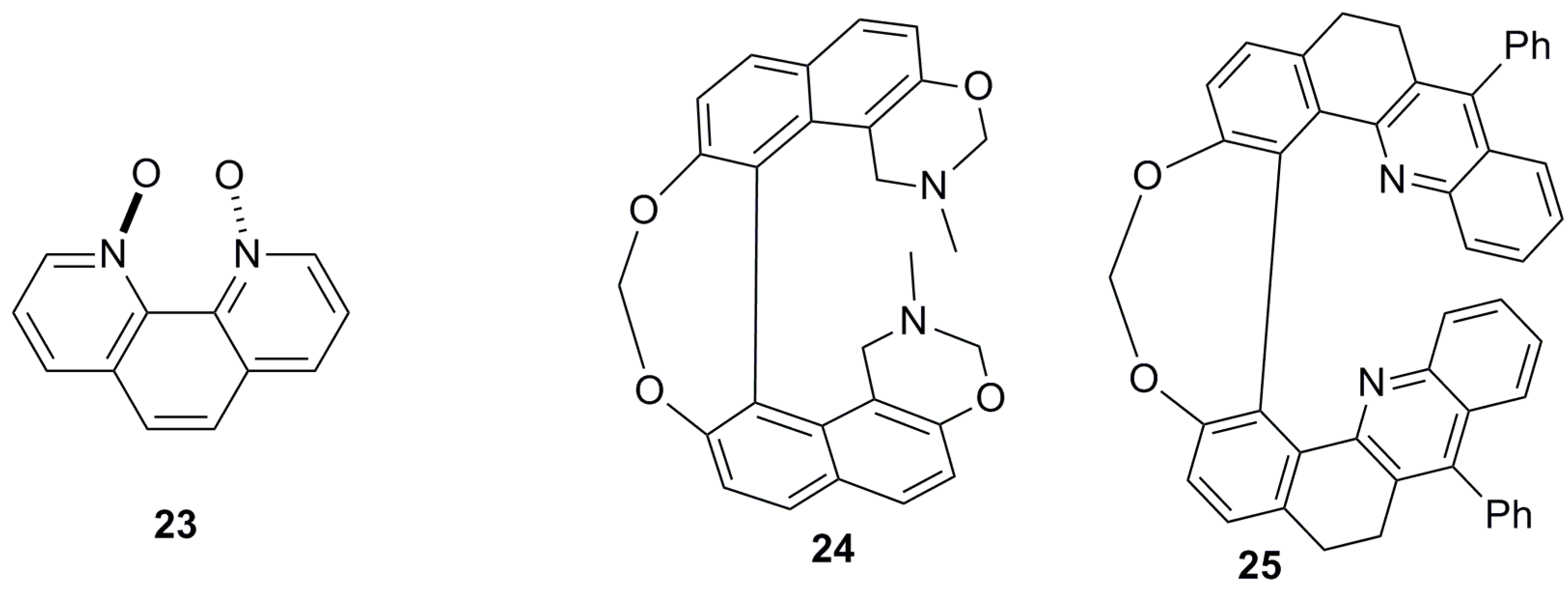
Besides the traditional definition of helicene molecules, there are several special cases which belong to this structural category but are not associated with the above-described major groups of carbo- and heterohelicenes; these are discussed below. Naphthalene 2 and phenanthrene 3 are the simplest ortho-fused flat molecules without any steric constraint and, hence, are not helicenes, whilst four fused aromatic rings induce a certain steric hindrance at the innermost peripheral hydrogen atoms, setting up the first member of helicene family. Thus, in [n]helicenes, n should be >3 to be classified as a helicene molecule. However, there are some exceptions to this general rule. Interestingly, phenanthroline-N,N-dioxide 23 [33] (Figure 5) is also classified as a helicene in the literature, despite the fact that n = 3. This is due to the fact that the N,N-dioxide functionalities at the peri positions in the bay area of the phenanthroline are able to introduce the corresponding spatial constraint to force the molecule to adopt a configurationally stable helical conformation.
Besides the well-defined helicene molecules, there also exist helical structures, such as 24 [34] and 25 [35], called helicene-like molecules or helicenoids (Figure 5). Helicenoids usually contain at least one saturated atom as a part of the ring system in the helicene-type core system. Thus, the saturated ring is not able to adopt a flat structure, resulting in the enhancement of helicity. For example, a series of 1,1′-Bi-2-naphthol (BINOL)-based helical systems, sometimes referred to as oxahelicenoids, have been reported so far as configurationally stable structures [34,35]. Other types of saturated hydrocarbon-containing helicenoids, which are actually dihydrohelicenes, are also well documented in literature as precursors for helicene synthesis.
3.2. Spatial Diversity
Besides the structural diversity described above, helicenes can also be categorized, based on their stereochemistry, into four different categories: achiral (flat structures), stereodynamically labile (when P and M spatial conformations are in fast equilibrium; for the P and M definitions, see Section 3.2.1), chiral (enantiomerically stable structures), and meso helicenes (helical structure with some symmetry elements). This categorization is highly important for their applications in chiral auxiliaries and for chirogenic systems.
3.2.1. Chiral (Configurationally Stable) Helicenes
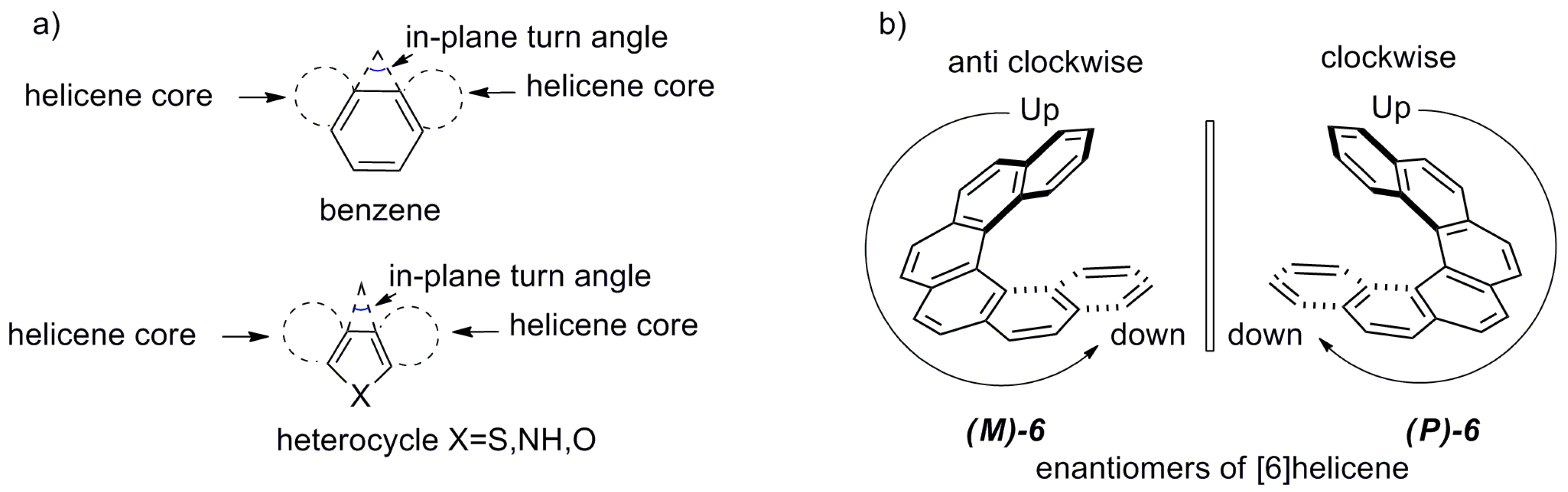
In order to understand the helicene spatial structures and this classification, the term “in-plane turn angle” needs to be introduced. Due to the ortho fusion mode, each aromatic ring contributes to the overall helicity of the molecule via an in-plane turn angle (Figure 6a). When the sum of the in-plane angles of the contributing rings becomes 360° or more, the helicene is forced to adopt a helically chiral structure caused by the corresponding steric clashes between the terminal/peripheral rings. Conventional six-membered (hexagon) aromatic rings such as benzene and pyridine have an in-plane turn angle of 60° [36]. However, in the case of five-membered heteroaromatic (pentagon) rings, the value is considerably smaller at 45° (thiophene), 35° (pyrrole), and 32° (furan) [36] due to the different geometric properties of aromatic rings as a result of the variable atomic sizes of heteroatoms and bond length of C–X bond (X = heteroatom S, N, O in the five-member aromatic rings, correspondingly). Therefore, the helicenes with at least one five-membered heteroaromatic ring need n > 6 to become intrinsically chiral, whereas for the helicenes containing all six-membered carbo/hetero aromatic rings, n = 6 is sufficient to make them helically chiral.
In general, a chiral helicene molecule exists in two enantiomeric forms. The right-handed helical structure (moving from up to down) is assigned the name (P)-enantiomer, whereas the left-handed helical structure (moving from up to down) is designated the (M)-enantiomer, according to the Cahn–Ingold–Prelog rule (Figure 6b). This type of helicene tends to give configurationally stable enantiomers as the racemization process is expected to go through the highly energetic, strained Cs-symmetrical transition state [37]. Along with this thermodynamic stability of enantiomers, helicenes also exhibit notable enhanced (chir)optical properties, making them suitable candidates for chiral auxiliaries and other chirogenic processes. Interestingly, in general, (P)-carbohelicenes are observed as dextrorotatory, whereas (M)-enantiomers display a levorotatory nature [38].
3.2.2. Achiral Helicenes
Another type of helicene is the achiral flat molecules; for example, ortho-condensed polyaromatic molecules consisting of four rings with at least one ring being a five-membered (pentagon) heteroaromatic ring, as in [4]heterohelicenes. Whilst these structures are less interesting from the viewpoint of chirality, they possess enhanced conjugation, electron transfer, and emission properties, making them suitable candidates for various material chemistry applications.
3.2.3. Stereodynamic Helicenes
Holding an intermediate position between the chiral and achiral helicenes are the stereodynamic helicene structures. The geometry of these molecules is close to flat, whilst the van der Waals radii of the innermost hydrogens touch each other, making the structure stereodynamic and nonresolvable in solution, owing to the low (P)–(M) interconversion energy barrier. These include helicenes consisting of five benzenoid (hexagon) rings or [6]heterohelicenes with two five-membered (pentagon) heteroaromatic rings. However, in a solid state, these structures are able to adopt a chiral helical conformation. The dioxa[6]helicene 16 [27] is a typical example of such a system.
Stereodynamic helicenes are also highly fluorescent in nature due to the extended conjugation, whilst lacking any intrinsic chiroptical properties in solution. Yet, applying host–guest chemistry with suitable enantiopure guests, the corresponding chiroptical properties can be induced and controlled in this type of helicene. Additionally, the stereodynamic helicenes can be made helically chiral by introducing a bulky substitution at the innermost (C1) carbon atom. Computational analysis confirmed that attachment of just one methyl group at the innermost position results in the same steric strength as introduction of one additional ortho-fused benzene ring. Thus, [5]helicene with a methyl group at the innermost position has a structure more helical than that of [6]helicene, thus increasing the racemization barrier for the former [37].
3.2.4. Meso Helicenes
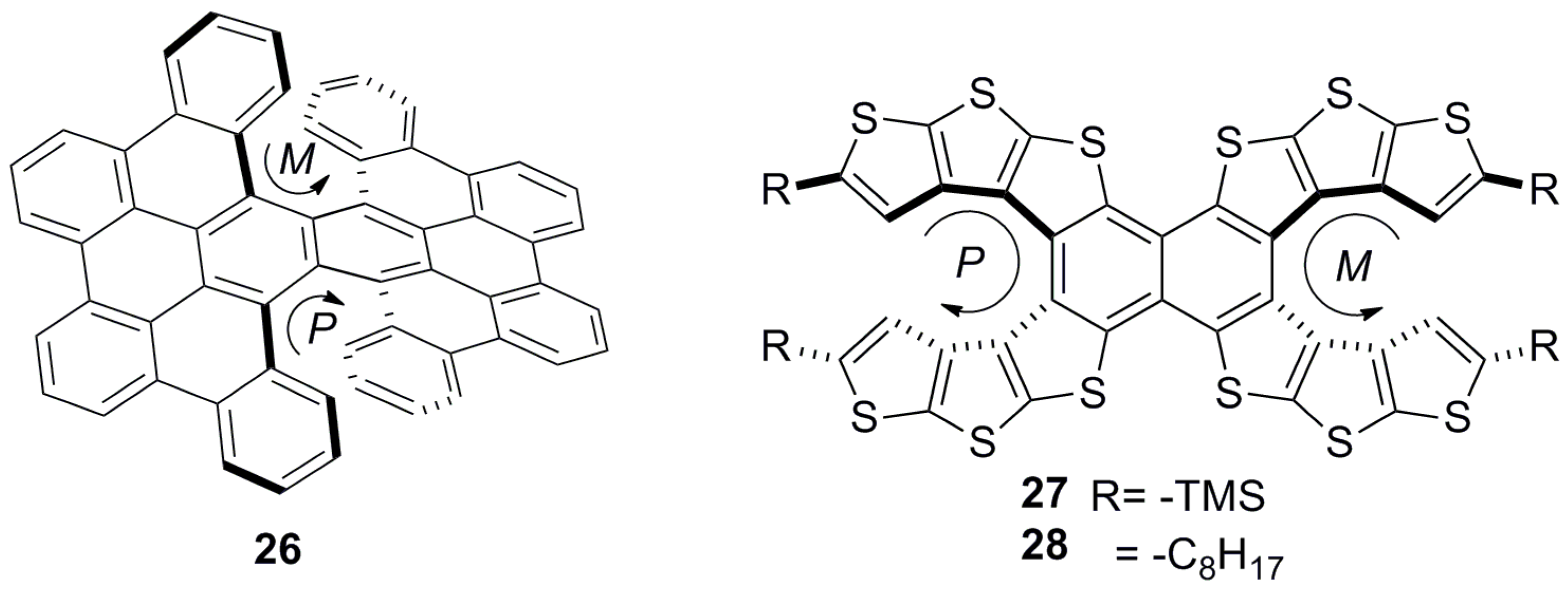
Meso helicenes are achiral, yet need to be classified separately. Two helicene fragments in one molecule, with the presence of a plane of symmetry, produce an achiral meso helicene. Each equivalent helical part of this molecule possesses the opposite helical sense, whilst the whole molecule is symmetrical. Potentially, it can be suitably desymmetrized by a covalent bond or supramolecular interaction, making it chiroptically active and, hence, usable in various chirogenic processes. However, this area has not been well investigated yet, as selective modification of only one helicene moiety out of two is an arduous task; this is due to the fact that both the helicene parts are chemically equivalent. For example, carbocyclic helicene 26 [39] and heterocyclic helicene 27–28 [40] are meso double helicenes (Figure 7). It should be noted that the double helicenes have three stereoisomers, out of which two are optically active ((P,P) and (M,M)) and one is optically inactive ((P,M) or (M,P)), defined as a meso structure.
4. General Description and Main Synthetic Approaches toward Helicene-Based Chirogenic Systems
The early pioneering work on helicene synthesis, resolution, and chiroptical properties has been performed by Newman [15,16,41], Wynberg [36,38], Martin [42], and Katz [26]. In general, classical methods of helicene synthesis include the following approaches: oxidative photocyclization, Diels–Alder reactions, and various aromatic and metal-catalyzed coupling reactions [2,3,4,5,6,11]. The synthesis of helicenes is always a challenging task for organic chemistry due to the steric factors involved and difficulty in controlling the regioselectivity of reactions. The same factors are also responsible for the unique reactivity of the helicene skeleton.
The current advances in synthetic chemistry, resolution, and chiral separation are essentially helpful in designing and obtaining novel enantiopure helicene molecules in sufficient quantities to be used for various applications. As BINOL, (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) (BINAP), and related biaryl systems have been successfully applied as ligands, catalysts, and hosts in stereodiscriminating processes [43,44,45], it is theoretically possible to thoroughly design helicene-based helical systems possessing advantageous chiral properties. Progress in this area is highlighted below with several selected examples.
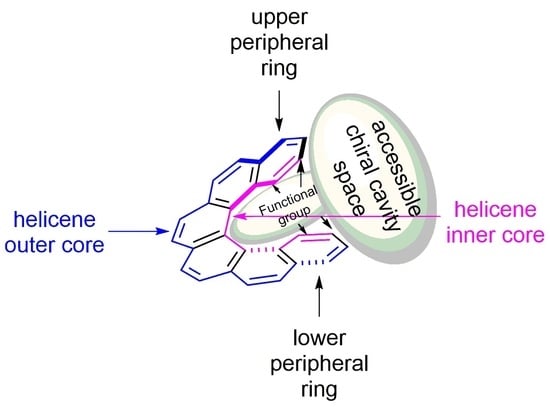
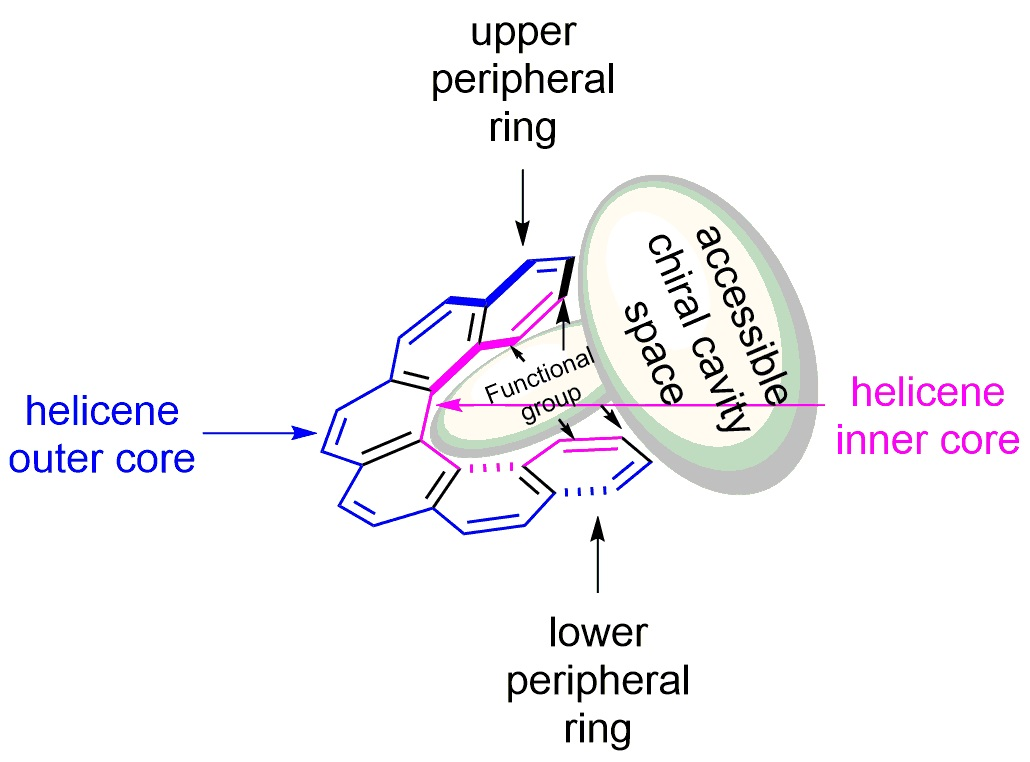
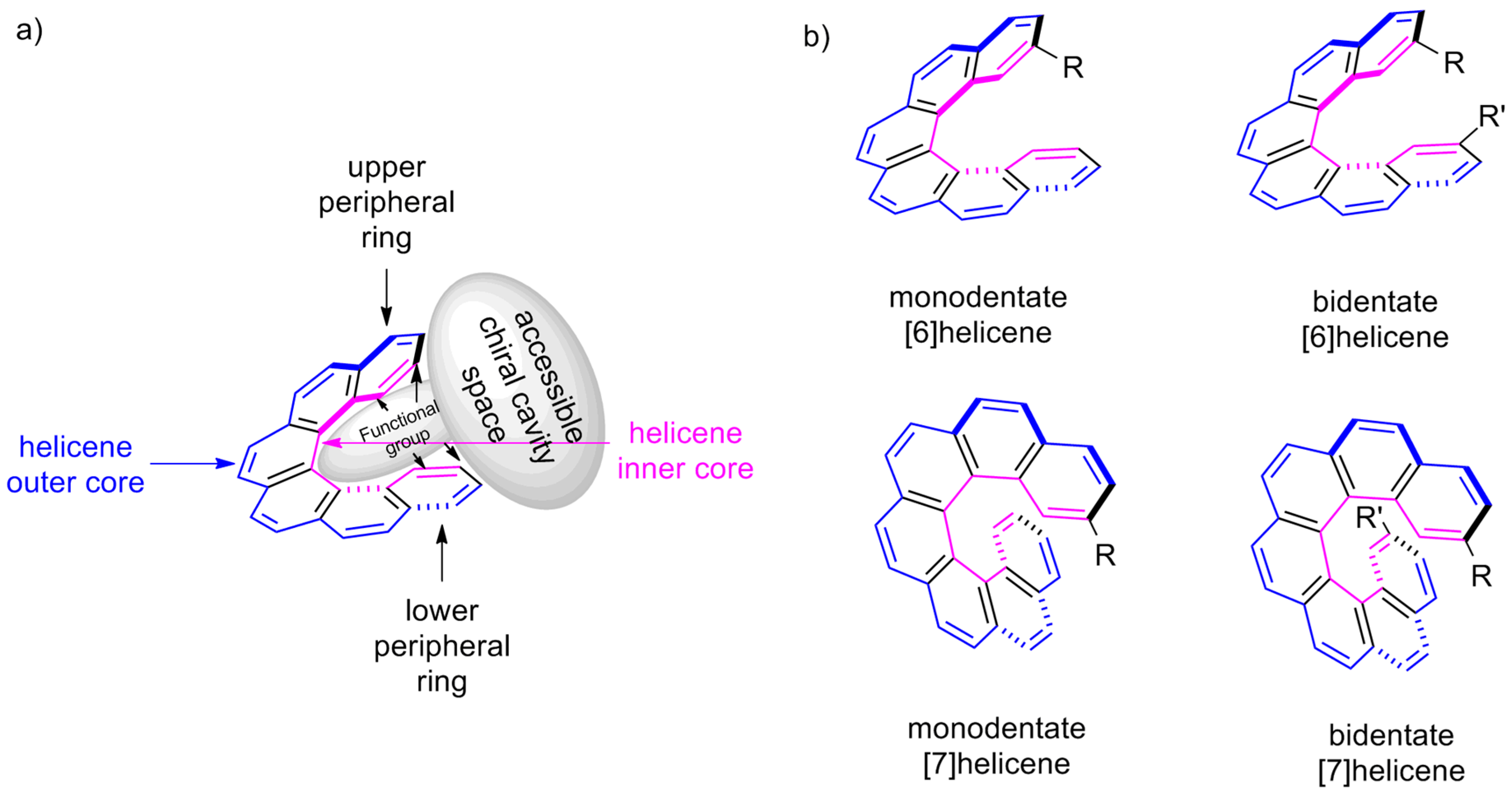
Let us look closely at a general structure of (M)-[6]helicene 6 (Figure 8a), where the benzene rings at the beginning and at the end of the helical arrangement are called the terminal or peripheral rings. It can be clearly seen that the helix experiences steric clashes towards the inner sides (interior), called the helicene’s inner core. This structural organization results in a helical chiral cavity. The restricted space available between and around the two peripheral rings can be explored effectively for various chiral discriminating processes. In general, the introduction of suitable functional groups at the peripheral rings can act as a binding site to hold the reactant or guest molecules to facilitate the enantioselectivity, where the chiral cavity space of the helicene is able to influence the corresponding (diastereomeric) transition state and/or complexation mode. In contrast, the opposite outer skeleton of the helicene, termed as the helicene outer core (exterior), can be functionalized and used to covalently link these molecules on surfaces and for other applications.
The tailor-made design of helicenes for a suitable application can be of two types; first, where the functionalization at one peripheral ring is sufficient (monodentate), and second, where functionalization on both the terminal rings is required (bidentate) (Figure 8b). The bidentate helicenes can be C2-symmetric (R = R’) or nonsymmetric (R ≠ R’) molecules. In accordance with the structural features and substituent patterns of helicenes, their efficiency in chirogenic processes is observed, as demonstrated throughout this review.
However, since the majority of recently published reviews [2,3,4,5,6] have focused on helicene synthetic chemistry, there is no need to describe this topic here in detail. Alternatively, this review addresses the structural design and stereochemical aspects of helicene chromophores suitable for various applications in chiral processes, whilst synthesis of these molecules will only be highlighted in brief upon necessity to emphasize their spatial and chiral properties.
5. Helicenes as Chiral Auxiliary/Reagent or Additive
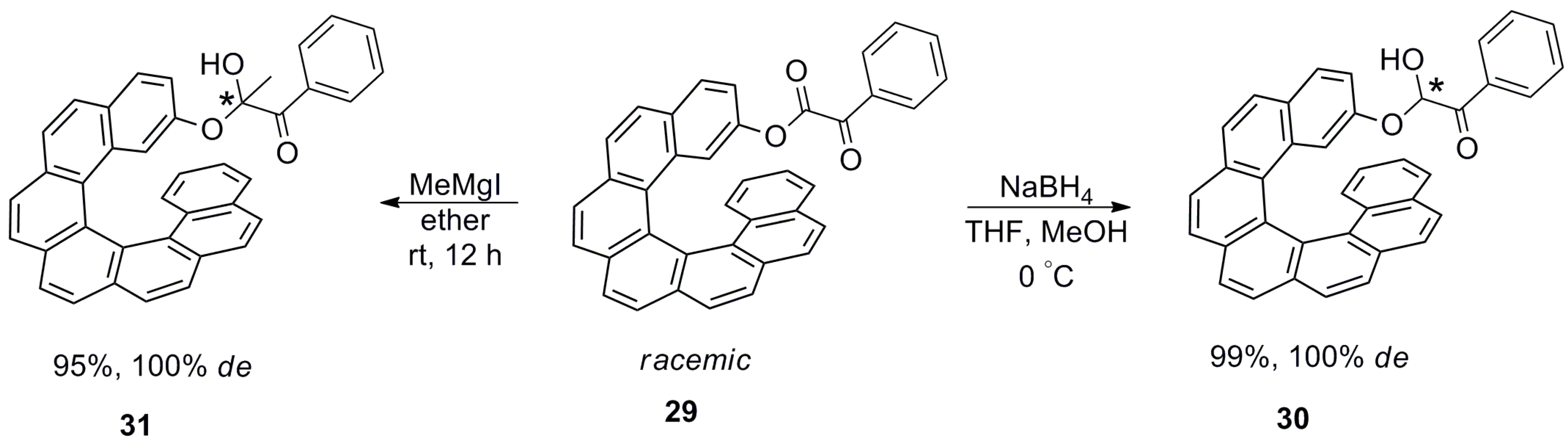
Due to the difficulties associated with the synthesis of helicenes and obtaining the corresponding enantiopure forms in sufficient amounts, their application as a chiral auxiliary in stoichiometric reactions is limited. However, one of the first results on the use of helicenes as chiral auxiliaries in few diastereoselective reactions was reported in 1985–1987 by Martin et al. with racemic 2-substituted-[7]helicene [46,47,48,49,50]. Hence, racemic 29 was employed as an inbuilt chiral auxiliary for the diastereoselective reaction of the carbonyl group of α-keto esters upon reduction with NaBH4, yielding 30 (99% yield; 100% de) [46], and on the nucleophilic addition of Grignard reagent to give 31 (95% yield; 100% de) [47] (Scheme 1).
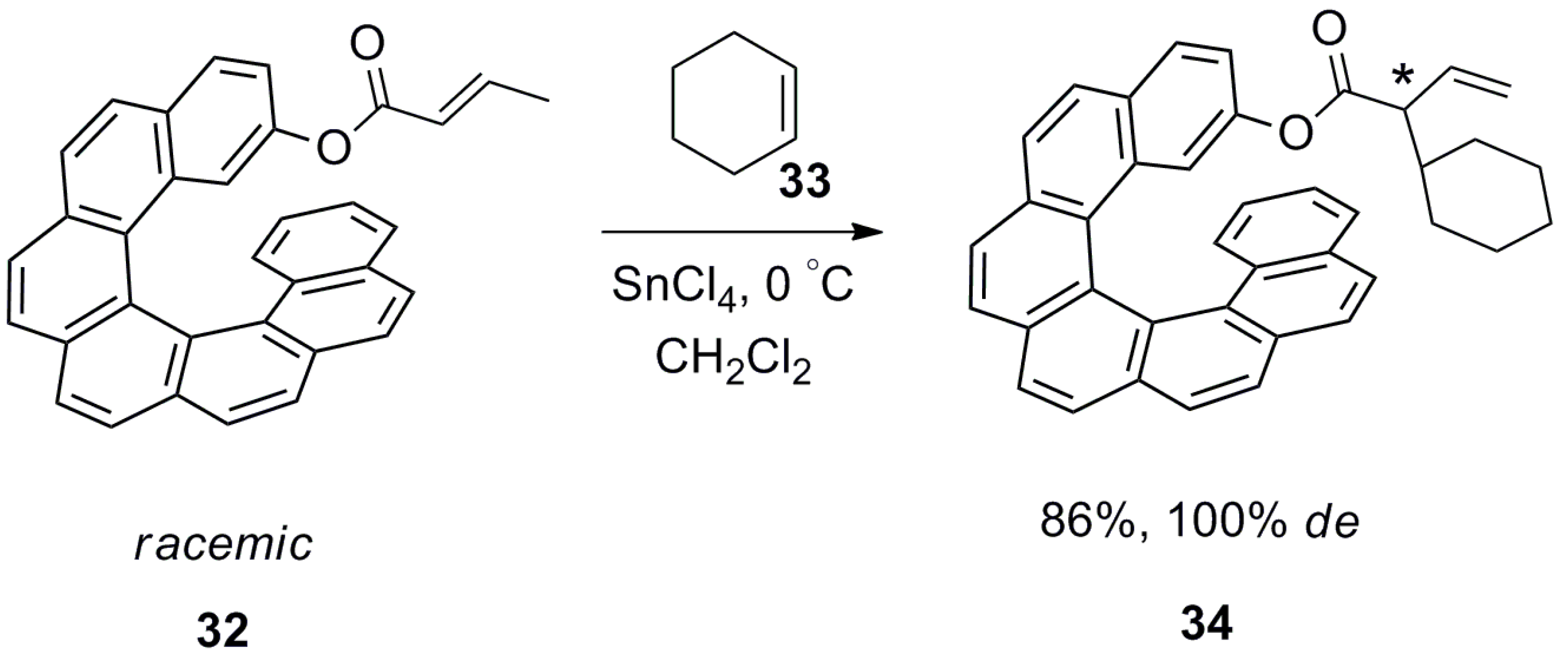
Although high diastereoselectivity (100%) was observed, the overall process requires attaching (protection) and detaching (deprotection) the helicene auxiliary, thereby making it difficult to scale up. Similarly, [7]helicene-attached unsaturated ester 32 was also utilized for the highly diastereoselective ene reaction with cyclohexene 33 in the presence of SnCl4 to afford 34 with high yield (86%) and 100% de [48] (Scheme 2).
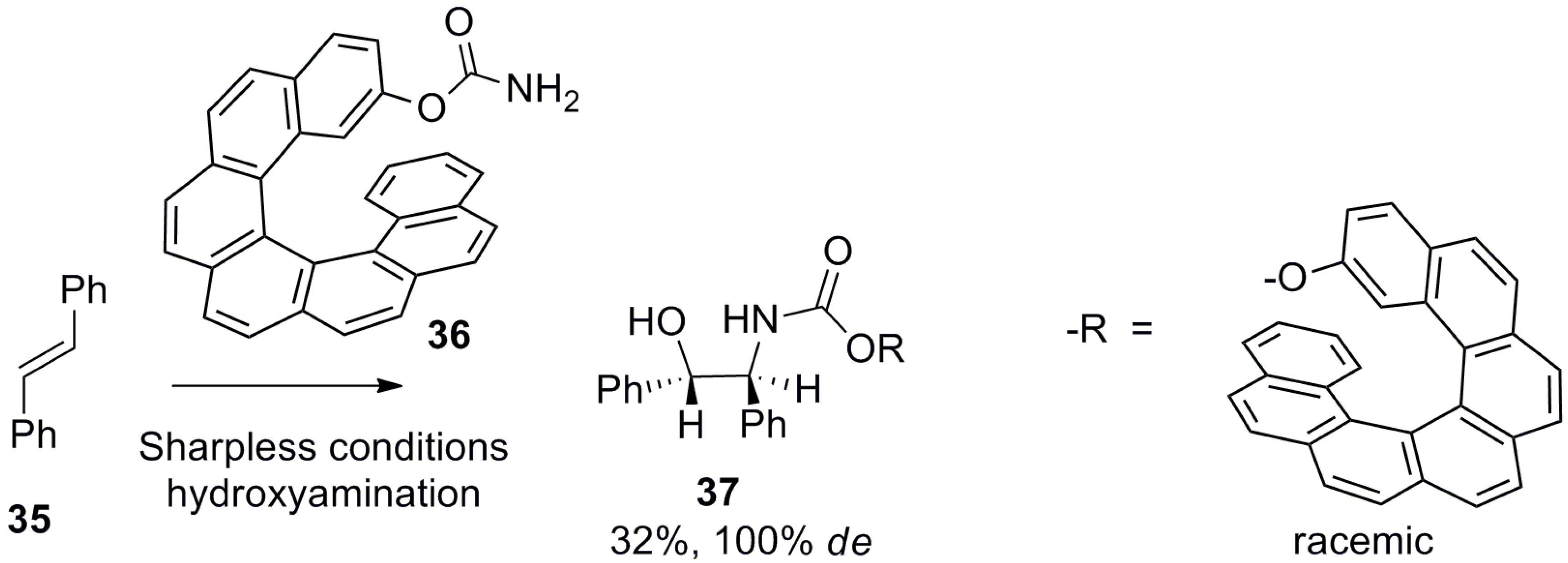
Further, hydroxyamination of E-stilbene 35 under the Sharpless condition by using helicene 36 as a chiral auxiliary resulted in 37 with 32% yield with 100% de [49] (Scheme 3).
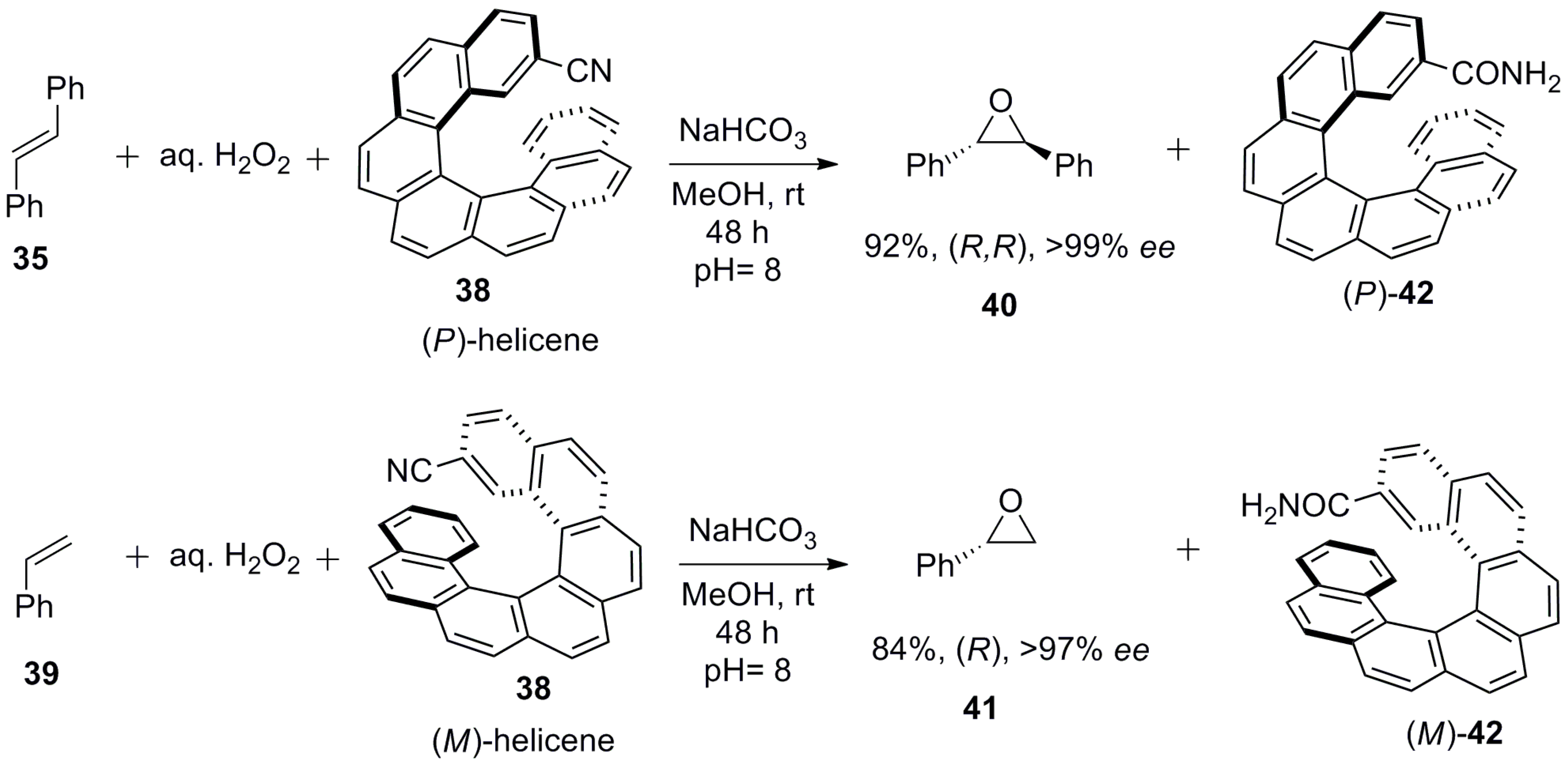
Another example is based on enantioenriched 2-cyano-[7]helicene 38 employed as a stoichiometric chiral additive (reagent) for the epoxidation of alkenes 35 and 39 using H2O2 as an oxidant to give enantiopure epoxides 40 (92%, 99% ee) and 41 (84%, 97% ee), respectively [50] (Scheme 4). The mechanism includes the initial reaction between enantiopure 2-cyano-[7]helicene 37 and H2O2 to generate in situ the corresponding chiral hydroperoxyimine, which serves as a chiral oxidizing reagent for the subsequent epoxidation of alkenes 35 and 39, whilst itself converting finally to [7]helicene-2-carboxamide 42.
These examples (Scheme 1, Scheme 2, Scheme 3 and Scheme 4) [46,47,48,49,50] are the first cases of helicenes being used as chiral auxiliaries resulting in a high asymmetric conversion (de or ee), yet noncatalytic in nature. These excellent results were due to the judicious selection of [7]helicene with corresponding functionalization at the 2-position (monodentate) (refer, Figure 8), which locates inside the helical chiral cavity with sufficient accessibility to the reactants. Besides this, a geometric feature of [7]helicenes is that the two peripheral rings are located one above another, hence inducing steric hindrance to one of the reacting prochiral faces and generating high selectivity via the stereospecific approach.
It is of note that most of the helicene-based catalytic research is focused on the use of [6]helicene and substituted [5]helicene-based systems. In cases where functionalization at both the rings is needed, substituted [5]helicenes and [6]helicenes are more suitable due to possibility of the geometrical accessibility of these groups (refer, Figure 8b). These groups are located on the same side; hence, they are capable of coordinating or bonding together with the reactive partners. However, this situation is difficult to obtain in [7]helicenes, as seven benzenoid rings give the in-plane turn angle of 420° (7 × 60°), forcing the functional groups present on the terminal rings to exist in opposite directions, thus unable to work co-operatively (refer, Figure 8b). Yet, when only one of the peripheral rings is functionalized and acts as an active catalytic site, the [7]helicene-based systems have an additional advantage over substituted [5]- and [6]helicenes. This is owing to the same in-plane turn angle property allowing another peripheral ring to block one of the prochiral faces of the reactant. Nevertheless, surprisingly, asymmetric catalysis with [7]helicenes is not well explored in comparison with that with [5]- and [6]helicenes.
6. Helicenes as Chiral Catalysts
Since various helicene-based phosphine-related ligands have been reviewed [2,5,7,8,9], only recent selected examples capable of delivering exceptionally high ee values are summarized here.
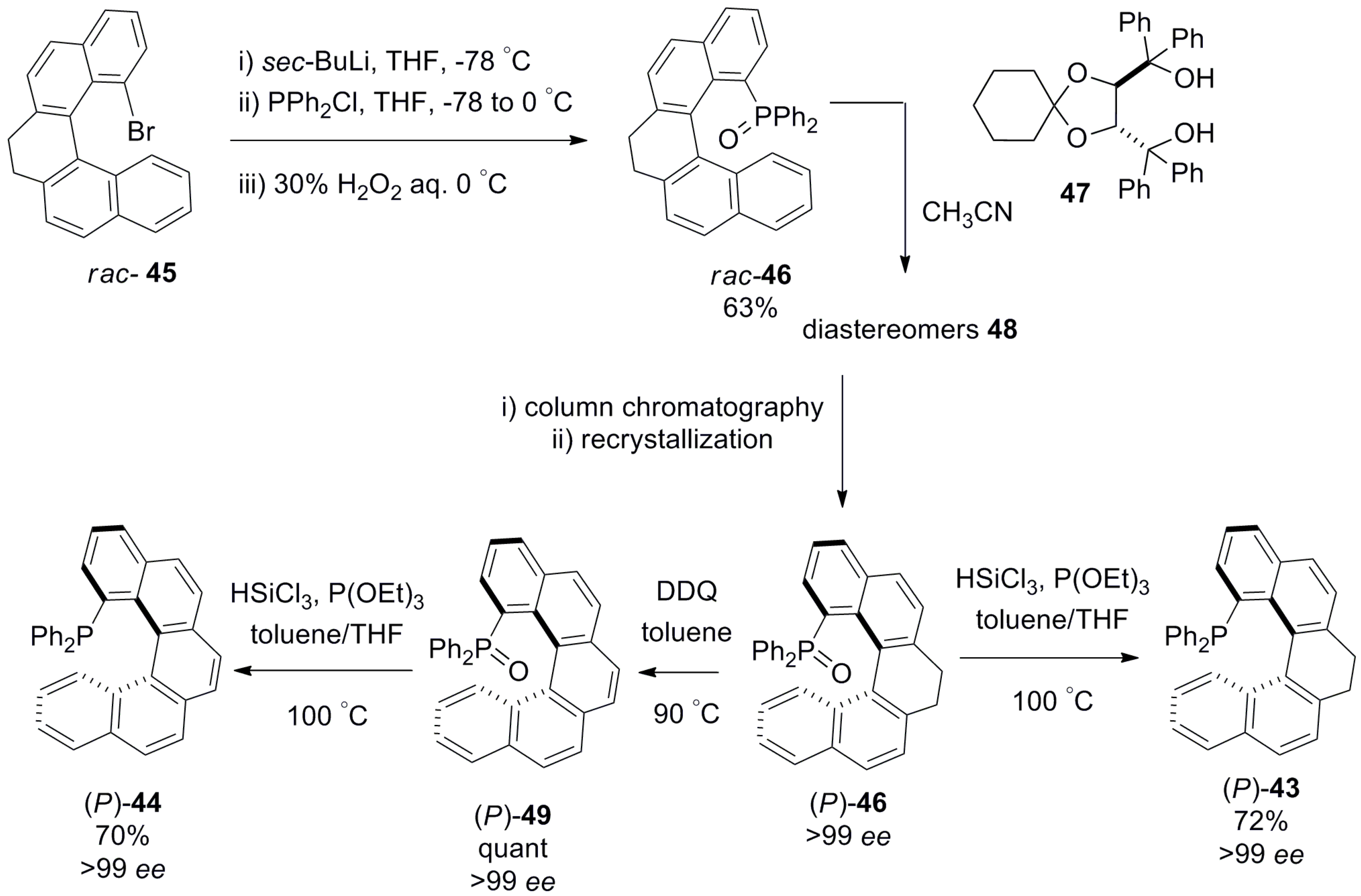
In 2016, the [5]carbohelicene-based phosphine ligands 43 and 44 were rationally designed for application in Pd-catalyzed asymmetric synthesis [51]. Both the systems were prepared from corresponding rac-bromo substituted 45 via lithiation and subsequent reaction with chlorodiphenylphosphine followed by oxidation with hydrogen peroxide to afford rac-phosphine oxide 46 with 63% yield. It was successfully resolved by using spiro-TADDOL (α,α,α,α-tetraphenyl-1,3-dioxolane-4,5-dimethanol) 47 as a resolving agent to form the insoluble diastereomer 48 with (P)-46, which in turn gave pure (P)-46 with greater than 99% ee on subsequent treatment. Then, (P)-46 was aromatized to 49 quantitatively by using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. Both the phosphine oxides 48 and 49 were reduced to the corresponding phosphines, 43 and 44, with trichlorosilane and P(OEt)3 with 70–72% yield and 99% ee (Scheme 5) [51].
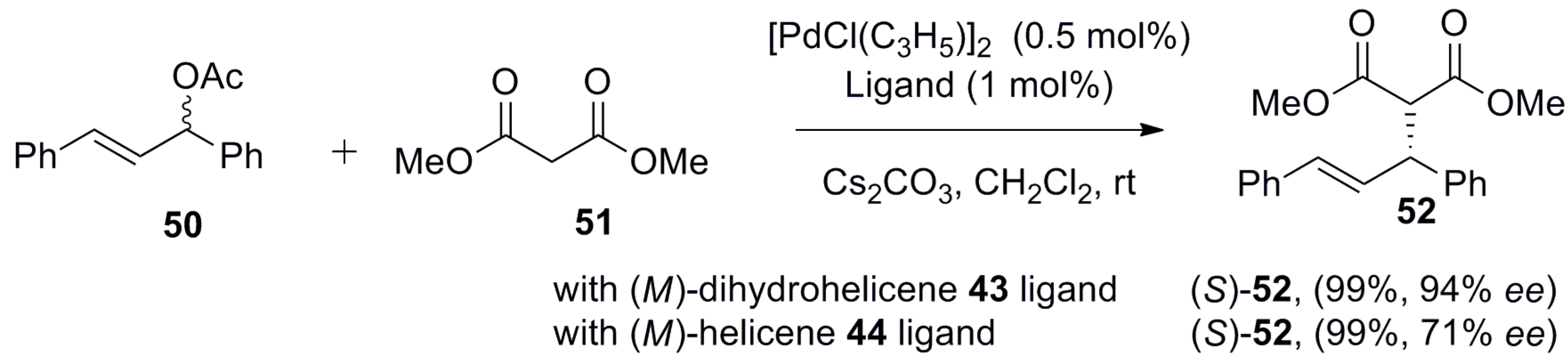
In general, unsubstituted [5]helicenes are configurationally unstable and tend to racemize easily. However, introduction of the diphenylphosphine group at the 1-position results in the configurational stability of 43 and 44. Of these two structures, the 7,8-dihydro-derivative 43 tends to be more helical due to the saturated ethane unit, which has greater conformational flexibility. From X-ray analysis, it was confirmed that the helical pitch diameter of dihydrohelicene 43 is 3.54–3.50 Å, which is larger than that of fully aromatic 44, found to be 3.39–3.34 Å. This rationally changed spatial arrangement has a noticeable influence on its application as a ligand owing to the favorable geometry and distance, which was clearly demonstrated by the metal–ligand and arene interaction. Hence, the double bond (C8a–C14b) is coordinated with the palladium metal center of ligand 44 in a side-on (η 2) fashion, which is not possible in the case of dihydrohelicene 43. This difference in geometric features controls the outcome of Pd-catalyzed reactions. For example, the alkylation of rac-1,3-diphenylallyl acetate 50 with dimethyl malonate 51 by using (M)-dihydrohelicene 43 as a chiral ligand was highly efficient, resulting in the corresponding (S)-enantiomer 52 with 99% yield and 94% ee (Scheme 6) [51]. In contrast, the fully aromatic (M)-helicene 44, whilst affording the same configuration of the product 52, gave similar yield, but only 71% ee (Scheme 6). This clearly demonstrates that the ligands’ geometry allowed them to opt for different transition states.
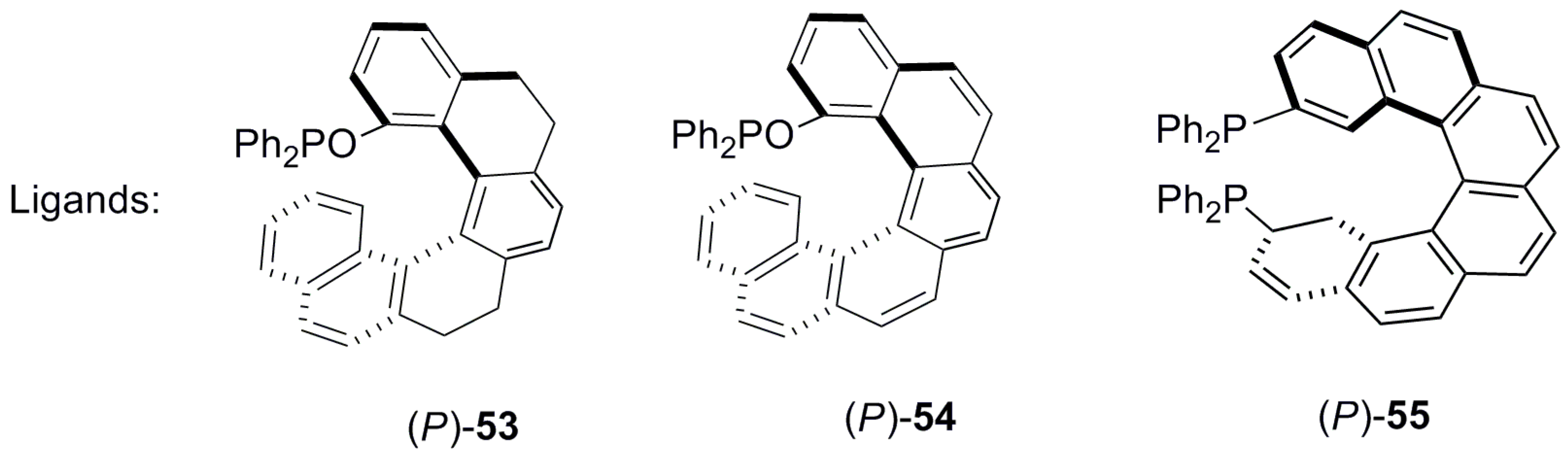
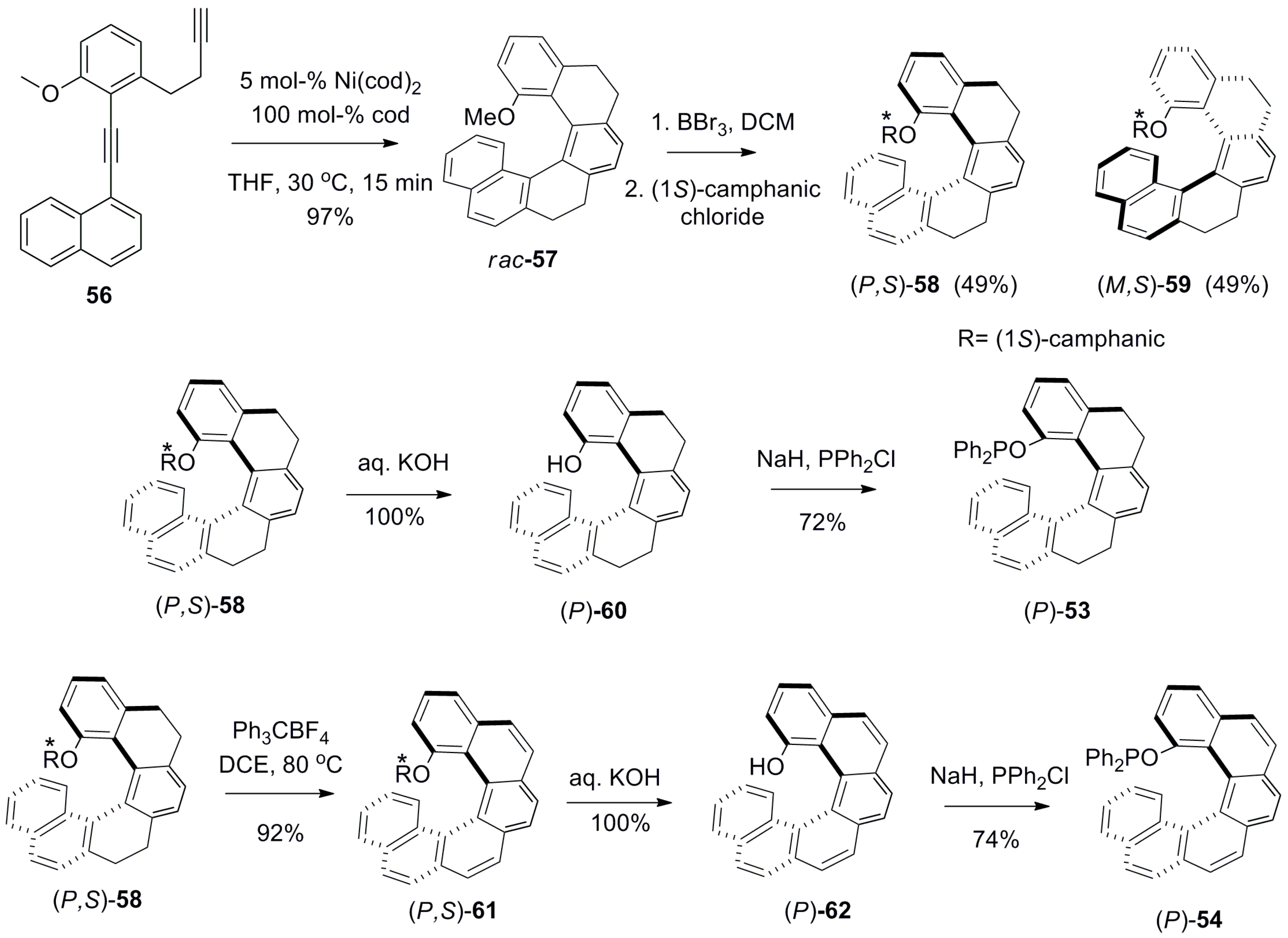
Similar types of C1 position substituted phosphinite ligands, helicenoid 53 [52] and helicene 54 [52] (Figure 9), were also prepared and successfully explored for the Pd-catalyzed asymmetric allylic alkylation between 50 and 51 (see Scheme 5). In both cases, the high yields and ee of 52 were obtained as with 53 (96%, 90% ee) and with 54 (97%, 99% ee) [52]. It is of note that this kinetic resolution reaction using helicene ligand 55 [53] was firstly reported in 2000 with 100% conversion and 81% ee (Figure 9). Although the results from using 55 are comparatively worse in terms of ee, the reaction requires less catalyst loading and proceeds faster compared with other ligands. Interestingly, 55, whilst being a bidentate ligand, still acts as a monophosphine (monodentate) ligand during the catalytic process owing to the large distance (6.481 Å) between the two phosphorus atoms; this was the subject of pioneering work by Reetz et al. [53]. Thus, one can see that all phosphine (43, 44) and phosphinite (53, 54) ligands prepared later, possess only a mono phosphorus atom and are suitable for application as monodentate ligands in this reaction.
The corresponding enantiopure 53 and 54 were synthesized using the Ni-catalyzed [2 + 2 + 2] cycloaddition reaction of alkyne 56 to give 57 (97%), and the subsequent demethylation and esterification as (1S)-camphanate to yield the corresponding diastereomers, 58 and 59. Further, the separated diastereomers were hydrolyzed with alkali to quantitatively produce the enantiomer 60, which, on subsequent reaction with chlorodiphenylphosphine in the presence of sodium hydride, resulted in the tetrahydro phosphinite ligand, (P)-53. For 54, an additional step of dehydrogenation using triphenylmethylium tetrafluoroborate (Ph3CBF4) was required to obtain the diastereomer 61 (92%), which upon hydrolysis gave [6]helicen-1-ol 62, which was finally converted to phosphinite 54 with 74% yield (Scheme 7) [52].
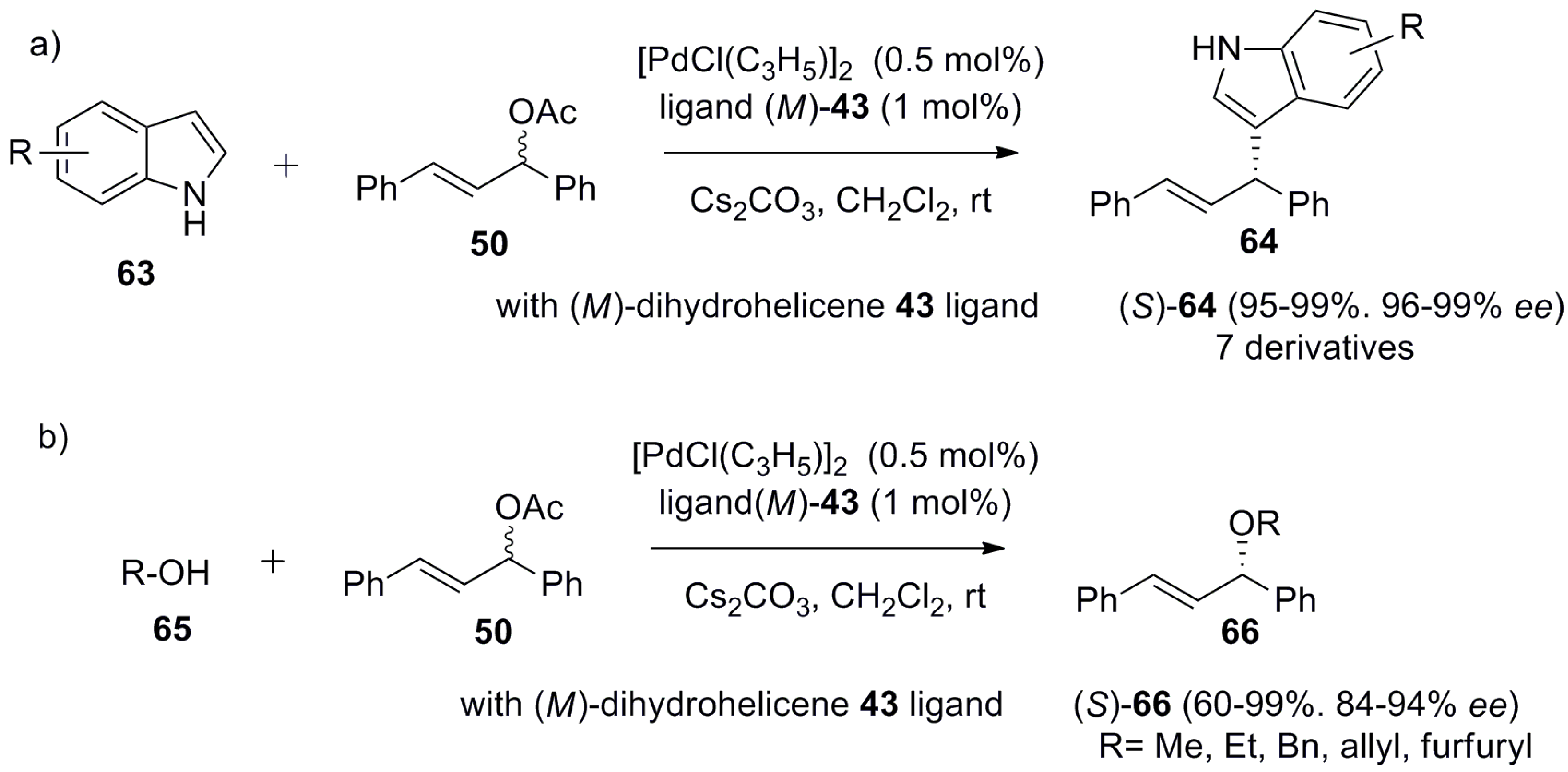
Inspired by the difference in the catalytic performance of 43 (94% ee) and 44 (71% ee) (Scheme 6) in Pd-catalyzed asymmetric allylic alkylation, the same group also applied these ligands in other Pd-catalyzed reactions. Thus, in further investigation, the dihydrohelicene ligand 43 was found to be highly useful in the asymmetric allylation of indoles 63 with 1,3-diphenylallyl acetate 50 to give the 3-alkylated indole product 64 in up to 99% ee, and in the etherification with alcohols 65 to yield 66 in up to 94% ee (Scheme 8) [51].
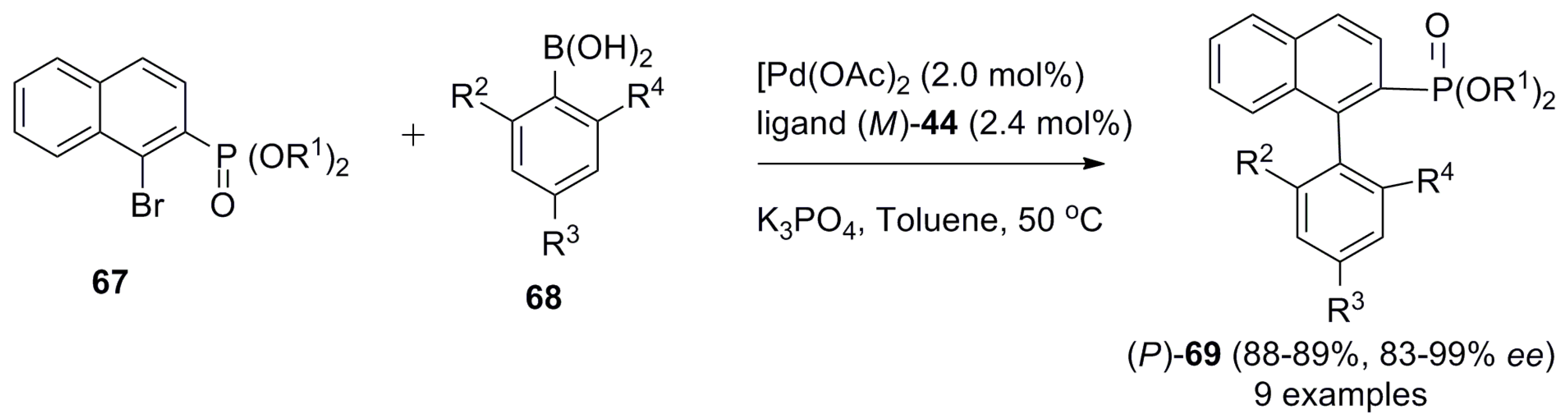
In the case of fully aromatic helicene 44, the ligand was highly effective in the stereocontrol of helical chirality in the Suzuki–Miyaura coupling reaction between bromide derivative 67 and boronic acid 68, resulting in up to 99% ee for chiral biaryl products 69 of (P)-helicity (Scheme 9) [51].
These examples (43 and 44) clearly show that the helicity and space available in the helical groove suitable for a specific transition state play a vital role in the stereoselective control of the reaction pathway. The DFT-based calculation at the B3PW91/6-31G* level (LANL2DZ for the Pd atoms) revealed that the transition state with minimum steric repulsions between the helicene backbone and the reactant group is responsible for the (S)-configuration products (52, 64, and 66) and P-configuration product (69) obtained with high ee [51].
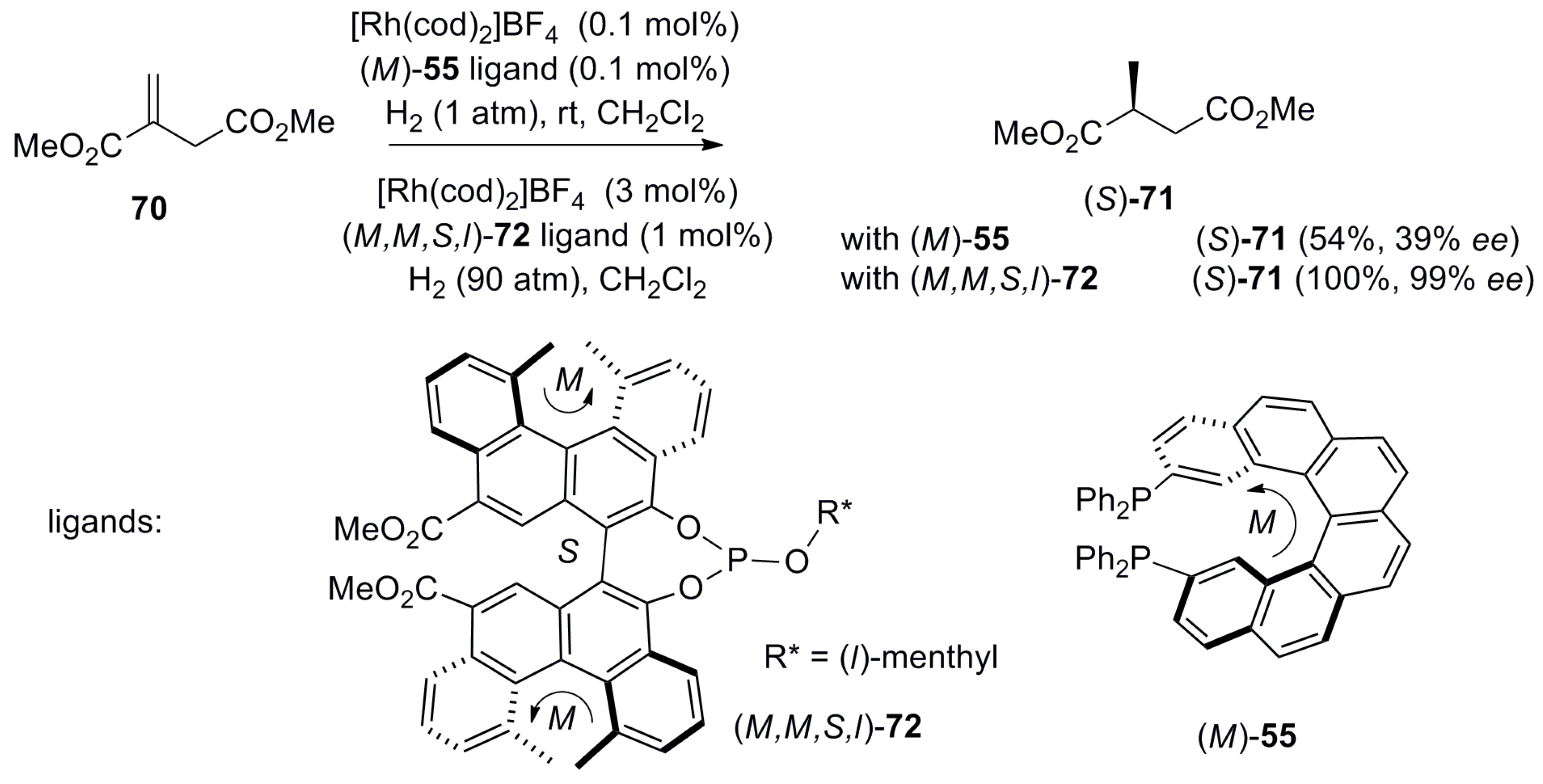
Helicene-based ligands have been also successfully investigated for the rhodium-catalyzed asymmetric hydrogenation of alkenes. In fact, the first enantioselective catalysis using helical diphosphane 55 was the asymmetric reduction of itaconate 70 as a model compound under mild conditions using in situ generated (M)-55 and [Rh(cod)2]+BF4−. This results in the corresponding diester (S)-71 with 54% yield and 39% ee product, as reported by Reetz et al. [54] (Scheme 10). The synthesis of 55 is based on photochemical cyclization with subsequent conversion of the corresponding helicenedibromide derivative to phosphine 55, followed by preparative chiral HPLC separation of the enantiomers [54]. Yamaguchi et al. improved the yield (up to 100%) and enantioselectivity up to 96% ee for (S)-71 using the (M,M,S,l)-configuration of the bis-helicenic phosphite ligand 72 [55] (Scheme 10). The excellent output in this reaction obtained with 72 was attributed to the matched (suitable) pair of asymmetries, i.e., two (M)-helical and one (S)-axial chirality. However, the central chirality derived from the l-menthyl part does not have much effect on the reaction ee outcome.
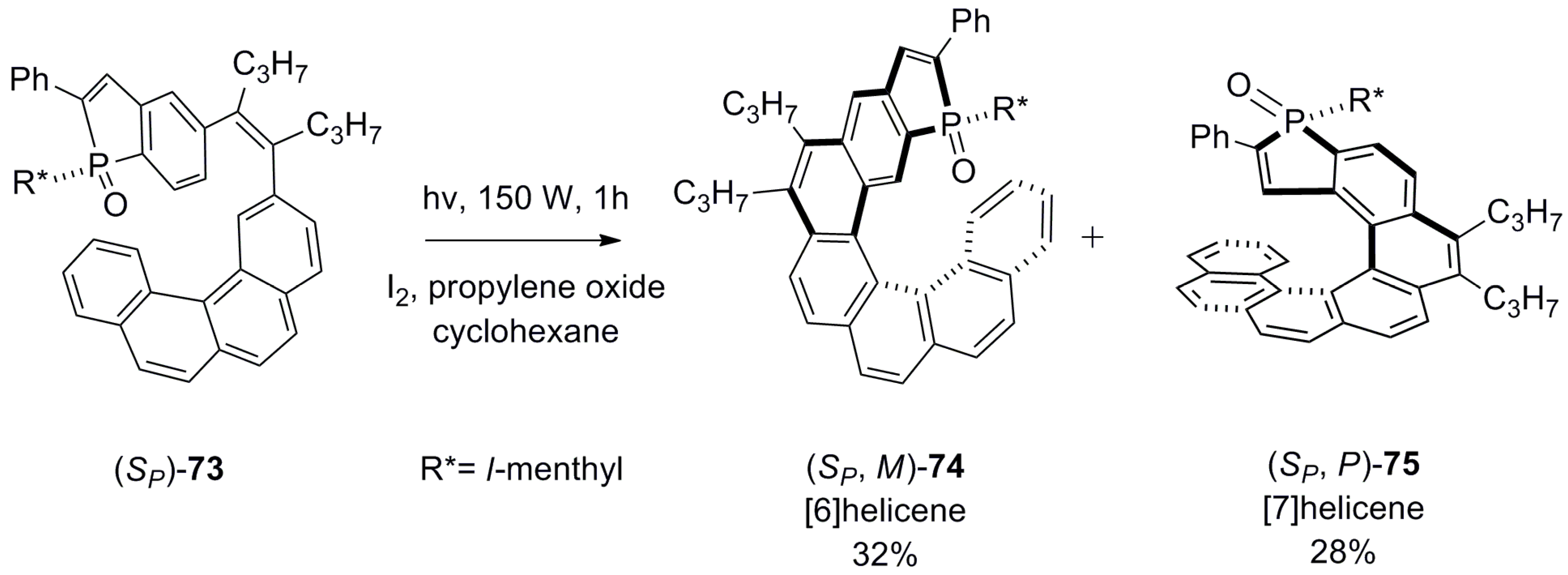
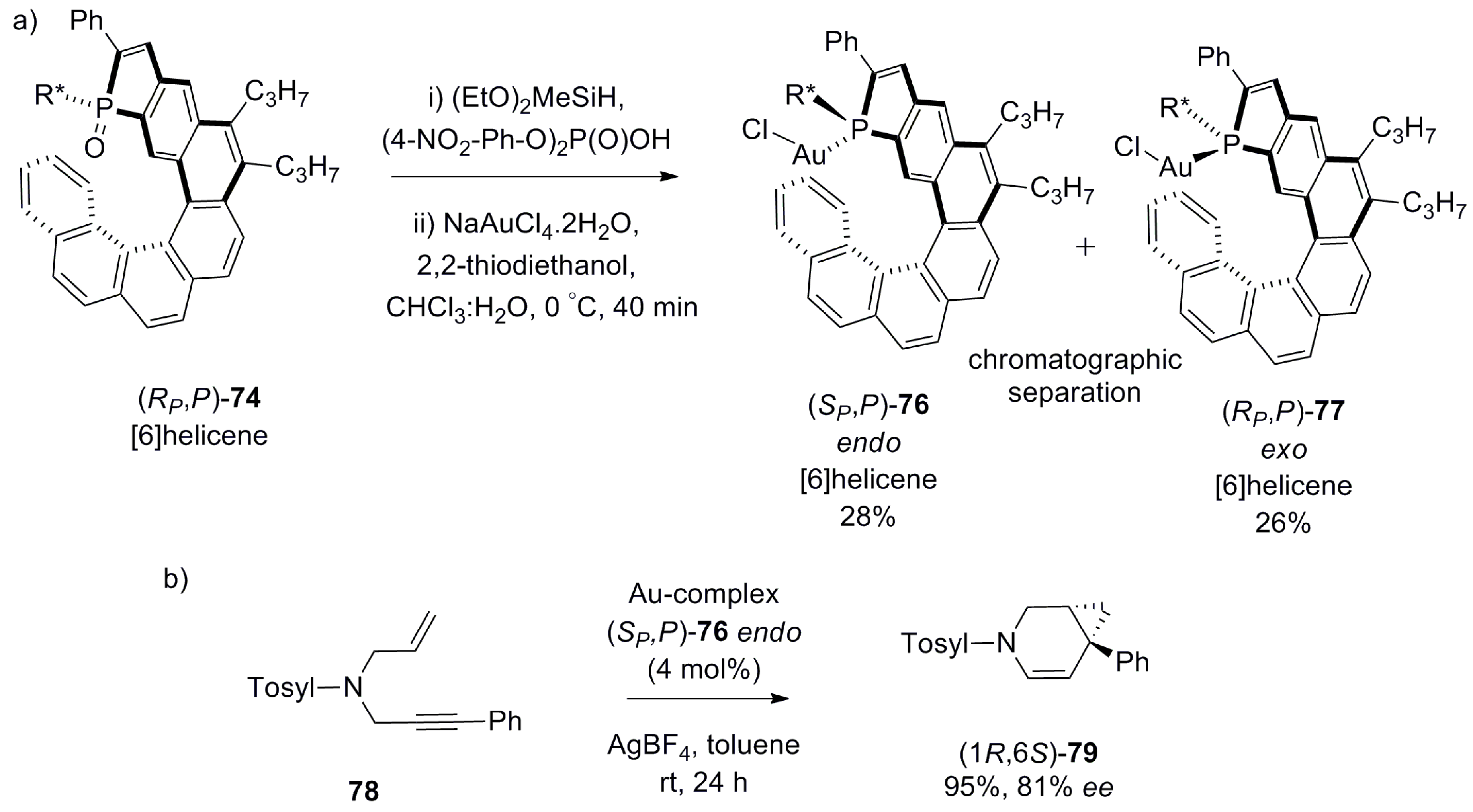
Another approach adopted was to design a helicene with the fused phosphole heterocyclic unit at one of the terminal rings where the phosphorus atom has an additional point chirality, denoted as SP or RP. A key synthetic step includes the diastereoselective photochemical cyclization reaction of a suitable (SP or RP) enantiopure phosphole-bearing stilbene derivative 73. The resulting regioisomeric [6]helicene phosphine oxide 74 and phospha[7]helicene 75 were chromatographically separated with 32% and 28% yield, respectively [56] (Scheme 11).
Subsequently, enantiopure 74 was reduced and in situ reacted with gold(I) salts to generate the phospha-substituted helicene–gold complexes, 76 (endo) and 77 (exo). In endo-76, the gold atom occupies the internal chiral cavity space, approaching towards the opposite terminal ring, whereas in exo-77, the gold atom is situated at the external face, being away from the helical moiety. Thus, chromatographically separated endo-76 effectively catalyzes the enantioselective cycloisomerization of N-tethered 1,6-enyne 78 to give bicyclo[4.1.0]heptane 79 with 95% yield and 81% ee, whilst exo-77 remained almost inactive [56] (Scheme 12). Meanwhile, various phosphole-containing ligands were also prepared and screened in similar reactions with success [8,9,57].
However, at present, the scope of the use of helicenes containing phosphorus functionality appears to be limited towards the metal-catalyzed reactions, as described above. A few other attempted asymmetric reactions such as ketoimine reduction (22% ee), addition of thioester to benzaldehyde (22% ee), and reductive aldol reaction afforded quite low ee values, with tetrathia[6]helicene diphosphine oxides as a Lewis base catalyst [58].
Besides these phosphorus-containing helicenes, heterohelicenes with a nitrogen atom at the innermost 1- or 2-position are also able to provide a chiral cavity suitable for various asymmetric organocatalytic processes, where the nitrogen atom plays a crucial role as a Lewis base or hydrogen-bonding auxiliary. The classical synthetic approach for azahelicenes having a nitrogen atom at the peripheral ring as the pyridine subunit is based on the preparation of the corresponding stilbene derivative followed by the photocyclization reaction.
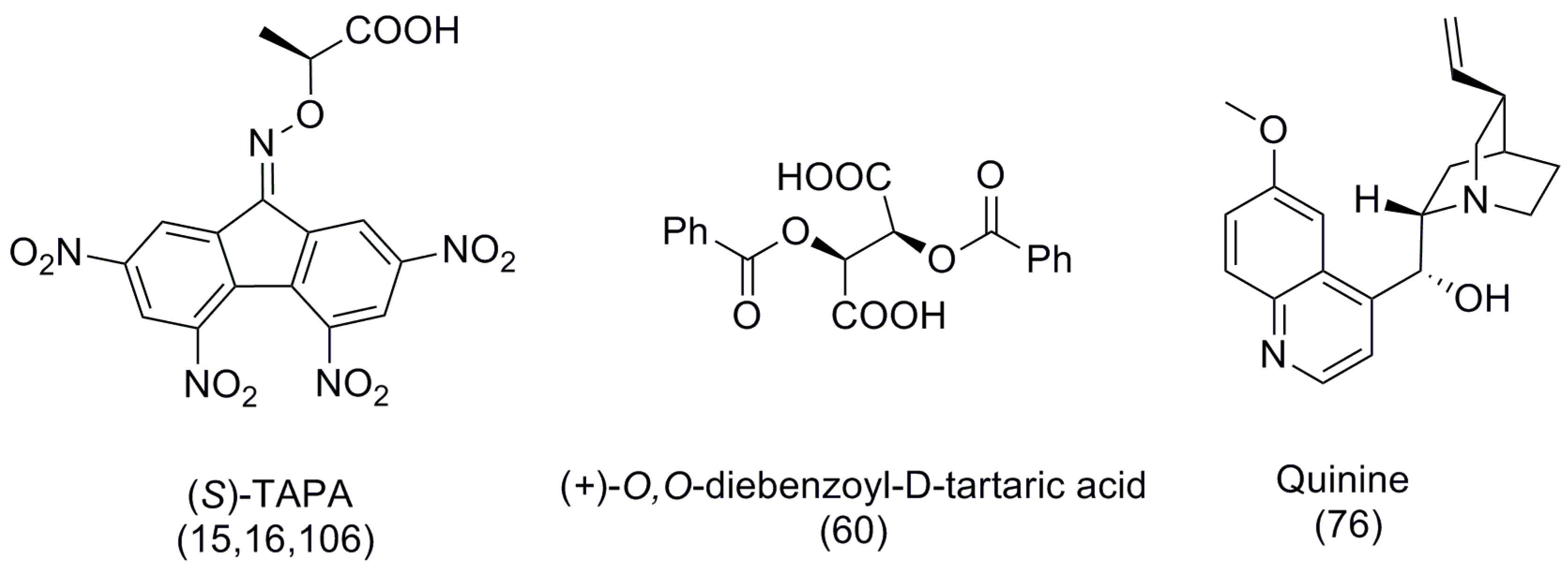
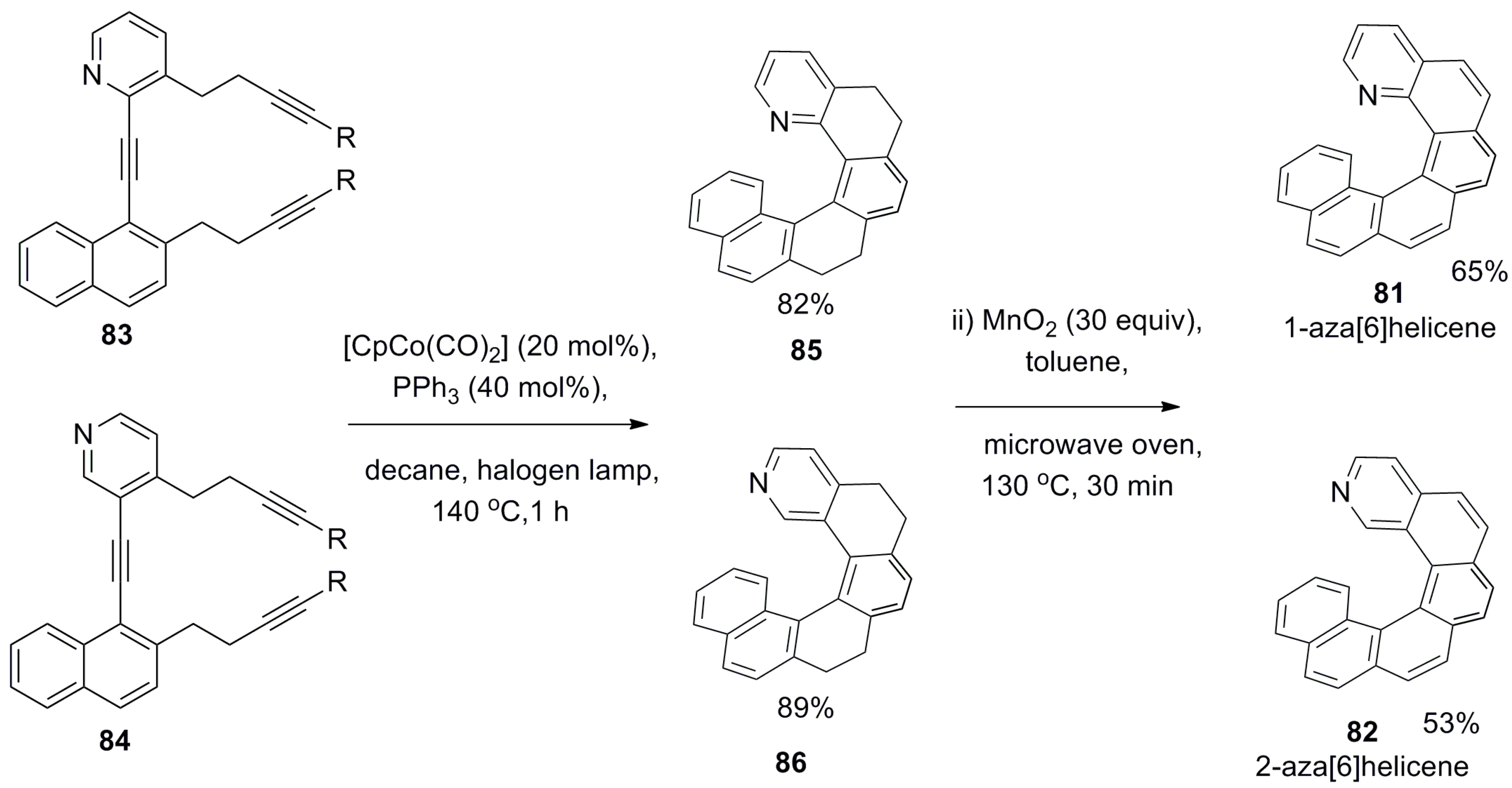
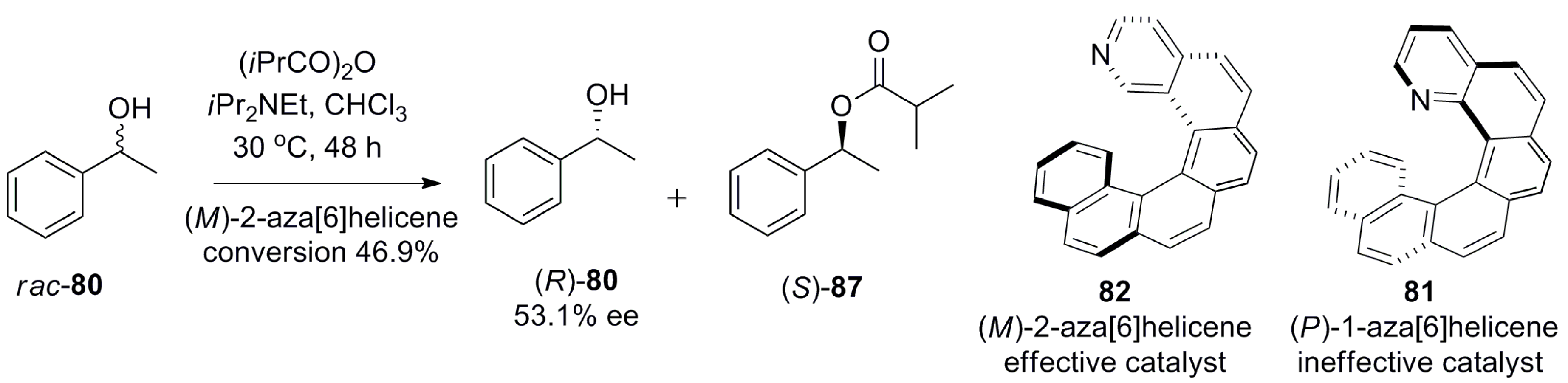
The first use of a pyridine-based helicene as a chiral organocatalyst for promoting the enantioselective acyl group transfer reaction and kinetic resolution of racemic phenylethanol 80 was reported by Stary et al. in 2009 [59]. The preparation of helicenes 81 and 82 [60] involves the [2 + 2 + 2] cyclotrimerization of regioisomeric aromatic triynes 83 and 84 using a Co(I)-catalyzed reaction as the major synthetic step to obtain tetrahydrohelicenes 85 (82%) and 86 (89%), respectively (Scheme 13). Finally, the MnO2-based oxidation resulted in rac aza[6]helicene 81 (65%) and 82 (53%). The optical resolution of helicene 81 was carried out through the diastereomeric salt formation with optically pure (+)-O,O′-dibenzoyl-d-tartaric acid, whereas enantiopure 82 was obtained by separation of the corresponding racemic mixture with chiral HPLC (Scheme 13) [60].
It was found that (+)-(P)-1-aza[6]helicene 81 is an ineffective catalyst for this process with <5% yield, as the nitrogen atom at the most sterically congested position has very limited accessibility for the reactant. However, the isomeric (−)-(M)-2-aza[6]helicene 82, due to the easily accessible nitrogen atom position, considerably improved the catalytic performance with 46.9% conversion to 87 and 53.1% ee for the unreacted starting alcohol 80 (R-configuration) with a selectivity factor of 7 for the corresponding (S)-enantiomer (Scheme 14) [59].
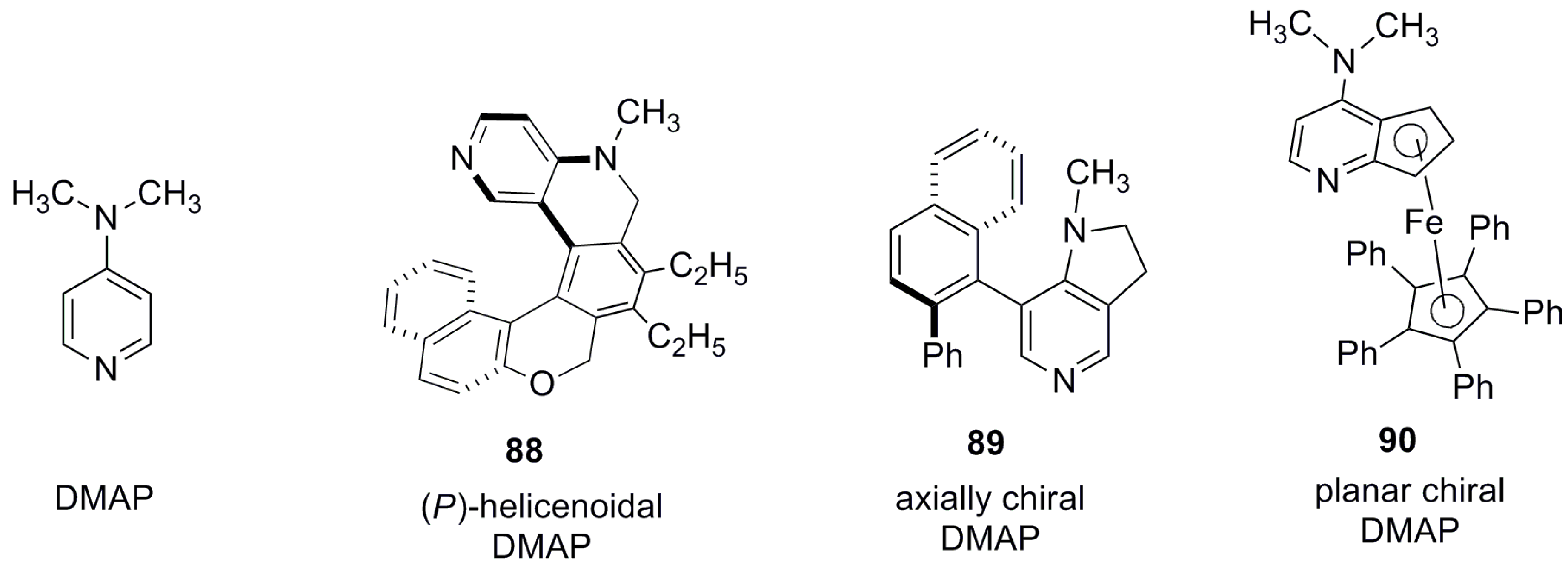
This example clearly demonstrates how the suitably tailor-made design of azahelicenes can provide a useful chiral pyridine-based nucleophilic organocatalyst comparable to other enantiopure amine-containing catalysts, whilst possessing different elements of asymmetry. In comparison to pyridine, dimethylaminopyridine (DMAP) and DMAP-containing compounds 88-90 [61,62,63,64] (Figure 10) are better nucleophilic catalysts for acylation of alcohols due to the enhanced nucleophilicity provided by the electron donating nature of the dimethylamino group at the para position. Thus, (P)-helicenoids 88 containing a p-dialkyl amino substituent turned out to be a more effective enantiodifferentiating catalyst for the resolution of the (S)-enantiomer of racemic 80 with the selectivity factor of 17, in 5 h, as compared to 48 h with a selectivity factor of 7 using (M)-82 as the catalyst.
The helicenoidal DMAP 88 has a nitrogen atom at the 2-position and saturated N-alkyl group, whilst the helicenoid core ensures two additional advantages. Firstly, it enhances the nucleophilicity of the pyridine nitrogen at the 2-position by its electron donating ability (+I effect), and secondly, it increases the conformational flexibility to accommodate reactants inside the helical chiral cavity for efficient enantiodifferentiating processes. The attached ethyl substituents at the central ring of the helical outer core were responsible for increasing the lipophilic character and, thus, the solubility in organic solvents for better performance [61]. The helicenoid DMAP 88 displayed a comparatively higher selectivity factor of 17–116, depending on the structure of the racemic aromatic ethanol. In comparison to 88, the axially chiral DMAP catalyst 89 [62,63] showed selectivity factors of 12–52 (in ether) [62] and 32–95 (in t-amyl alcohol) [63] for enantioresolution of different racemic aromatic ethanols. However, the planar chiral DMAP catalyst 90 [64] displayed the smaller selectivity factor of 8.9–29. It is of note that, with planar chiral DMAP 89, the acylating agent was acetic anhydride, whereas, in other cases, bulkier isobutyric anhydride was used. Thus, the helical DMAP 88 appears to be highly superior to 90 under similar experimental conditions.
Indeed, the first use of unsubstituted aza[6]helicene 81 and 82 as an organocatalyst for the kinetic resolution of alcohols rationalized the corresponding steric constraint imposed at the 1-position for a bulky acyl moiety and its subsequent group transfer reaction to rac-alcohols. However, the additionally functionalized 1-aza[6]helicene was found to be an effective asymmetric catalyst for various reactions [65,66], probably due to the facile access to reactants in the chiral cavity in the transition state, as demonstrated in the following examples.
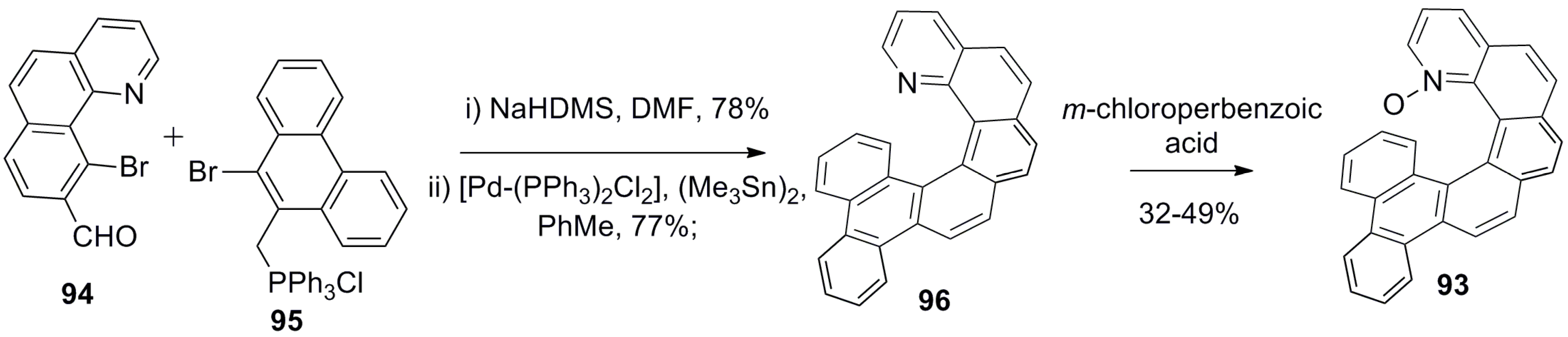
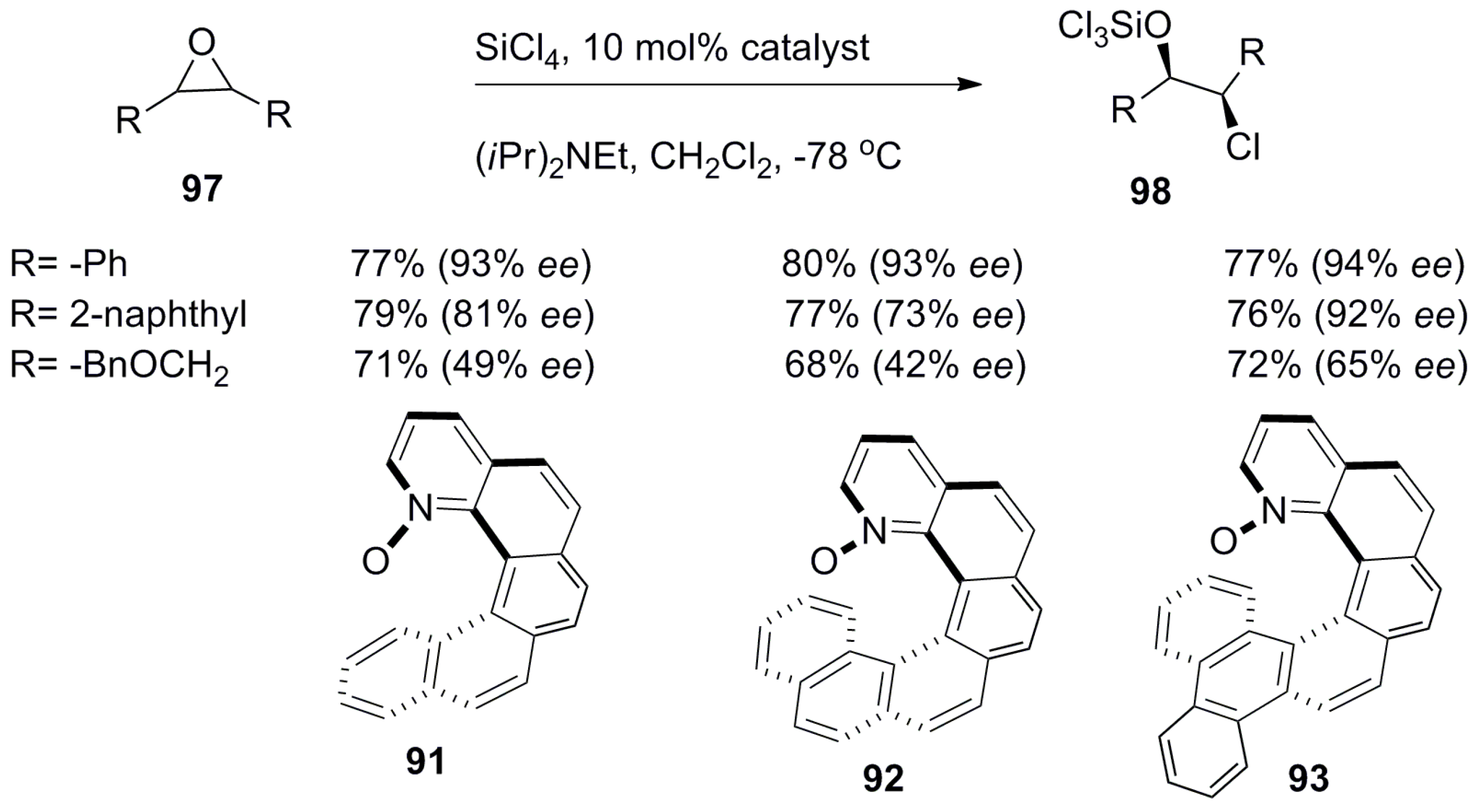
The azahelicene-based systems were converted to the corresponding N-oxides and investigated for the asymmetric ring opening of epoxides with chloride as a nucleophile. For example, 1-aza[6]helicene-N-oxide derivatives 91–93 (Scheme 15 and Scheme 16) [67] were prepared in the racemic form using a general three-step synthetic methodology, and their enantiomers were resolved over chiral HPLC [67]. The representative synthesis of 93 starts from the benzoquinoline aldehyde unit 94 and corresponding bromo-substituted phosphonium salt 95 in three steps, where the initial step involves the Z-selective Wittig condensation followed by the palladium-catalyzed Stille–Kelly coupling reaction to result in azahelicene 96. Finally, oxidation with meta-chloroperbenzoic acid gave the desired helicene-N-oxide 93 with 32–49% yield (Scheme 15).
Then, the enantiopure (P)-helical ligands 91–93 were screened for the desymmetrization of meso epoxide 97 with chloride as a nucleophile to obtain 98 with satisfactory yields and ee (Scheme 16) [67].
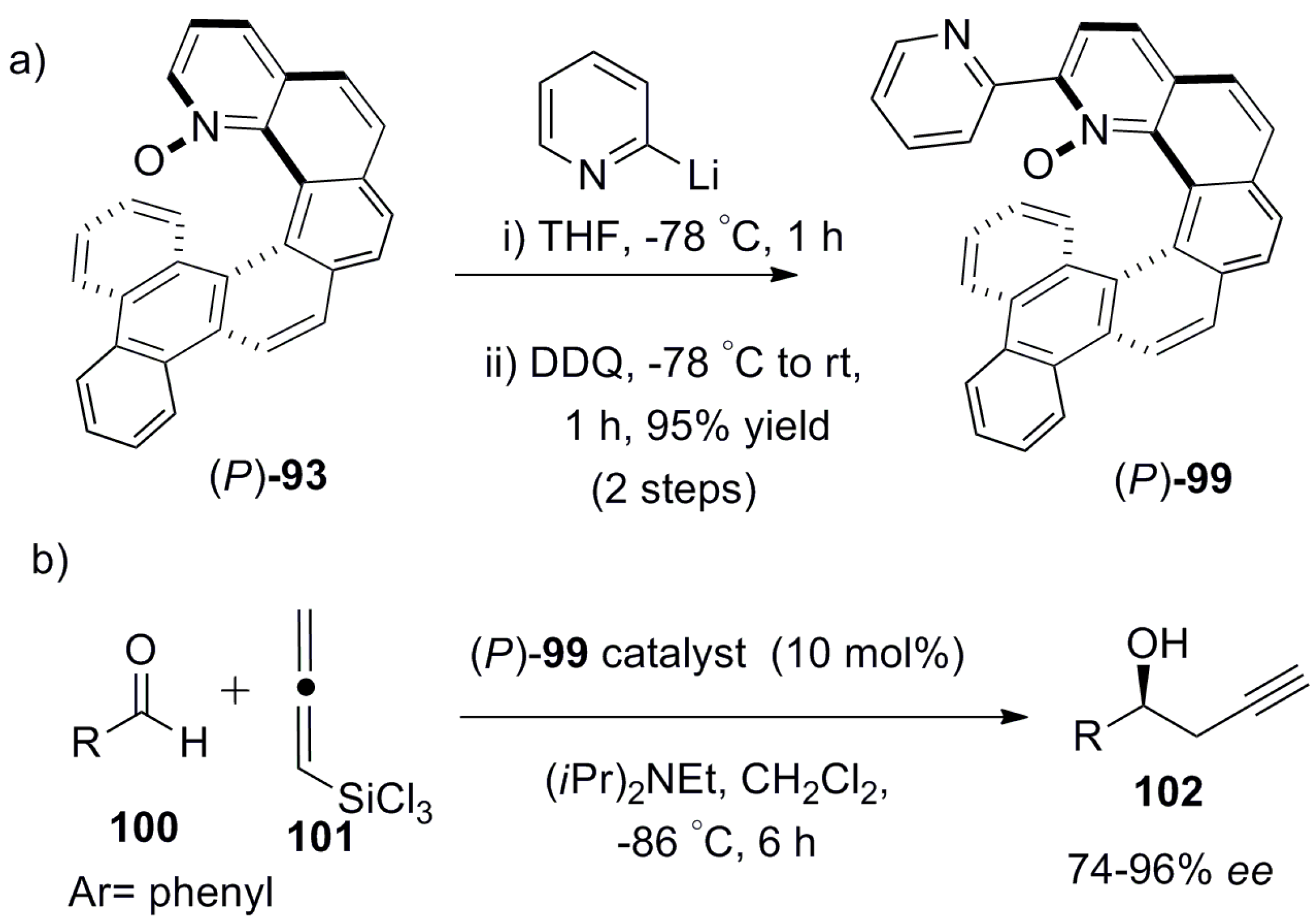
Similarly, enantiopure helical 2,2′-bipyridine-N-monoxide 99 was prepared in two steps from enantiopure 93 with 95% yield (Scheme 17a) and applied as a Lewis base type catalyst for the highly enantioselective reaction between aldehyde, 100, and allenyltrichlorosilane 101 to obtain enantiomerically enriched homopropargylic alcohol 102 [68]. The reaction displayed high conversion (78–97%) and 74–96% ee for various ortho- and para-substituted aromatic aldehyde derivatives, with 18 examples in total (Scheme 17b). Further, the catalyst 99 can be recovered (~80%) without any loss of activity and selectivity after the reaction, making it highly attractive for industrial application.
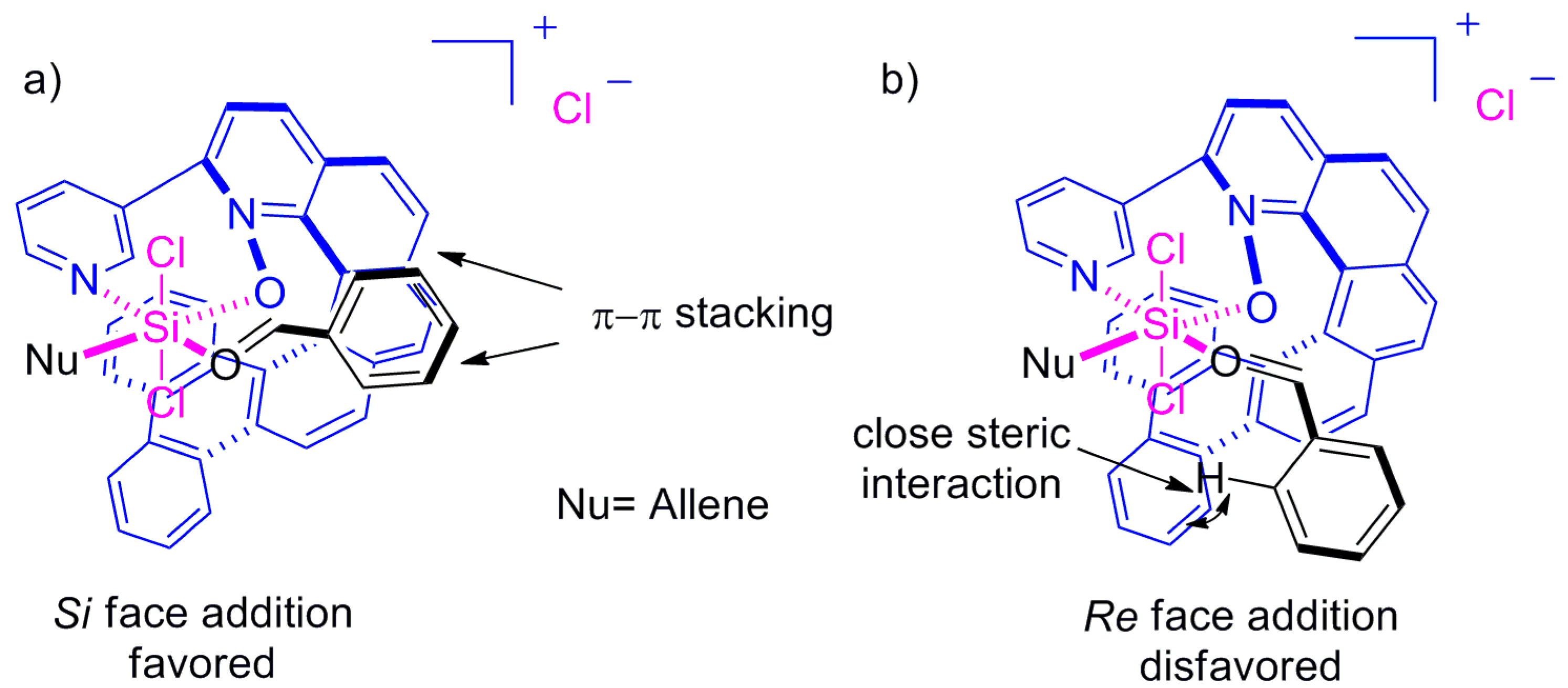
The (S)-alcohol 102 was the major product with (P)-helicene 99 as a catalyst. It was also observed that ortho-substituted aldehydes 100 always provided higher ee than the corresponding para-substituted derivatives, regardless of the functional group attached. This key experimental observation indicated that, out of two possible transition states (Figure 11a,b), in one of them, the hydrogen or substituent at the ortho position of aldehyde 100 results in steric clash with catalyst 99, which is responsible for the stereoselectivity. Thus, the suggested transition state model showed preference for the Si-face addition with π-π stacking between the bound aldehyde 100 and the bezofused helicene framework of 99 (Figure 11a), whereas the Re-face approach is disfavored due to the steric hindrance in the transition state (Figure 11b), where the reactants 100 and 101 and catalyst 99 are coordinated through the silicon atom. A separate DFT-based computational study also supported this mechanism [69].
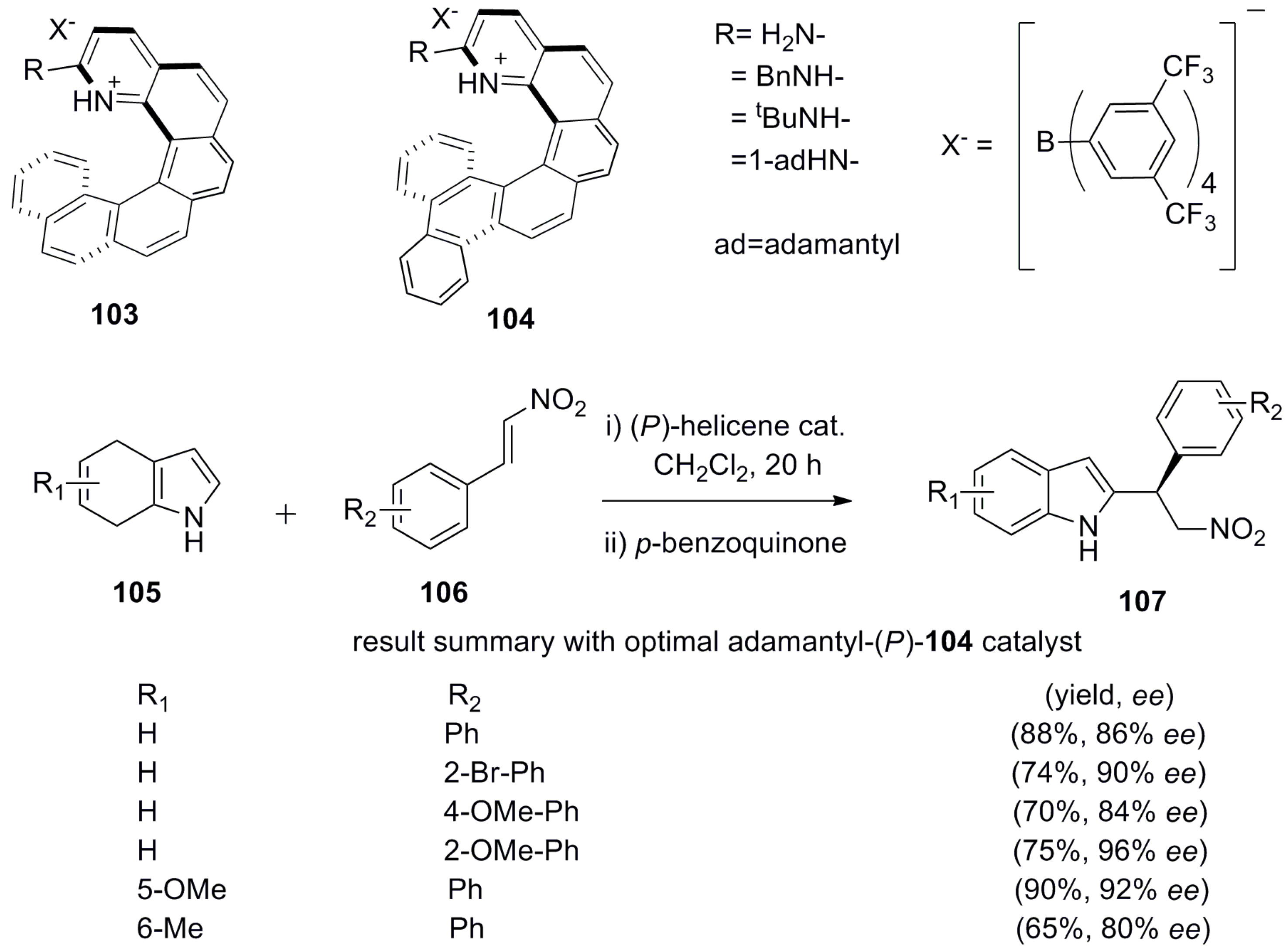
Azahelicenes have also been investigated as hydrogen bond donor chiral catalysts. The asymmetric addition of azole nucleophiles to nitroalkenes using various hydrogen bond catalysts is known. In this respect, aminopyridinium salts are also capable of activating nitroalkenes through the hydrogen bond donating ability to the nitro group. For this purpose, aza[6]helicene derivatives 103 and 104, having a 2-aminopyridinium ring at the peripheral position, have been prepared [70]. Hence, the asymmetric addition of 4,7-dihydroindole 105 to nitrostyrene 106 using chiral (P)-helicenes 103 and 104 as catalysts, followed by oxidative aromatization, afforded exclusively β-nitro-indol-2-yl 107 with relatively high yield and ee (Scheme 18).
It was observed that the increased bulkiness of the amino group gives a higher ee. Thus, the results of (P)-104 in terms of ee ratio were as follows for the corresponding R: H (69:31) = Bn (69:31) < t-Bu (92:8) = 1-adamantyl (92:8). Further, the yields were noticeably improved from 79% (for the t-Bu derivative) to 88% (for the adamantyl derivative) under similar conditions; hence, adamantyl-containing (P)-104 is the optimal organocatalyst for this reaction (see the corresponding results summarized in Scheme 18). Further, the enhanced effectiveness of 104 in comparison with 103 indicates that the benzofused helicene framework affects the ee value by covering the space beneath the two hydrogen bonds [70], and is thus similar to the case shown in Figure 11.
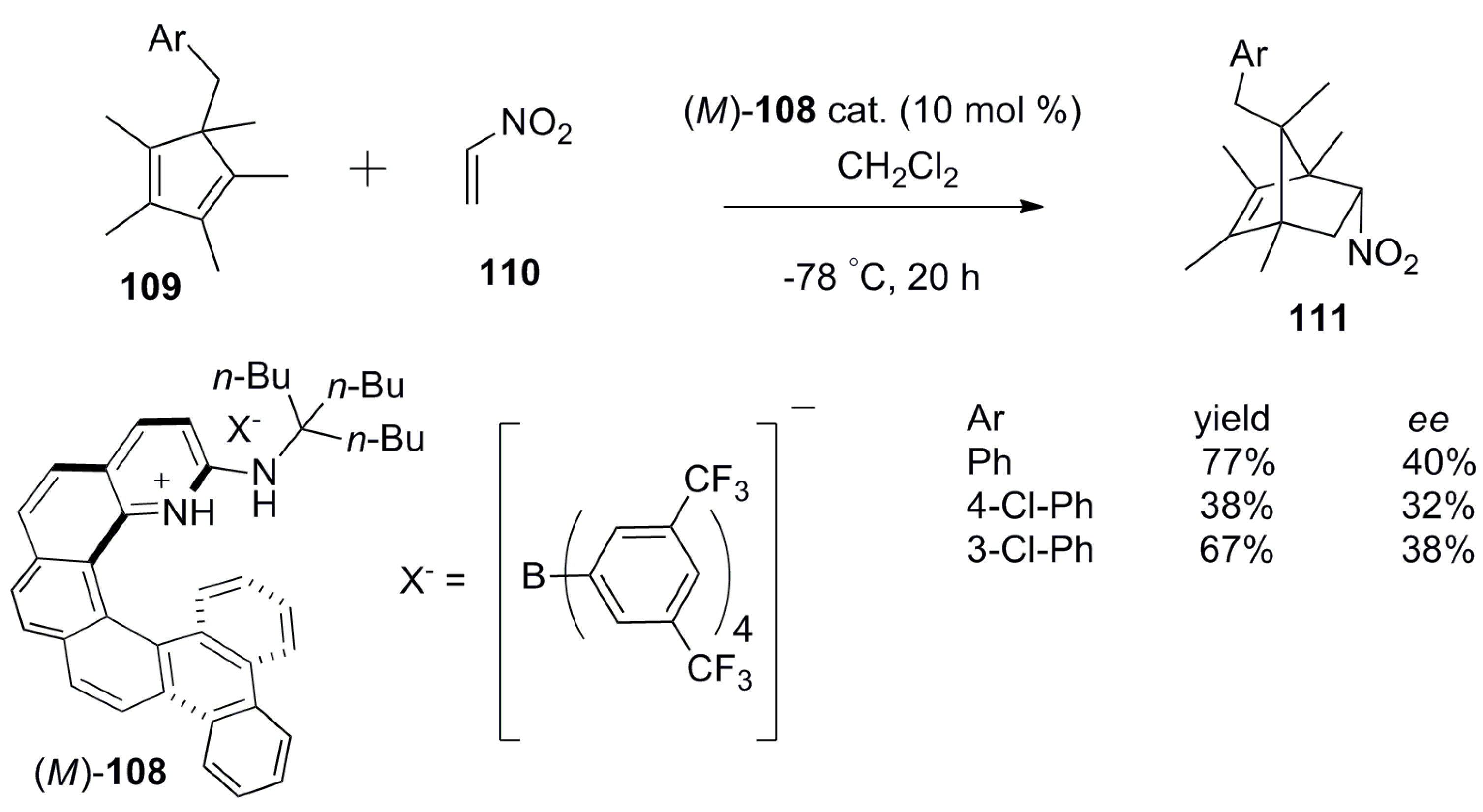
Another example of the activation of nitroalkenes via the hydrogen bonding donor characteristics was demonstrated by using aminopyridinium-based (M)-[6]helicene 108 in the asymmetric Diels–Alder reaction between cyclopentadienes 109 and nitroethylene 110 to give the cycloadduct product 111 in moderate 30–40% ee [71]. A traditional hydrogen bonded catalyst generally requires the complementary donor–acceptor units in both reactants to orient specifically for asymmetric induction in the addition reaction. However, here is a case where one counterpart is a nonfunctionalized cyclopentadiene 109, unsuitable for hydrogen bonding. Therefore, the (M)-helicene catalyst 108 used has a aminopyridinium peripheral ring which only binds to nitroethylene 110 (dienophile) through the corresponding hydrogen bonding. The stereospecificity of this reaction was achieved in the following manner: the helical backbone of (M)-108 blocks one of the faces of 110, leaving another face accessible to 109 (diene), hence making the whole reaction enantioselective in nature (Scheme 19).
Most of the successful examples of azahelicenes (91–93, 99, 103, 104, 108) as organocatalysts described here have a nitrogen atom at the 1-position of the innermost part of the helicene with additional substituents at the 1- or 2-position to modify the reactivity as well as the helicity (chiral cavity space), to facilitate accessibility of the reactant in a stereospecific manner, and to enhance asymmetric output.
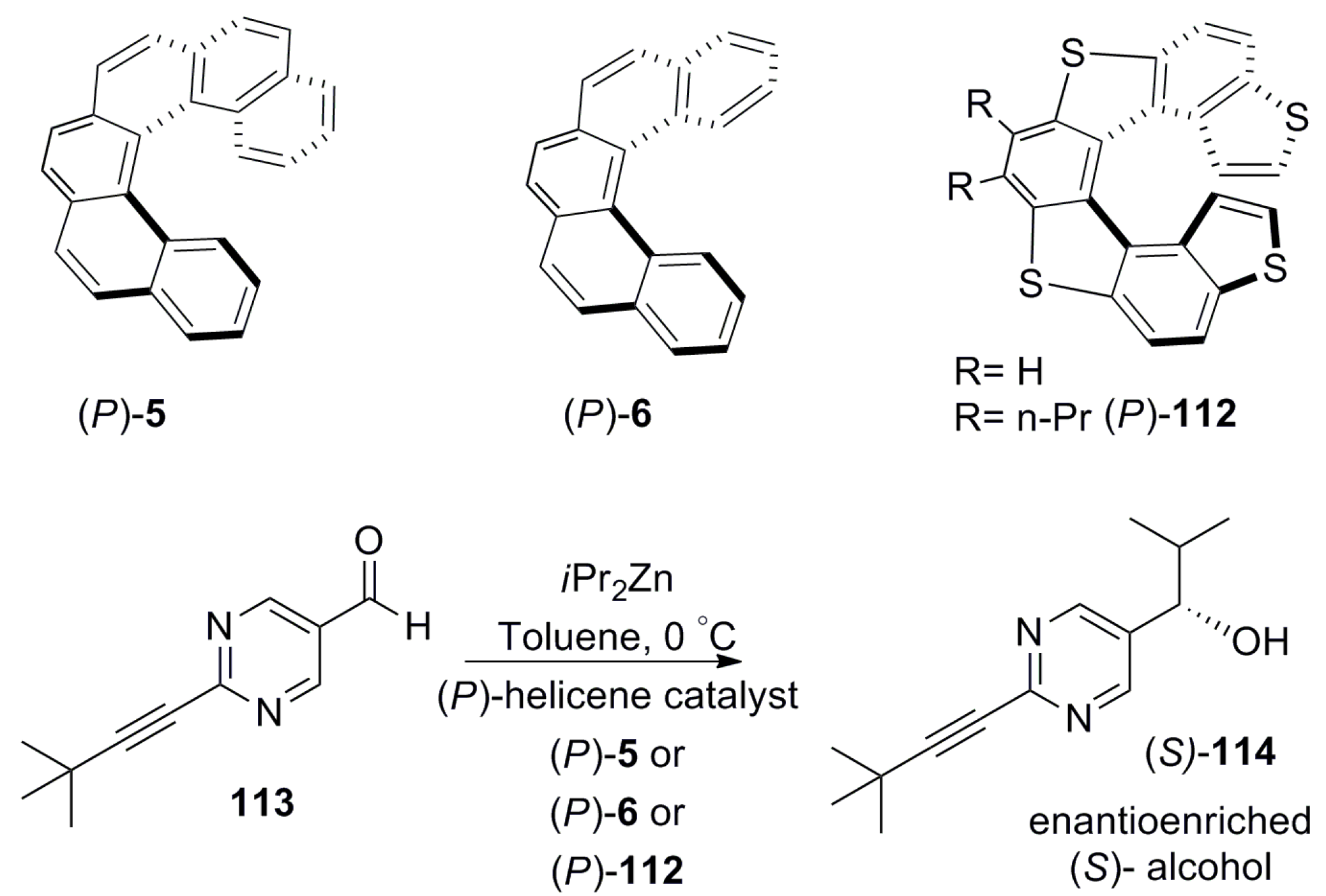
Carbohelicenes 5 and 6 and heterohelicene 112, devoid of any functional group, are also able to act as chirality inducers in selected asymmetric reactions. Soai et al. have successfully used electron-rich nonfunctionalized carbohelicenes 5 and 6 [72] and heterohelicene 112 [73] for the nucleophilic addition of diisopropylzinc to electron-deficient pyrimidine-5-carbaldehyde 113. In this case, the chirality induction was a result of two main reasons. Firstly, the formation of a charge transfer complex between helicenes 5, 6, and 112 and the carbonyl group of 113 results in the blocking of one reactive face while another face is left to react. Secondly, the final alcohol product 114 obtained can catalyze this reaction by itself, leading to overall chirality amplification to yield up to 95–99% ee (Scheme 20). In all these cases, the use of (P)-enantiomers of 5, 6, and 112 as catalysts resulted in the (S)-configuration of 114, whereas the (M)-enantiomer produced the corresponding R-configuration. Further, even carbohelicenes 5 and 6 with a very low ee such as 0.13% and 0.54% were also able to act as chiral inducers, ensuring considerable stereospecificity and affording 56% and 60% ee of 114, respectively, as a result of chirality amplification by asymmetric autocatalysis [72].
This is an interesting example where chiral inducers with a very low ee generate significantly enhanced ee of the product. This type of chiral amplification is a “soldiers-and-sergeant” principle and often observable in the field of supramolecular chirogenesis [1]. However, this scope appears to be limited in the case of asymmetric autocatalysis.
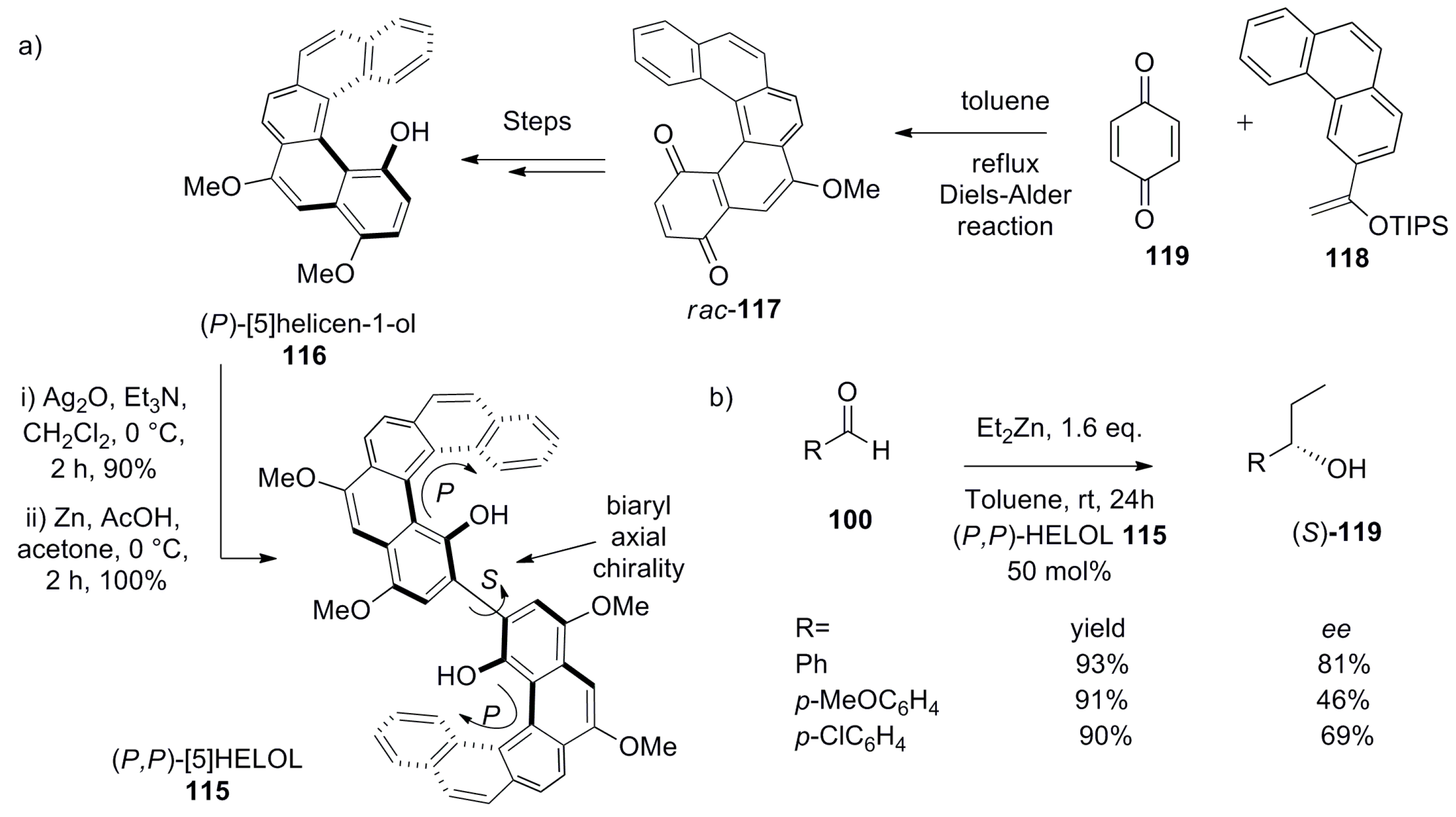
Indeed, the asymmetric nucleophilic addition of an organozinc compound to the carbonyl group has been mainly investigated with the use of biaryl-based systems such as BINOL and 2,2′-Diphenyl-(4-biphenanthrol)—axially chiral diol systems capable of coordinating to Lewis acids. Inspired by this, Katz et al. developed the bis[5]helicene-based diol system ([5]HELOL) 115, which contains two identical units of (P)-[5]helicene-1-ol 116 connected by a single biaryl bond at the 2-position. Interestingly, the hydroxyl group is attached at the 1-position (the innermost position of [5]helicene), which makes 116 configurationally stable, and hence provides increased steric hindrance for racemization. The HELOL 115 was synthesized in a total of eight steps on the multigram scale with an overall 44% yield (Scheme 21a) [74]. The helicene backbone 117 was synthesized by Diels–Alder reaction between protected enol ether 118 and quinone 119. The enantiopure (P)-[5]helicene-1-ol 116 was oxidatively coupled using Ag2O, to selectively give (P,P)-[5]HELOL 115, with only ~2% of the meso isomer. The energy barrier for the racemization of 115 (ΔH‡ = 106 ± 3 kJ mol−1 and ΔS‡ = −10.0 ± 0.1 J mol−1 K−1) as determined by 1H NMR was found to be even higher than that of (P)-[5]helicene 116 (ΔH‡ = 95.8 kJ mol−1 and ΔS‡ = −17.2 J mol−1 K−1) [74] with the optical rotation value of +2580 in acetonitrile (c = 0.01 M). Further, the thermal racemization at 45 °C did not occur in 72 h as indicated by circular dichroism (CD) spectroscopy. Additionally, in degassed acetonitrile solution, it was configurationally stable for six days at room temperature as also analyzed by CD measurement. The enantiomeric stability toward conversion to the meso isomer and racemization made it a suitable candidate for screening in asymmetric catalysis.
The catalyst 115 was screened for diethylzinc addition to a series of aromatic aldehydes 100. However, for appreciable catalytic activity, catalyst loading as high as 50 mol% was needed to obtain moderate to good ee values of the alcohol products (S)-119 (Scheme 21b). Under similar conditions, the (R)-(+)-BINOL gave lower yield (56%) and ee (34%) for (R)-119 in comparison to (P,P)-(+)-HELOL 115 giving (S)-119 (93% yield, 81% ee). This high and opposite stereospecificity is apparently due to a greater steric hindrance of HELOL 115 in comparison to BINOL upon chelation of the zinc atom of the reactant diethyl zinc. Additionally, this crowding in 115 is also able to prevent the phenomenon of zinc–oxygen coordination-based aggregation, responsible for catalyst deactivation, making 115 comparatively more reactive than BINOL [74]. Further, the opposite stereochemical outcome of alcohol products 119 obtained with (P,P)-(+)-[5]HELOL 115 as a catalyst and (R)-(+)-BINOL as catalyst is in accordance with the stereochemistry of the biaryl bond present in HELOL 115, which appears to be of (S)-configuration (see structure 115 in Scheme 21a).
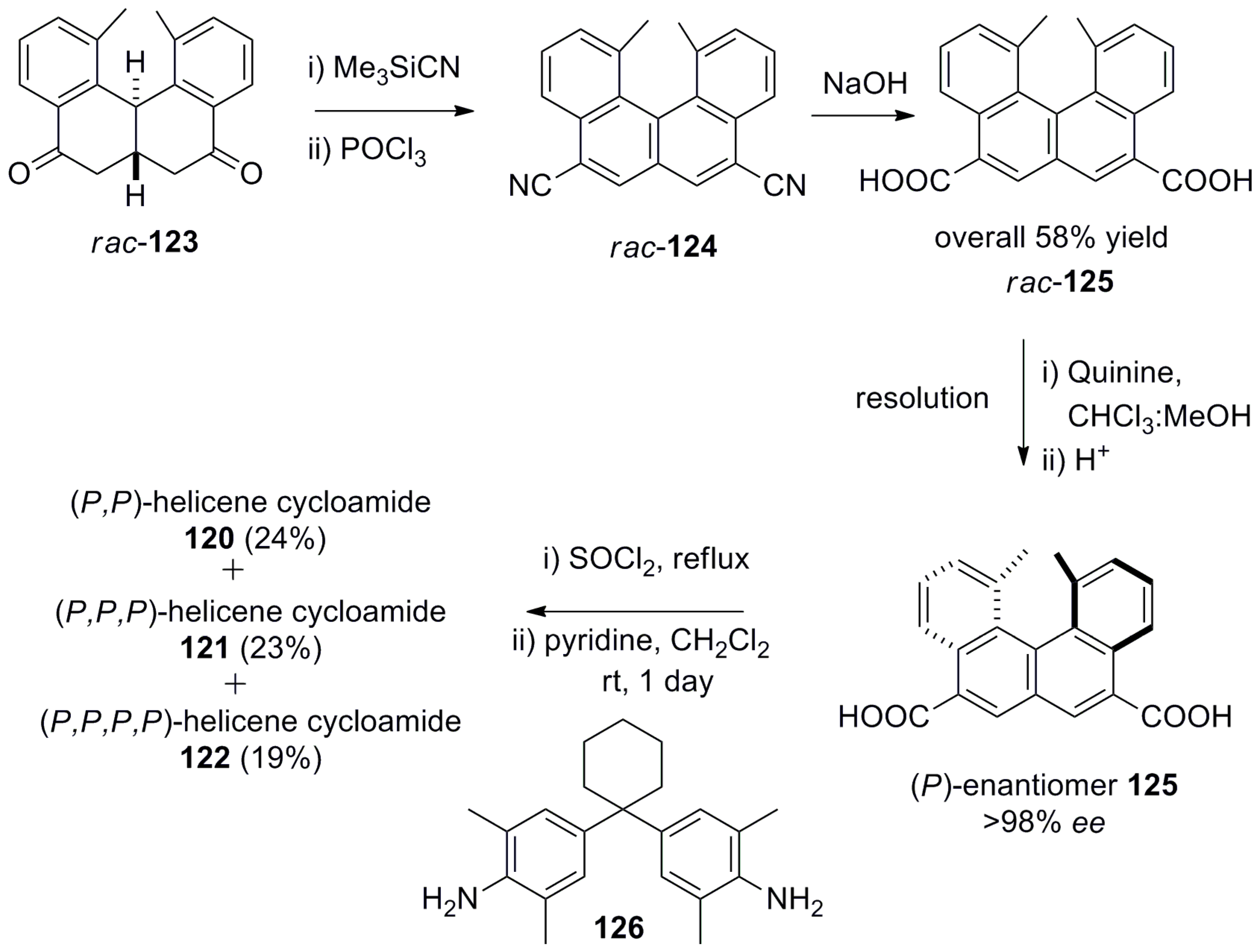
Whilst unsubstituted [4]helicene 4 is a planar and achiral structure, the introduction of two methyl groups on both the peripheral rings at the innermost positions gives rise to a helical structure with configurational stability. In this respect, Yamaguchi et al. have extensively explored the synthetic chemistry and applications of functionalized dimethyl[4]helicene in different fields [75]. Thus, (P,P)-[4]helicene cycloamides 120–122 were prepared from well-known rac-diketone 123 via dicyanohelicene 124, which was converted to helicene diacid 125 by alkaline hydrolysis. Then, helicene diaicid 125 was resolved by using the chiral base quinine through diastereomeric salt formation. Finally, (P)-125 was converted to acid chloride and then coupled with a suitable diamine linker 126 to obtain cycloamides dimer 120 (24%), trimer 121 (23%), and tetramer 122 (19%) (Scheme 22) [76].
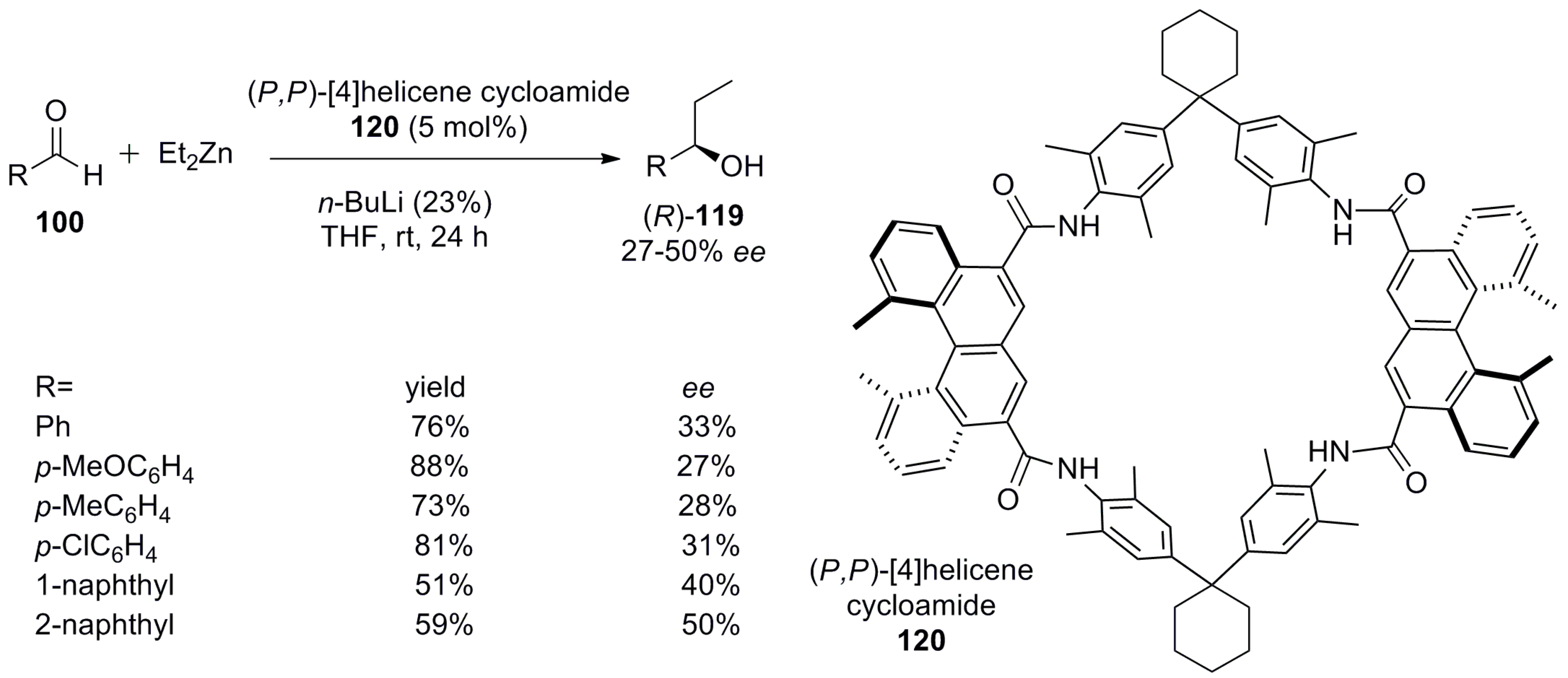
Interestingly, in this example, the functionalized amino groups are present at the outer core of the helicene but not at the helical chiral cavity as in previous examples; this makes it remarkably different. Yet, when it is dimerized with a suitable linker such as cycloamide 120, the chirality of the helicene is transferred to a macrocycloamide, which essentially provides a chiral pocket for the asymmetric catalytic transformations. Macrocycloamides such as (P,P)-120 have been applied for the asymmetric nucleophilic addition of diethyl zinc to aromatic aldehydes 100 with good yields (51–88%) and moderate enantioselectivities (27–50% ee) upon use of up to 5 mol % catalyst (Scheme 23) [76].
It has been clearly demonstrated that helicene-based asymmetric organocatalysis is a highly promising and exciting field in terms of the enantioselectivity obtained with the lowest catalyst loading. Thus, this area of chemistry has exciting prospects to explore other well-known asymmetric reactions by using helicenes as organocatalysts, with foreseen advances in terms of the ee values in comparison to conventional organocatalysts. For the most asymmetric organocatalysts, it is possible to design and develop helicene-based helical counterparts which are expected to give improved reactivity and stereoselectivity. However, the design of smart catalysts and their multigram synthesis in the enantiopure form still remain a major challenge to further investigations.
7. Helicene in Supramolecular Chirogenesis
Besides asymmetric catalysis, helicenes are efficiently used in a modern branch of chiral science called supramolecular chirogenesis, which covers all aspects of asymmetry induction, transfer, amplification, and modulation through noncovalent interactions. In numerous chromophoric systems [1] available to date, helicenes are also successfully employed. Indeed, helicenes have turned out to be one of the most attractive chromophores, which are suitable for several chirogenic applications including chiral recognition, sensors, and self-assembly, and are applicable to various areas ranging from classical solution chemistry, nanoparticles, and polymers, up to the sensing of biologically relevant molecules. Owing to their conjugated aromaticity, these chromophoric systems can be conveniently studied by diverse spectroscopic methods, such as UV, fluorescence, NMR, CD, etc., which are described below with selected examples.
7.1. Helicene-Based Chiral Recognition of Small Molecules
Since chirality is an essential part of life on Earth, its recognition and sensing play a vital role in such important fields of human activity as scientific research and modern technologies. Therefore, various methods of obtaining enantiopure molecules by optical resolution and by asymmetric synthesis are attractive areas of chemical research. Besides preparation, the analysis of all types of chiral molecules is also an equally important part of physical–organic chemistry. So far, existing analytical methods are based on the enantiodifferentiation procedure conventionally performed by using chiral HPLC, NMR, fluorescence, and CD techniques. In this respect, chiral helicenes are highly prospective host molecules to serve as specific chirality sensors.
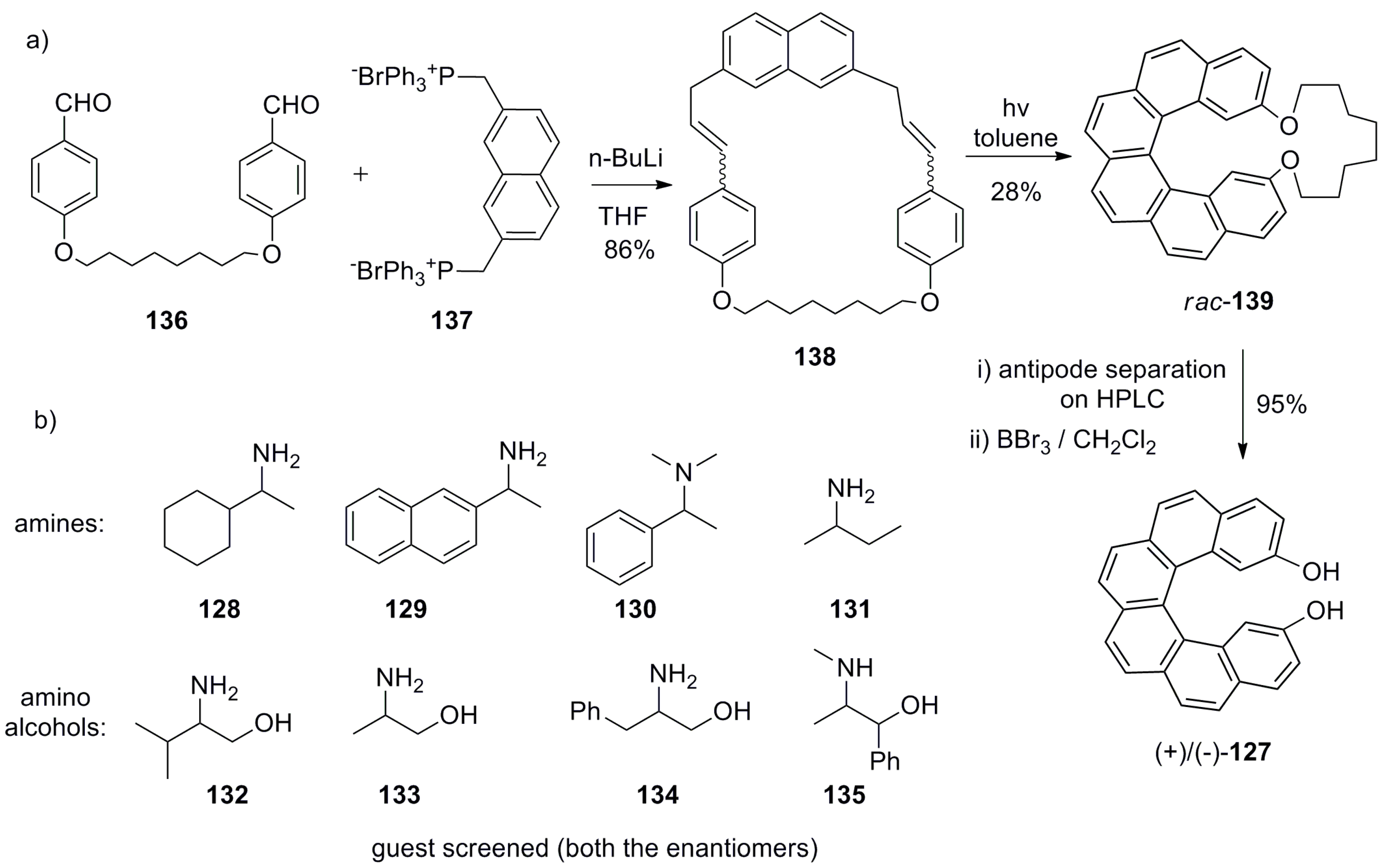
One of the first reports of such an application was published in 2001 by Reetz et al. [77]. To this end, 2,5-dihydroxy[6]helicene 127 (HELIXOL) was prepared for chiral recognition of amines 128–131 and amino alcohols 132–135 by using fluorescence as a detection tool (Scheme 24). The synthetic strategy includes classical Wittig olefination and photochemical cyclization steps. The condensation of 136 with 137 gives cyclic 138, ether linkage of which ensures structural control during the double photocyclization reaction to obtain helicene cyclophane 139. The chiral HPLC separation of racemic 139, followed by the deprotection of the ether group using BBr3, affords enantiopure HELIXOL 127 (Scheme 24).
It was found that the host 127 served as an excellent fluorescent sensor, able to display enantioselective quenching by several amines and amino alcohols through the hydrogen bonding interactions. The best results were obtained for alaninol 133 with enantioselective factor α = KR/KS = 2.1 upon using the levorotary (M)-127. Although the results obtained with 127 for chirality sensing are moderate, this work is a pioneering contribution for the use of helicenes for the purpose of chirality sensing. Further studies have demonstrated that this new, emerging area is challenging and highly prospective in chemical sciences.
Meanwhile, HELIXOL 127 is a bidentate compound with two hydrogen bonding units (phenolic groups) in the chiral space cavity, which can be used for stereodiscriminating recognition (Figure 8). Thus, it was assumed that amino alcohols may form the corresponding 1:1 host–guest complex, resulting in stronger fluorescence quenching than amines. Indeed, the best result among amines and amino alcohols was obtained for alaninol 133 due to the optimal steric matching in the chiral space available, while displaying the strongest fluorescence quenching output.
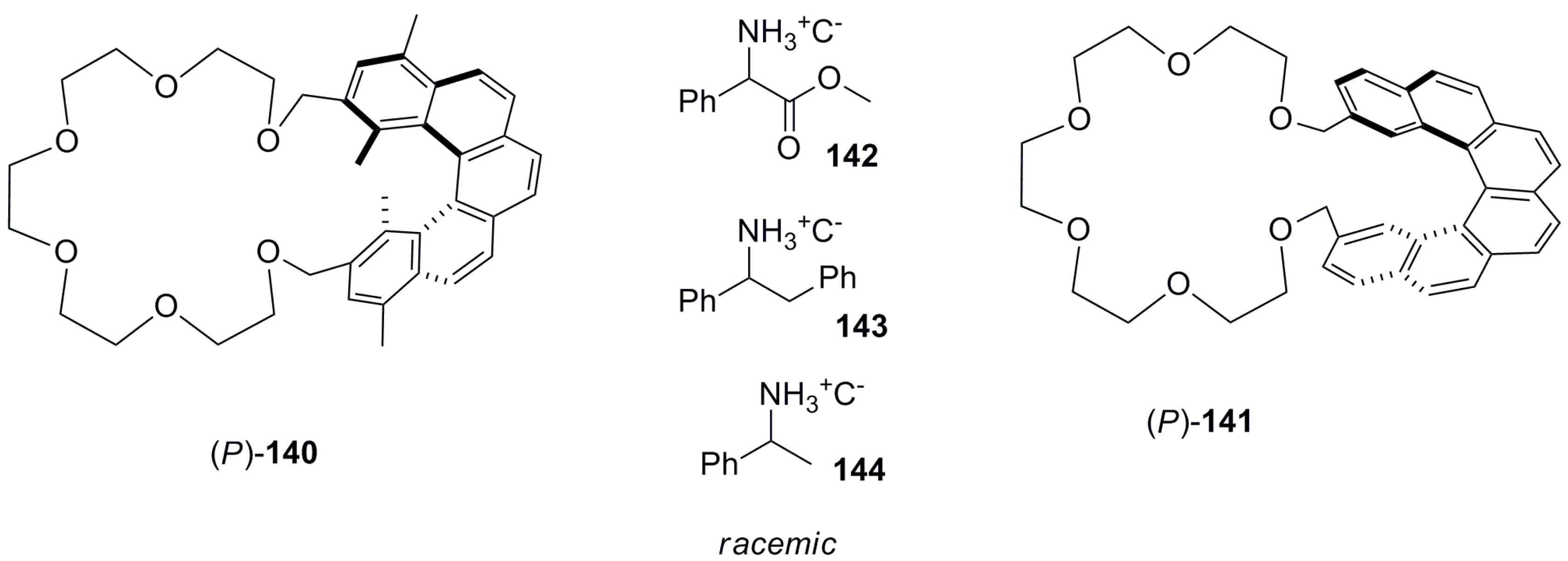
Another approach was demonstrated by the application of crown ether-based systems, which are known to bind various cations ranging from metal ions to organic ammonium ions. Hence, the helicene-based crown ethers 140 and 141 were firstly prepared in 1983 by using 1,14-dimethyl pentahelicene and heptahelicene backbones [78,79]. The synthesis was performed by a classical strategy via Wittig condensation followed by photochemical cyclization. Finally, the construction of crown ethers and subsequent resolution of enantiomers using a chiral column packed with (+)-poly-(triphenylmethyl methacrylate) with methanol as an eluting solvent resulted in enantiomers of 140 and 141. Then, enantiopure 140 and 141 were investigated towards application in the chiral discrimination of racemic amine salts 142–144 (Figure 12). This process was based on the selective differential transports of recognizable enantiomers from an aqueous solution to the corresponding organic solvent (CHCl3) by a liquid–liquid extraction procedure. The 1H NMR analysis of the extracted organic layer displayed two distinct –CO2Me signals of the representing diastereoisomeric complexes with the guest, 142. Finally, the amount of transported guest was analyzed by a reverse extraction back to an aqueous acidic phase followed by the measurement of its UV absorption.
It was observed that the [5]helicene-based crown ether 140 showed higher enantioselective extracting ability than the [6]helicene-based crown ether 141. For example, (M)-140 was able to transport 6% of (S)-142, with optical purity of 75% in 6 h, comparatively better than (M)-141 with 2% transport of 26% optical purity of opposite enantiomer (R)-142 in the same time. The observed opposite enantioselectivity extraction using helicenes 140 and 141 with the same helical sense was rationalized by the phenomenon of changing the crown ether conformation [80]; this is evident in their CPK models. Additionally, the inner methyl groups also assist in the chiral recognition process, as suggested by the same models [78,79].
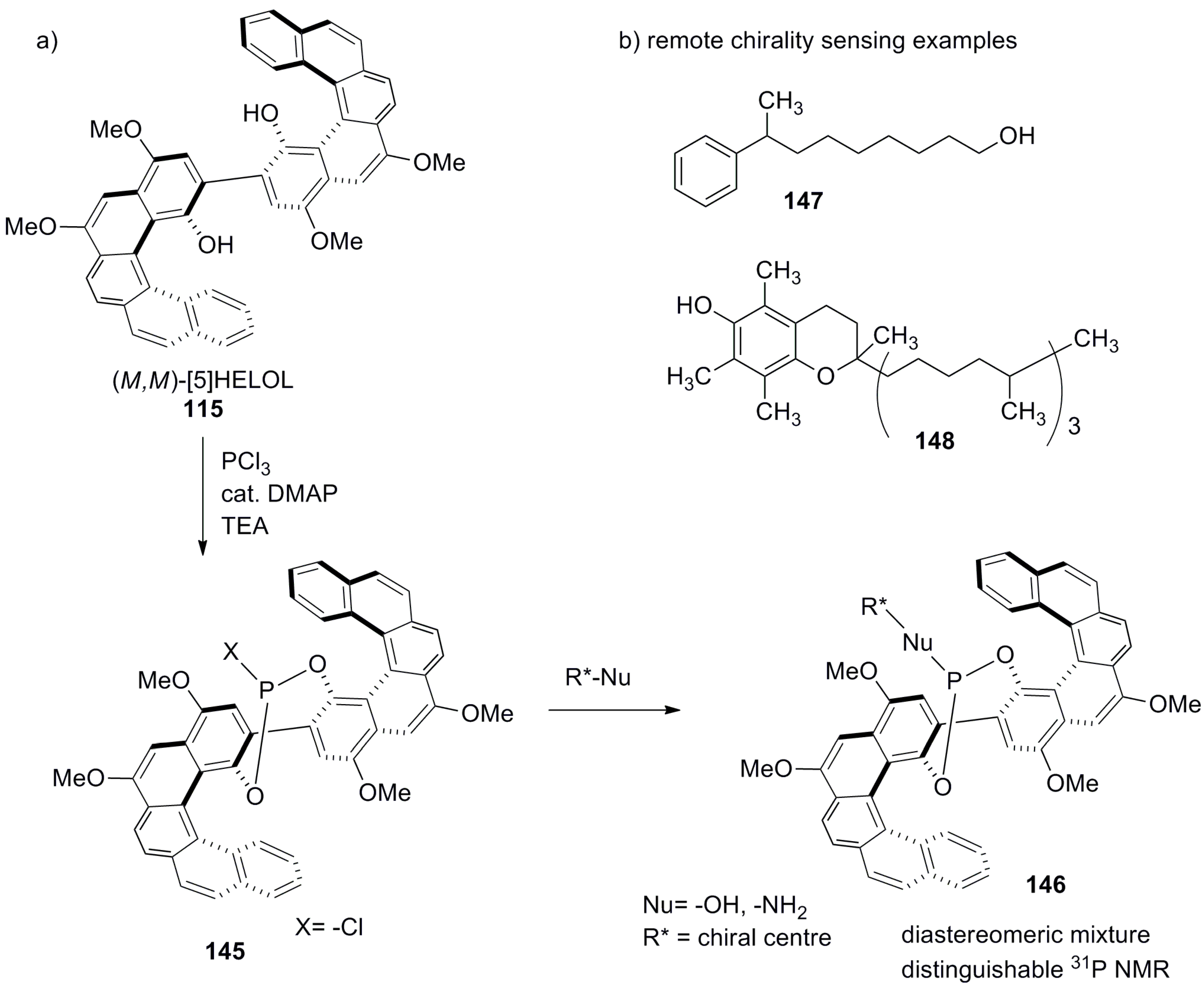
Another approach to chiral recognition was undertaken by Katz et al. via developing the helicene-based biaryl diol compound named (M,M)-[5]HELOL, 115 [81], which was based on enantiopure 1-hydroxy substituted [5]helicene (see 116 in Scheme 20). The in situ generated chloro phosphite derivative 145 was used as a chiral NMR derivatizing agent to discriminate alcohols or amines serving as nucleophilic guests. From the 31P NMR analysis of diastereomeric mixture 146, the enantiomeric excess of guests (alcohols or amines) was easily determined. An interesting feature of this sensor is its ability to sense a remote chirality, which locates far away from the phosphite group. This is related to the fact that the combined inner cavity formed by individual helicene units and the biaryl bond yields an extended chiral space in 115. For example, this chiral space is able to recognize the asymmetric centers of 8-phenylnonanol 147 (as far as up to seven methylene units) and vitamin E 148 (Scheme 25). Additionally, the recognition ability of this sensor was related to 100% abundance of phosphorus as NMR active nuclei with high sensitivity and accuracy in measurement; thus, the enantiomeric excess of up to 3% can be easily analyzed [81]. The scope of this HELOL host and its structural analogue were further extended towards other nucleophiles such as alcohols, phenols, amines, and carboxylic acids, also containing a remote chirality, under similar methodology to the 31P NMR-based enantiodiscrimination [82].
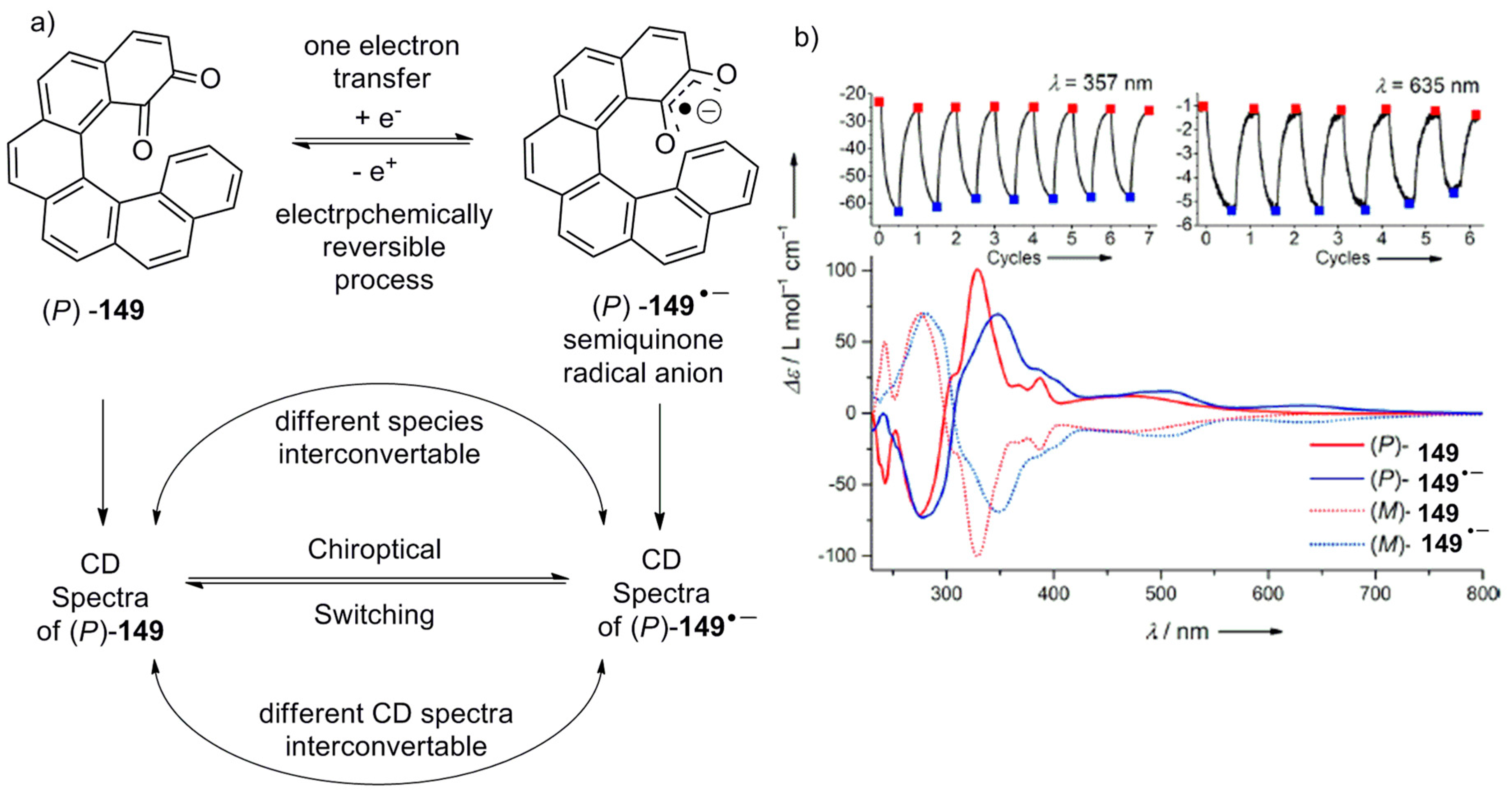
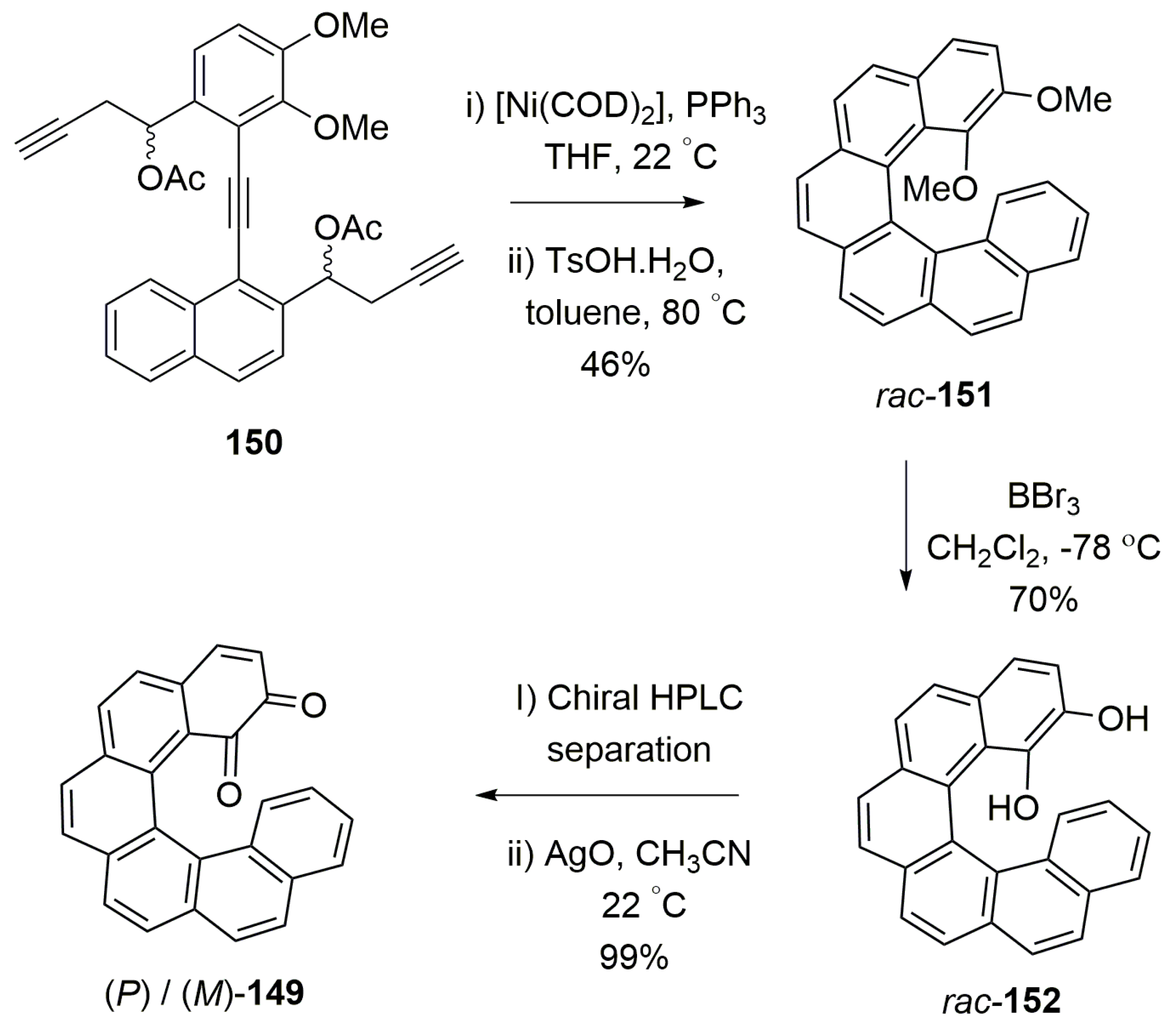
A new chiral application as a chiroptical switch along with chiral recognition properties was demonstrated using enantiomerically pure [6]helicene o-quinone 149. The synthesis was carried out by using the Nickel-catalyzed [2 + 2 + 2] cyclotrimerization reaction of triyne 150, followed by the deprotection of the methoxy group in 151 to result in helicene diol 152 in a racemic form. The enantiomers of 152 were separated on chiral HPLC, followed by the silver oxide-mediated oxidation, to give the quinone 149 in quantitative yield (Scheme 26) [83].
In general, quinones are electrochemically redox active molecules which undergo reduction as diol anions and re-oxidation back to quinone, with the corresponding measurable structural transformations in chromophoric absorption (Figure 13a). Therefore, the helicene-based quinone 149 was also tested for the same, with an additional possibility of chiroptical switching as observed by CD measurement. Both the enantiomers of 149 exhibited a one-electron reduction process to form the semiquinone radical anions, 149●−, as evident from cyclic voltammetry studies with E1/2 = −1.00 V (vs Fc+/0, ΔEp = 0.07 V) in acetonitrile, with a reversible electrochemical behavior. Since 149●− is a stable species, its UV and CD measurements were performed by using an optically transparent thin layer electrochemical cell. It was observed that the helicene quinone 149 is able to be reversibly reduced to the radical anion 149●− and re-oxidized over several cycles, and is an example of chiroptical switching to be conveniently monitored with CD spectroscopy at different wavelengths (Figure 13b) owing to the distinguishable spectral profiles of 149 and 149●−. However, as there is no change in the (P) or (M) helicity of the chromophore upon electrochemical transformations, the Cotton effect (CE) signs of 149 and 149●− remained the same.
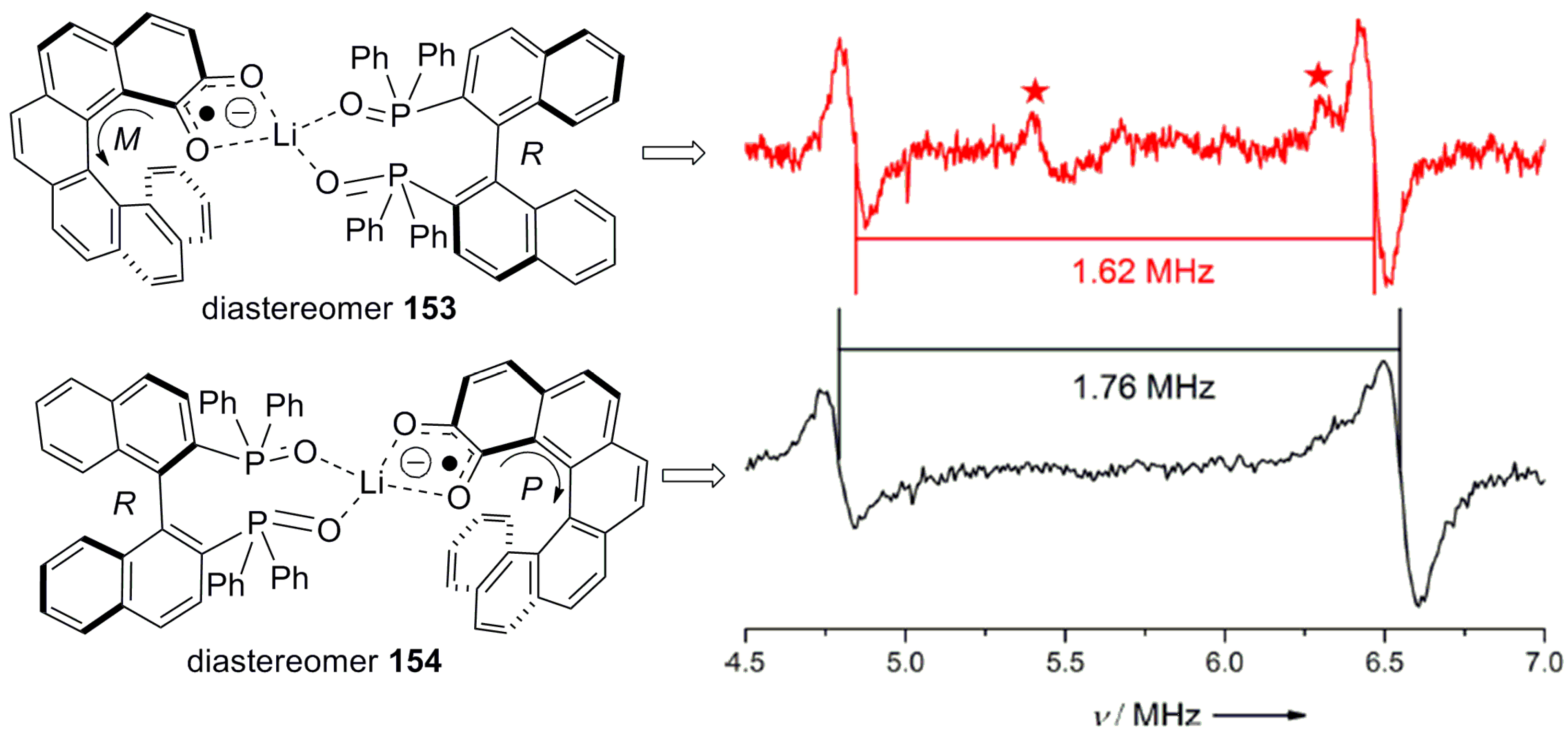
Besides electrochemical switching, 149●− was tested for chiral recognition against 2,2′-bis(diphenylphosphanyl)-1,1′-binaphthyldioxide (BINAPO) using electron paramagnetic resonance and electron–nuclear double resonance (ENDOR) spectroscopy. The diastereomeric species formed by coordinated lithiated semiquinone radical anions [Li+{(±)-149●−}] and enantiomers of BINAPO generated noticeable changes of the lithium and proton hyperfine coupling measured by ENDOR spectroscopy. The diastereoisomeric complex 153 exhibits a hyperfine coupling constant (A) value for 7Li at −1.62 MHz, in comparison to −1.76 MHz for another diastereomeric complex 154, indicating the appreciable difference between the diastereomers (Figure 14). It is of note that the racemic sample gives an average value of −1.69 MHz.
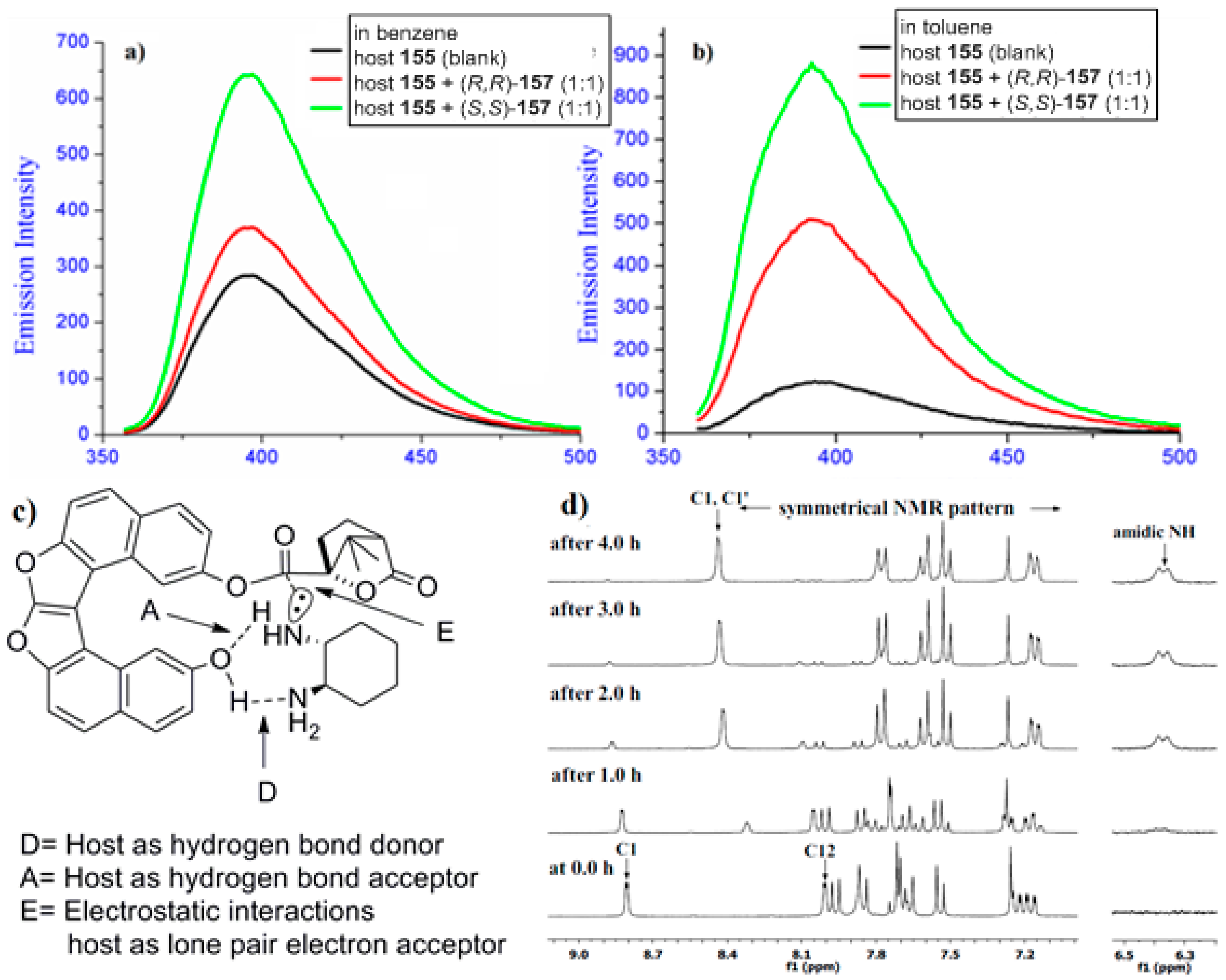
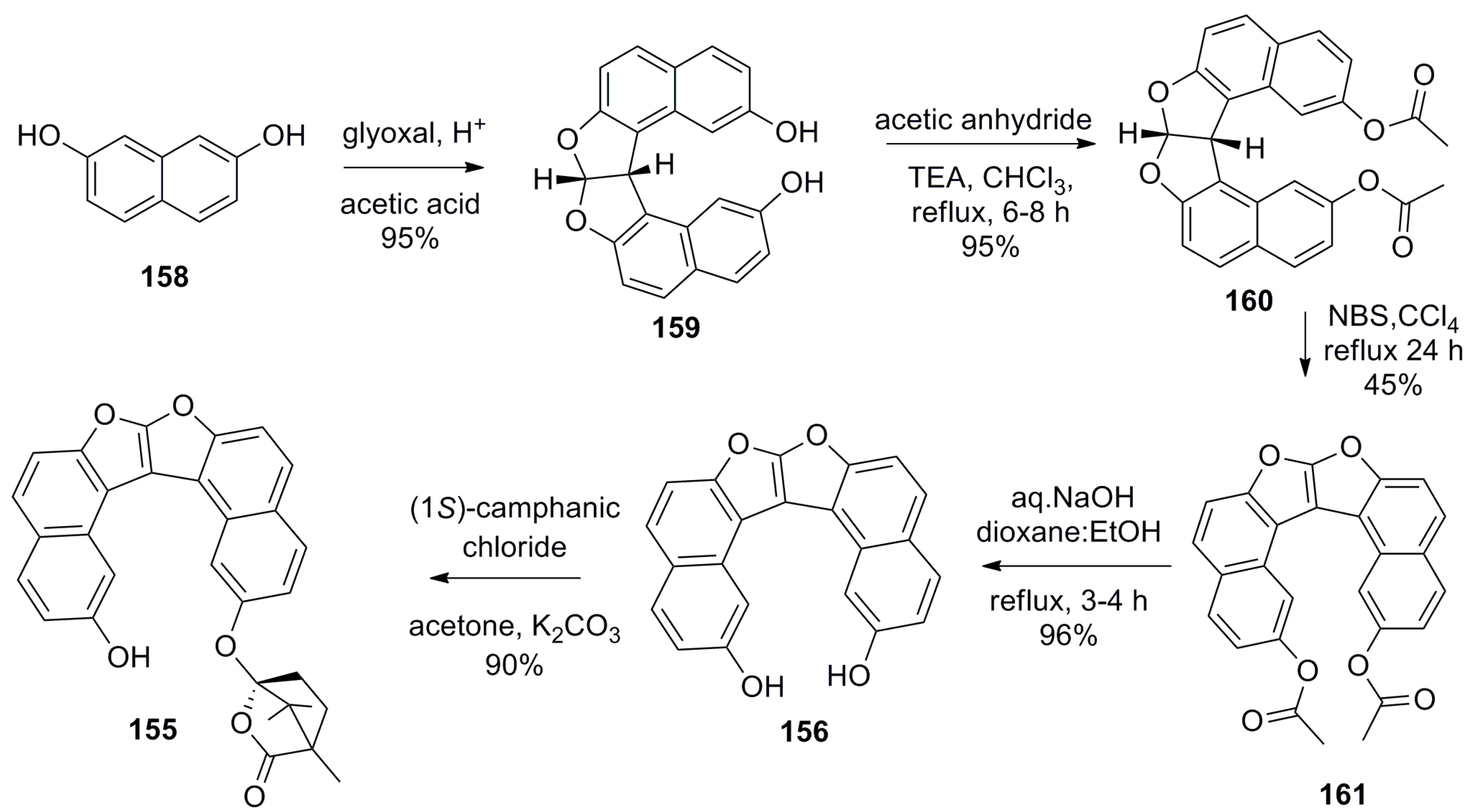
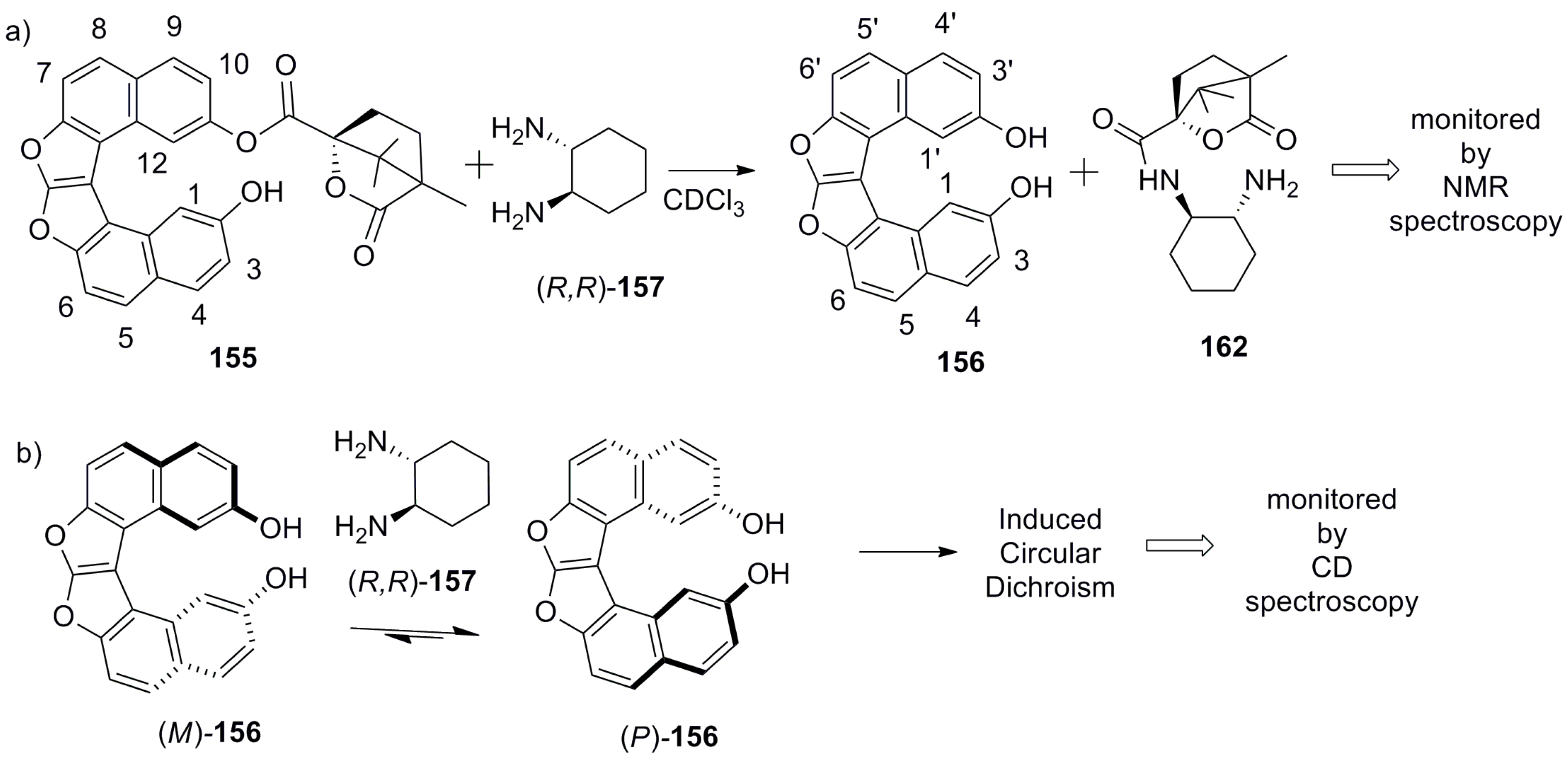
The previously described examples, 127 [77] and 115 [81], represent the inbuilt chiral helicene chromophoric hosts for the chiral recognition of a general class of chiral molecules using some selected analytical techniques such as fluorescence and NMR spectroscopy. An essentially different approach has been recently developed by Karnik et al. on the basis of a new supramolecular chirogenic system, 155, the key element of which was a stereodynamic dioxa[6]helicene diol 156 chromophore. This system was applied as a specific sensor, suitable for chirality sensing of trans-1,2-cyclohexanediamine 157 and monitored by various spectroscopic methods [84]. The helicene 156 was synthesized from 2,7-dihydroxynaphthalene 158 [85]. The first step involved acid-catalyzed condensation between two molecules of 158 and glyoxal to obtain dihydrofurofuran 159 (>95%), followed by the protection of phenolic groups to give acetate 160 (95%). Then, one-pot benzylic bromination and dehydrobromination (aromatization) of 160 using N-bromosuccinamide led to 161 (45%), followed by basic hydrolysis resulting in dioxa[6]helicene diol 156 (96%). Finally, selective mono esterification with chiral (1S)-camphanic chloride in acetone afforded the desired host, 155, with 90% yield (Scheme 27) [85].
Indeed, the parent dioxa[6]helicene diol 156 is almost a flat stereodynamic-type fluorescent compound, which was selectively modified with a chiral (1S)-camphanate group to give 155, leaving one of the (phenolic) hydroxyl groups free. This inherently free phenolic group and (1S)-camphanate chiral handle on the opposite peripheral ring provided a suitable sui generis chiral space for guest 157. In particular, this perfect stereospecific matching results in an effective turn-on fluorescent sensor for the (S,S)-enantiomer of 157 with a relatively high enantioselective factor, α = KSS/KRR = 6.3 (in benzene). The favorable hydrogen-bonding interactions in nonpolar media (benzene and toluene) are responsible for the efficient chiral recognition observed by fluorescence spectroscopy (Figure 15a,b). In nonpolar media, the donor–acceptor type cooperative hydrogen bonding between host 155 and guest 157 (Figure 15c) resulted in a high turn-on enantioselective fluorescence response. The enantiodiscrimination behavior was found to be solvent-dependent as the polarity of solvent can influence the extent of hydrogen bonding between the host and guest. In turn, this induces a unidirectional switch in the helical conformation of the helicene chromophore, resulting in reverse selectivity for the (R,R)-enantiomer of 157 in acetonitrile, THF, and chloroform [84].
Further, the NMR studies performed in deuterated chloroform exhibited the chiral camphanate group transfer reaction (Scheme 28a) with the reaction rate of 5.83 × 10−6 and 5.97 × 10−7 mol dm−3 s−1 for (R,R)-157 and (S,S)-157, respectively, as monitored by 1H NMR (Figure 15d). This tenfold increase of the reaction rate for the (R,R)-enantiomer allowed the host 155 to behave as a kinetic resolving agent for (R,R)-157, with up to 68% de at room temperature within 4 h (Figure 15d) [84].
Indeed, this is a rare example of the sui generic host–guest pair as evident from the fact that diamine 157, upon supramolecular interaction with the parent stereodynamic helicene diol 156 (Scheme 28b) is able to induce a specific helicity and shift the stereodynamic equilibrium of 156, as monitored by CD spectroscopy in the region of helicene chromophore absorption.
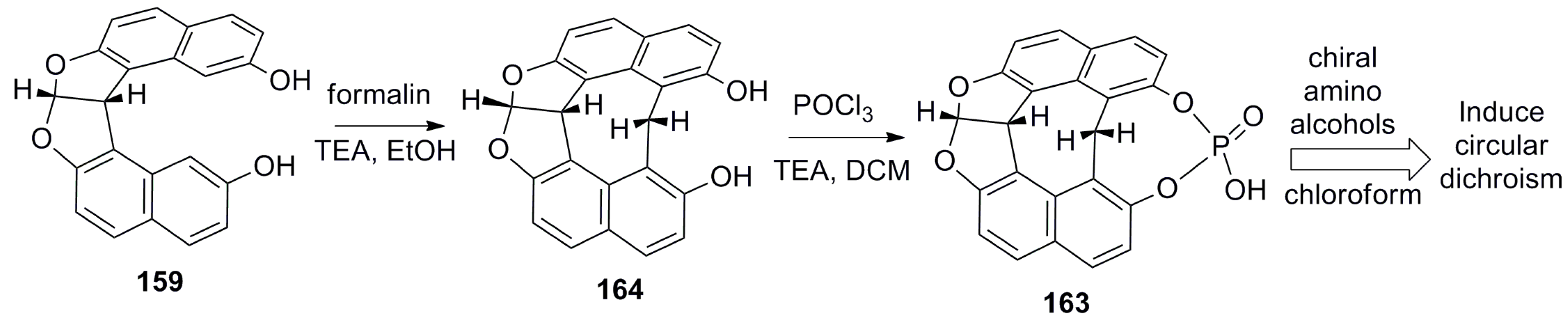
Further, the same group also developed Cs-symmetric rigid achiral organophosphoric acid 163 in two steps, starting from dihyrofurofuran helicene 159 as a supramolecular chirogenic system for the absolute configuration determination of 1,2-amino alcohols by induced CD (ICD) studies [86]. The two-step synthesis includes the reaction of dihyrofurofuran helicene 159 with formaldehyde to form an additionally bridged diol, 164, and subsequent reaction with POCl3 to obtain the target, 163 (Scheme 29).
Interestingly, due to the Cs-symmetric nature of phosphoric acid, 163 has two tautomeric structures. This structural feature of 163, in combination with the substituent bulkiness of the guest amino alcohols, together decide the preferred mode of two-point hydrogen bonding on equilibration, which gave a resultant ICD signal correlating with the absolute configuration of the guest [86].
7.2. Polymeric Helicene-Based Chiral Recognition
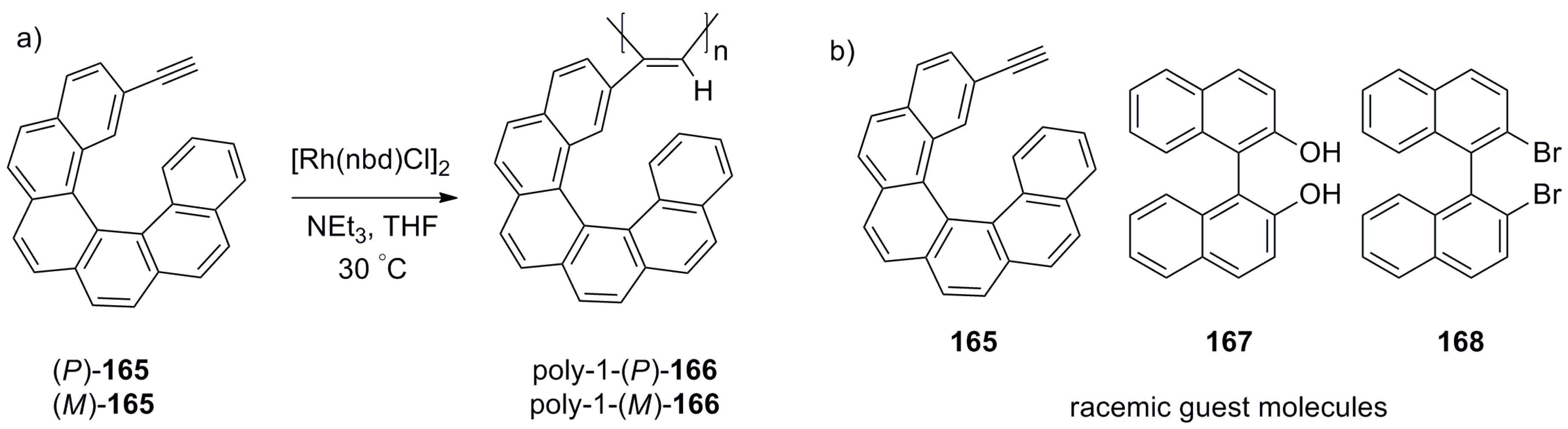
Polymer chains can also serve as suitable carriers for chiral helicenes, especially in the case of prospective applications. Thus, using 2-acetylene-[6]helicene 165, Yashima et al. prepared a series of polymeric helicene pendants, poly-1-166, via the rhodium-catalyzed polymerization reaction (Scheme 30) [87]. This polymerization reaction gave stereoregular (cis-transoidal) polyacetylenes bearing the corresponding helicene pendants 166 with (P)- and (M)-polymeric chains with average molecular weight Mn = 2.6 × 103. However, the formation of a precipitate during the reaction makes it difficult to reproduce. The careful analysis of the CD spectra of monomer 165 and polymers 166, and their corresponding differential CD spectra, suggests that the helical polyacetylenes with the optically active pendant group exist as a single-handed structure, apparently due to the attractive intramolecular π-π interactions.
The chiral recognition abilities of these polymers were studied in the context of the enantioselective adsorption of three racemic guests, namely, parent helicene 165, BINOL 167, and 2,2′-dibromo-binaphthalene 168. The enantiomers were adsorbed by the polymer in methanol solution and desorbed in a mixture of methanol/THF [1:1, v/v] followed by chiral HPLC analysis. The poly-1-(P)-166 preferentially adsorbed (P)-helicene 165 with 3.6% ee; both have similar configurations. As expected, in the case of 166 with opposite absolute configuration, the adsorption preference was observed for (M)-helicene 165 with a similar 3.7% ee. However, interesting results were obtained for binaphthyl derivatives 167 and 168, where poly-1-(P)-166 selectively adsorbed the opposite (S)-enantiomers with up to 36% ee, whilst poly-1-(M)-166 showed similar selectivity towards the (R)-enantiomers of 167 and 168. The calculated separation factor was sufficiently high (1.71–2.18), indicating that these polymers can be used as a stationary phase for the practical separation of racemic mixtures.
7.3. Helicene-Based Chiral Recognition via Nanoparticles
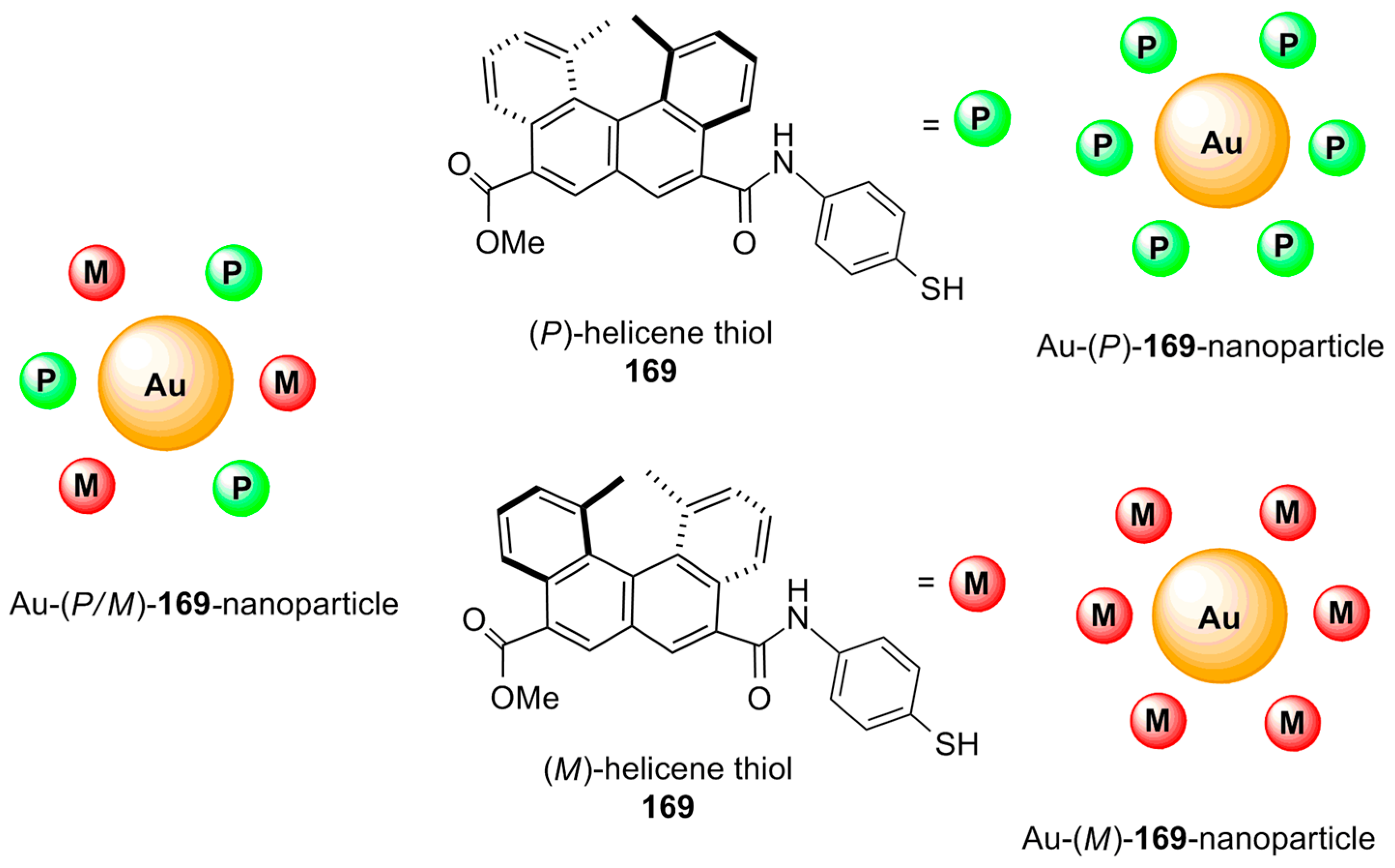
Another prospective approach for chiral recognition by helicene structures is based on nanomaterials. In this respect, individually racemic, (P)-, and (M)-helicene thiols 169 were grafted onto gold nanoparticles of 5–22 nm [88] in a mixture of ethanol/water (1:3, v/v) under sonication to form the corresponding Au-(P/M)-, (P)-, and (M)-169-nanoparticles (helicene-grafted nanoparticles) (Figure 16). These helicene-based nanoparticles show chirality-based inter(nano)molecular attractive interaction, resulting in the corresponding aggregation phenomenon followed by a time-dependent precipitation process, as analyzed by UV spectroscopy. All three types of nanoparticles dispersed in DMF gave a stable solution for a period of one week. The addition of the DMF solutions to an aromatic solvent such as benzonitrile resulted in different colors due to the surface plasmonic bands of the gold nanoparticles, as studied by UV–vis spectroscopy and a dynamic light scattering technique. Additionally, the time-dependent aggregates were of different sizes, based on the type of helicene nanoparticles present. The observed chiral recognition induced by the homo- (Au-(P/M)-169 nanoparticle : Au-(P/M)-169 nanoparticle, Au-(P)-169 nanoparticle : Au-(P)-169 nanoparticle, and Au-(M)-169 nanoparticle : Au-(M)-169 nanoparticle) and hetero self-aggregation (Au-(M)-169 nanoparticle : Au-(P)-169 nanoparticle) of the helicene nanoparticles was found to be enhanced according to the following sequence: Au-(P/M)-169 nanoparticle : Au-(P/M)-169 nanoparticle < Au-(M)-169 nanoparticle : Au-(M)-169 nanoparticle = Au-(P)-169 nanoparticle : Au-(P)-169 nanoparticle < Au-(P)-169 nanoparticle : Au-(M)-169 nanoparticle. This tendency relates to the interaction strength between the corresponding nanoparticles, which is controlled by stereochemical matching.
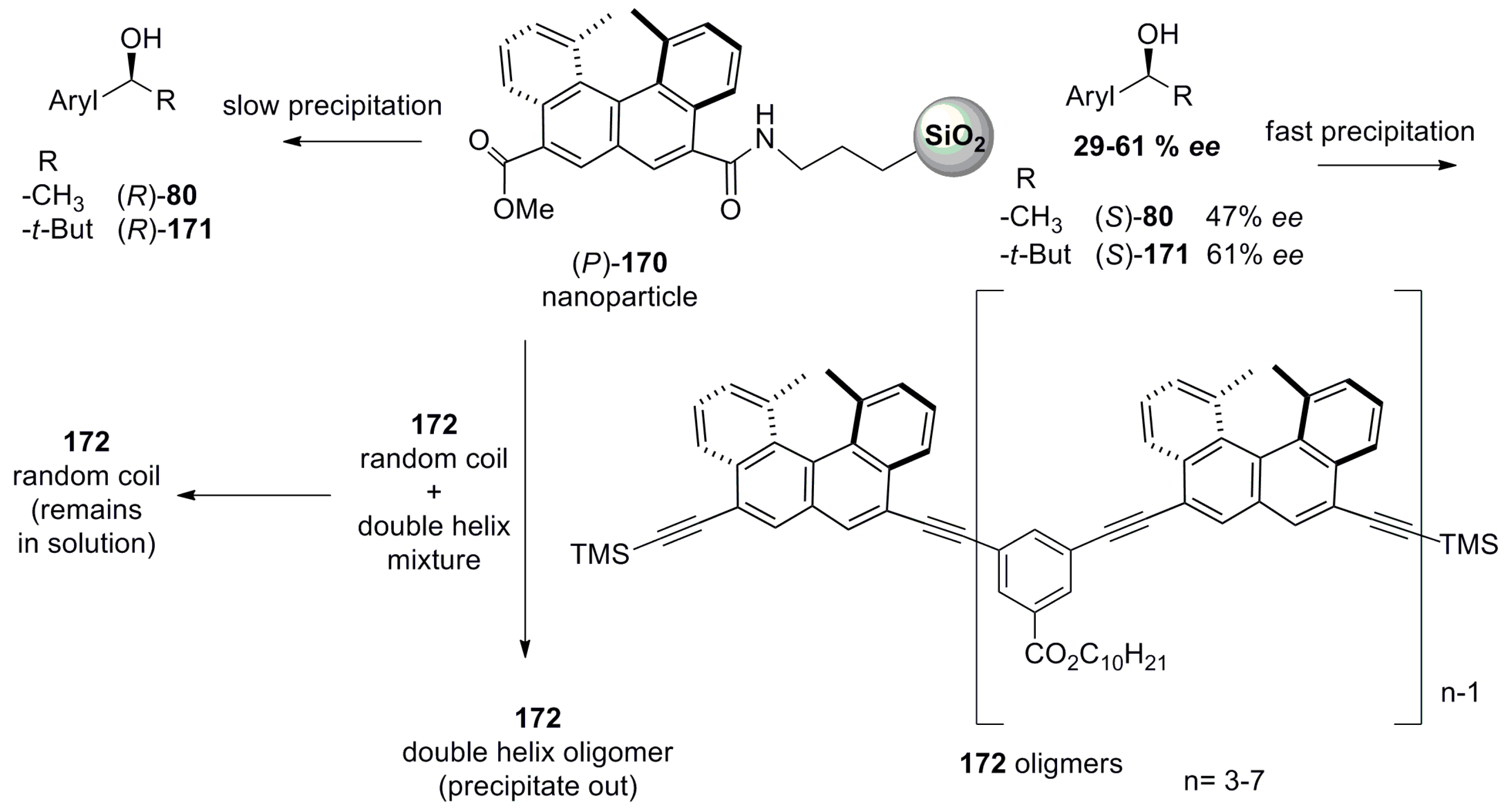
Another example of optically active nanoparticles 170 with 70 nm diameter size was obtained by grafting (P)-1,12-dimethyl-8-methoxycarbonylbenzo[c]phenanthrene-5-carboxyamide on 3-aminopropylated silica (Figure 17) [89,90]. These nanoparticles were prepared by reacting 3-aminopropylated silica nanoparticles with the corresponding (P)-helicene carboxylic acid chloride by refluxing in isopropyl ether for 4 hours using ethyldiisopropylamine as a base.
It was found that these nanoparticles enantioselectively precipitated at a faster speed with (S)-configuration of aryl alcohols 80 (47% ee) and 171 (61% ee) [89] in m-bis(trifluoromethyl)benzene (Figure 17). To elucidate the corresponding ee values, the precipitate was centrifuged and separated, followed by mixing in 2-propanol. Then, the insoluble material (nanoparticles) was removed and the organic layer was concentrated and analyzed with chiral HPLC, indicating the optical resolution of up to 29–61% ee for various aromatic alcohols for the (S)-configuration [89]. The mechanism of this stereoselectivity was related to chiral recognition on the basis of the electrostatic aggregation phenomenon. This nanoparticle-based kinetic resolution requires a relatively smaller amount (10 mol %) of -nanoparticles 170 as the resolving agent compared with the requirement of conventional 0.5 equivalence, with providing additional possibility for resolving noncrystalline substances, making it an attractive and greener method of choice.
Further, the helicene-based nanoparticles 170 were able to recognize the shape of (P)-ethynylhelicene oligomers 172 in solution [90]. It preferentially forms the precipitate with a double helix of the oligomeric structure of 172 in comparison to its random coil structure in trifluoromethylbenzene (Figure 17). The analysis was carried out by CD spectroscopy, where the double helix oligomer shows a strong positive CE followed by a negative one, whilst in the case of the random coil oligomer, a weak CE of opposite sign was observed. This phenomenon was related to the chiral shape recognition on the surface of the nanoparticle by stereospecific absorption, resulting in the visualized selective aggregation-based precipitation, which is useful for the separation of the double helix structure of 172 from its random coil conformation [90]. These examples clearly show the potential of helicene-based chiral nanomaterials for applications in the resolution of chiral molecules ranging from a simple alcohol to complex polymeric structures.
7.4. Helicene-Based Chiral Recognition Involving Biologically Relevant Molecules
The stereodynamic helicenes are also effectively employed for the induced CD-based sensing, where a chiral guest can induce a specific helicity via covalent or supramolecular interactions, thus generating the corresponding CD signals in the helicene absorption region. The guests range from simple organic molecules (Scheme 28b) to complicated biological molecules, as described here.
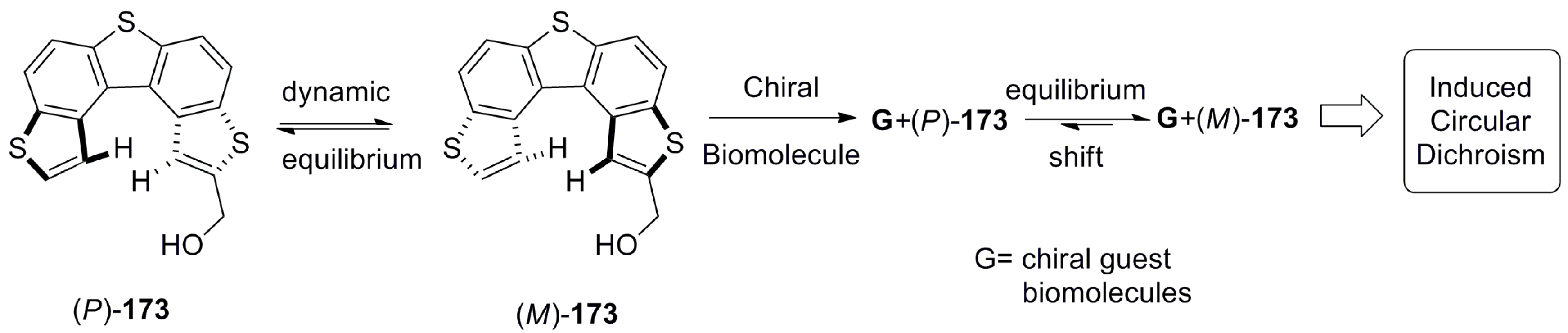
Thus, Yamada’s group explored hydroxymethyl trithia[5]helicene 173 [91,92,93,94,95,96], which is a stereodynamic compound. Both the (P)- and (M)-enantiomeric structures are configurationally labile, nonresolvable, and exist in a fast equilibrium. However, due to the presence of an electron-rich aromatic chromophore and polar hydroxymethyl group capable of hydrogen bonding, suitable complexation of the chiral guest (such as biomolecules) [91,93,94,95,96] is able to shift the equilibria to one of unidirectional helicity, which, in turn, can be monitored by CD spectroscopy (Figure 18).
The corresponding host–guest studies were successfully implemented with serum albumin [91], (R)-2-(2,4,5,7-tetranitrofluoren-9-ylideneaminooxy)propionic acid (TAPA) [92], alkyl b-d-pyranoside [93], and bilayered phosphatidylcholine vesicles [94,95,96] using CD spectroscopy as a major tool. As the extent of helicity induced in 173 by these guest molecules is small, the resultant CD signals are also small. Yet, as CD is a highly sensitive technique, even with a small signal, it can be applied for understanding molecular behavior and chirality in solution.
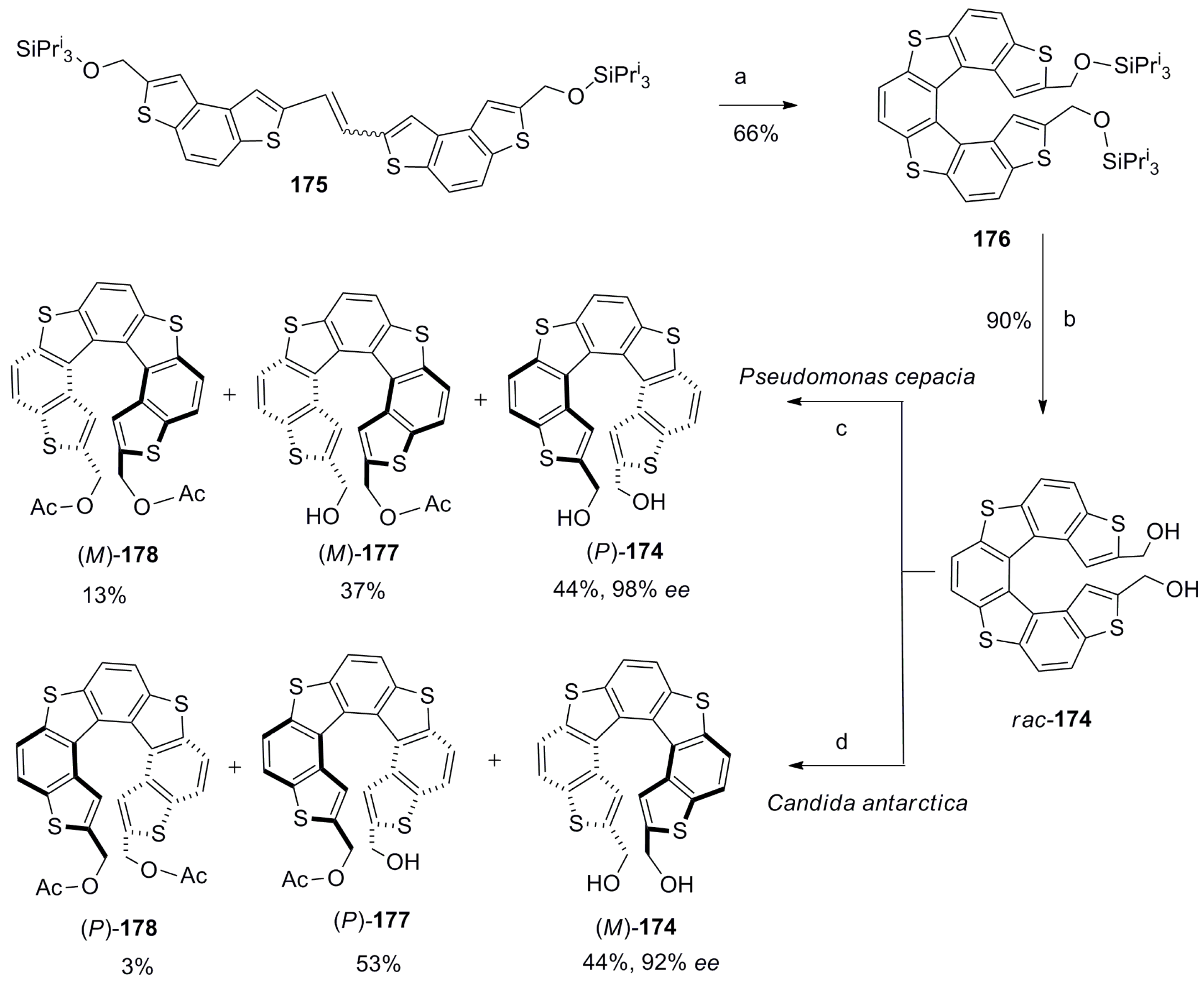
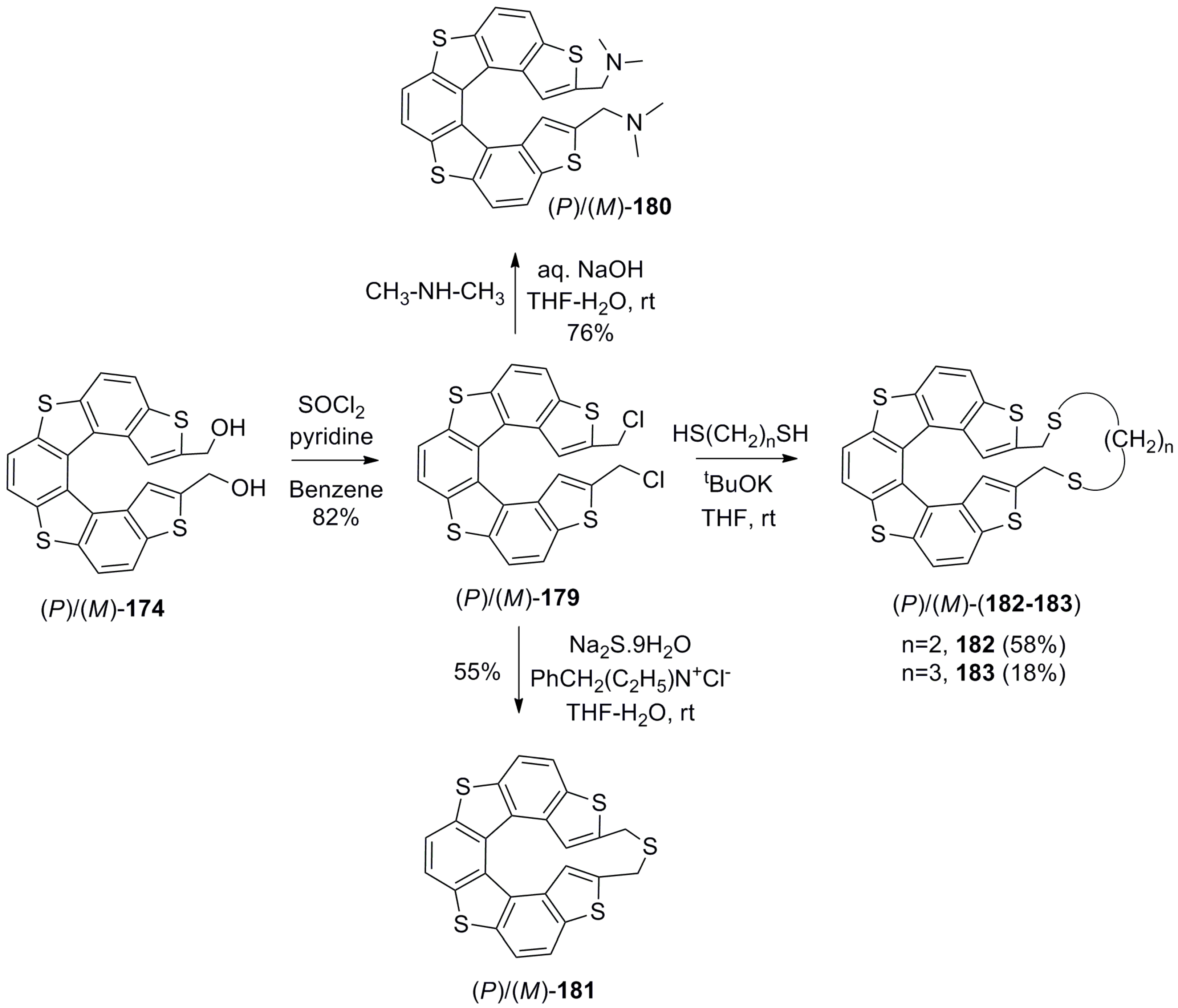
Tanaka et al. synthesized bis(hydroxymethyl)thia[7]helicene 174 to study its further synthetic chemistry and corresponding applications in the chirality recognition of biological molecules [97,98,99,100]. Hence, the helicene 174 was synthesized in a total of nine steps with an overall 33% yield, where the photochemical cyclization of alkene 175 was a key step to give silyl group protected rac-176. Further, deprotected racemic 174 was optically separated using the lipase-based enzymatic kinetic resolution method. For example, with the use of Pseudomonas cepacia enzyme, (P)-174 (44%, 98% ee) was obtained as a slow reactive isomer with simultaneous formation of the corresponding mono- and diacetates, (M)-177 and (M)-178. Similarly, the use of Candida antarctica enzyme resolved opposite (M)-174 (44%, 92% ee) with the formation of mono- and diacetates, (P)-177 and (P)-178 (Scheme 31) [97].
Further, enantiopure alcohol 174 was converted to its dichloride 179, which was converted to 180 [98] and cyclic thia ethers 181–183 [99] using suitable synthetic procedures (Scheme 32). These helicene molecules were explored in terms of possible chiral recognition of biologically relevant molecules such as DNA [98,100].
It is well known that, under physiological conditions, a DNA duplex exists as a mixture of the right-handed B-DNA and left-handed Z-DNA forms. The supramolecular association of chiral C2-symmetrical 180 with DNA revealed that the (P)-enantiomer of 180 displays chiral selection for binding Z-DNA over B-DNA [98]. The corresponding binding constants were found to be (8.0 ± 0.5) × 104 M−1 and (1.4 ± 0.3) × 104 M−1 for Z- and B-DNA, as measured by fluorescence titration. Besides, this interaction can be clearly monitored with CD spectroscopy, where, upon binding Z-DNA, there is a considerable decrease in the signal intensity of (P)-180 of up to 70% at 330 nm at neutral pH. Under similar conditions, B-DNA failed to show any significant change in the CD spectra of (P)-180, indicating (P)-helicity-based recognition of Z-DNA. In contrast, although the (M)-180 also showed a 20% reduction, it was observed upon binding both B- and Z-DNA, thus indicating the lack of its discrimination ability.
Additionally, it was observed that the (P)-180 not only selectively binds Z-DNA but is also able to force B-DNA to adopt an opposite left-handed helical form, like Z-DNA. Apparently, the methylene-N,N-dimethylamine functionality in (P)-180 plays a crucial role in this behavior, as the same helicene backbone but with hydroxy methyl substituents, as in the case of 174, did not show any binding response towards B- and Z-DNA. It is likely that the protonated form of amino substituents in 180 becomes a crucial site for binding DNA, whilst the helicene chirality is an important factor in the enantioselective recognition of left-handed helical Z-DNA.
Further extending the chiral recognition research of biologically important molecules resulted in the development of cyclic helicenes with thiaether linkage, 181–183 (Scheme 32) [99,100]. The (M)-enantiomers of 181–183 turned out to be potential inhibitors against telomerase enzyme activity based on the chiral and steric matching, while binding to the G-quadruplex superhelix structure. The recognition phenomenon is based on the chiral space available at the cleft-pocket and the presence of Z-DNA assisting this helicene binding. Initially, 180 was studied and exhibited no chiral selectivity. This result led to the assumption that the dihedral angle in helicene chromophores plays a key role in the interaction efficiency. On this basis, three different cyclic helicene thiaethers, 181–183, having different length of linkers, were designed to yield the dihedral angles of 22°, 53°, and 58°, respectively.
It was shown that the (M)-enantiomer of 181 with the smallest interplanar angle was able to inhibit the telomerase enzyme activity efficiently, presumably as a result of the chiral and steric matching. However, the exact binding mode of this helicene to the G-quadruplexes is not fully understood yet. Here, again, the binding studies relied on CD and fluorescence spectroscopy [100].
In 2013, chiral [5]helicene 184, containing methyl groups at the innermost positions, was suitably connected with the spermine moiety 185 at the outer sphere of the helicene backbone to act as a host capable of recognizing the DNA conformation [101]. This host was synthesized by the Suzuki–Miyaura coupling of 8-methylnaphthalene-2-boronic acid 186 with dibromo-maleimide 187 to obtain 188, followed by photocyclization to result in racemic 189 with 92% yield. The enantiomers of 189 were separated on a chiral column followed by further transformation to 190 and, finally, to the target enantiopure 184 (Scheme 33) [101].
The designing concept was such that the cationic spermine unit is able to provide the corresponding electrostatic interactions with the DNA’s phosphate backbone of the minor groove, whereas the chiral helicene is expected to form end-stacking mode complexes, altogether resulting in a strong chiral recognition. Indeed, it was demonstrated that (P)-184 binds preferentially to B-DNA over Z-DNA. The opposite trend was observed with (M)-184, which displayed strong binding for Z-DNA over B-DNA, as studied by performing CD, UV melting temperature, surface plasmon resonance measurements, and isothermal titration calorimetry. From the examples of 180 [98] and 184 [101], it appears that the cationic functional groups assist in binding with DNA through electrostatic interactions; along with this, the chiral helicity-based steric fit allows selectivity. The (P)-180 prefers Z-DNA, whereas in the case of 184, the (M)-enantiomer binds strongly to Z-DNA. As structures, tetrathia[7]helicene 180 having functional groups on the terminal rings (in chiral cavity space) and 1,14-dimethyl[5]helicene 184, where spermine 185 is situated at the helicene outer core, are completely different in terms of the functional groups and their positions, therefore the exact mode of binding with DNA cannot be generalized and is still not completely understood.
Chiral cationic [4]helicenes 191 (Figure 19), having methoxy substituents at the innermost position, were also able to bind with DNA molecules, as evident from a noticeable change in absorption, fluorescence, and CD spectra obtained during the corresponding host–guest studies [102]. It was concluded that the molecular plane of the helicene dye lies essentially parallel to the DNA bases, implying intercalation mode rather than groove binding. Further, the chiral selectivity based on association constant evaluation indicates that the (M)-enantiomer of 191 interacts more strongly with DNA than the corresponding (P)-enantiomer of 191.
Yang et al. developed chiral 3-aza[6]helicene-modified β-cyclodextrin 192 by reacting 6-deoxy-6-iodo-β-cyclodextrin 193 with (P)/(M)-helicene 194, resulting in two water soluble cationic charged diastereomeric species—(P)-192/β-cyclodextrin, and (M)-192/β-cyclodextrin (Scheme 34)—with different fluorescence behavior [103]. The concept was to use water-insoluble 3-aza[6]helicene 194 in aqueous media in combination with the β-cyclodextrin asymmetry for the chiral recognition of amino acids in water.
In polar solvents such as methanol, ethanol, and water, the helicene chromophore occupies the β-cyclodextrin cavity to a different extent in two diastereomers: (P)-192/- and (M)-192/β-cyclodextrin—favoring monomeric forms rather than self-aggregated species. Both the diastereomers were investigated for complexation towards proteinogenic amino acids by using the fluorescence technique in an aqueous buffer of pH 7.3. It turned out that hydrophobic interactions appeared to be a dominant binding mode with additional electrostatic forces between the cationic charge of helicene and carboxylate anion of corresponding amino acids; besides this, π-π interaction may also contribute in the case of aromatic guests. The chiral discrimination abilities were also found to be highly dependent on pH. A relatively high L/D selectivity of amino acids, up to 12.4, was observed as a consequence of the synergetic effects of the helicene auxiliary and β-cyclodextrin cavity [103].
7.5. Helicene-Based Chiral Recognition via Self-Assembly
A new approach for chiral recognition via self-assembly was applied by Yamaguchi et al. on the basis of functionalized chiral [4]carbohelicene. The overall studies demonstrated that the helicenes exhibit a noncovalent chiral recognition behavior via different functions, such as charge transfer complexation, crystallization, homocoupling reaction, layer structure formation, self-aggregation, and even upon double helix formation [75]. Their observations indicated that the right-handed helix structure favors the same helicity of its counterpart, which is not generally observed in the case of point chirality (i.e., an (R)- or (S)-host does not always prefer a guest of the same absolute configuration). Out of several examples, the following one is sufficient to understand this helicity matching phenomenon.
Tetranitro, dicyano-substituted [4]helicene 195, being an electron-deficient system, strongly interacts with diamino-substituted [4]helicene 196 via a charge transfer complex [104], the formation of which was monitored by UV and NMR techniques. Particularly, in the UV spectrum, a charge transfer band was observed at 500–800 nm in THF. The association constant of the complex with the same configuration—(M)-195 and (M)-196—was found to be 12.2 M−1; this was noticeably higher than the constant 10.2 M−1 observed for the opposite configuration—(P)-195 and (M)-196 (Figure 20). Further, the NMR-based NOE experiments revealed that the face-to-face syn-conformation of two 1,12-dimethyl groups was preferred over the anti-orientation in the corresponding charged transfer complexes [104].
In related studies, a series of racemic [7]helicene derivatives, 197–199, containing pyridinone rings at both the terminals, were prepared using a classical methodology of Wittig olefination and photochemical cyclization, and were characterized both in solution and in solid state [105]. Despite a variety of self-assembly modes according to the stereochemical and topological relationships, these helicenes formed only enantiomerically pure dimers held together by two pairs of the cooperative hydrogen bonds. The self-assembly process was found to be enantiospecific in solution and diastereoselective in solid crystal (Figure 21).
The terminal pyridinone rings were capable of hydrogen bonding via the corresponding amidic N–H functionalities. The solution-phase NMR studies indicated that the helicenes exist as enantiomeric dimers. For example, the NH proton in 2-quinolinone 200 (10−4–10−2 M), as a reference, appeared at 12.9 ppm, whilst the position of this proton in 198 was downfield-shifted by 1 ppm in dilute solution (>10−5 M), clearly indicating the cooperative hydrogen bonding and self-assembly phenomenon. In turn, the chemical shift was found to be concentration-independent in CDCl3 and THF. In pyridine, as expected, it was concentration-dependent, as pyridine itself is able to compete for the hydrogen bonding. Additionally, the dimeric binding model was confirmed by dilution experiments with the association constant of 207 M−1. The molecular modelling studies also indicated that each dimer is preferably constructed from a monomeric species of the same helicity.
The X-ray crystallographic structure of 199 showed only homochiral dimers; where the R-group (acetyl) is present in the cis-orientation to ensure the diastereoselective recognition process during crystallization. These enantiopure dimers are held together by four strong intermolecular hydrogen bonds between two terminal pyridinones.
8. Enantiopure Helicenes: Enantioselective Synthesis, Chiral Separation, and Racemization
The chiroptical properties and applications discussed above prompted the development of efficient synthetic procedures to obtain enantiopure helicenes in sufficient quantities required for these investigations [2,4]. Although this topic is not directly related to the category of helicene applications, it is an important background for the whole helicene chemistry; hence, a brief description should be included as a special section. The major methods of obtaining enantiopure helicenes from a racemic mixture are based on optical resolution by chiral HPLC [60,77,78,83,101], diastereomeric salt formation [51,60,76] (for representative resolving agents, see Figure 22), chromatographic separation of diastereomers [52,56,74], and enzymatic-based approaches [97].
For example, racemic nonfunctionalized helicenes were successfully resolved through charge transfer complexation with electron-deficient molecules, such as TAPA [15,16] or HPLC-based separation with the use of silica coated with TAPA [106]. In the case of functionalized helicene-containing phenolic groups at the terminal ring, (1S)-camphanic chloride acts as the best resolving agent for chromatographically separable diastereomers obtained. Furthermore, their absolute configuration determination was carried out by the 2D ROESY NMR technique [52,74,107]. Particularly, it was observed that in 19 examples of helicenes studied, (M,S)-diastereomers were less polar, whilst (P,S)-diastereomers were more polar, hence eluting as the first and second fractions, correspondingly, upon the chromatographic separation on silica gel [107]. Another resolving agent, which is also frequently employed for optical resolution, is an l-menthyl derivative [4,56]. This approach has a distinct advantage of attaching and detaching a chiral auxiliary before and after the diastereomeric separation, making the whole procedure recyclable and highly efficient. Interestingly, in the case of azahelicene 201 containing the chloro group as a substituent on the outer peripheral pyridine ring, the use of Pd-catalyzed Buchwald–Hartwig coupling with (S)-phenylethyl amine gave chromatographically separable diastereomers 202 and 203 (Scheme 35) [108,109]. However, the absolute configuration of the 2-ethylhexyl alkyl chain was not discussed in the paper [108].
Besides the optical resolution of racemates, another approach to enantiopure helicenes is based on the stereospecific synthesis, where the originally chiral precursors or reagents are converted into the corresponding helicenes with unidirectional helicity [110]. In particular, the asymmetric synthesis of helicene can be performed by using various synthetic methods, such as photocyclization, Diels–Alder reaction, or metal-catalyzed annulation, in the presence of chiral auxiliaries attached to the reactant(s). These topics have been comprehensively covered in previous reviews [2,4]. Therefore, only a few of those recently published examples of asymmetric helicene synthesis are highlighted below.
In 2016, Sako et al. successfully used the modified BINOL-based vanadium(V) complex 204, capable of functioning both as redox and Lewis acid catalysts, for the enantioselective synthesis of oxa[9]helicenes 205 starting from polycyclic phenols 206. The reaction proceeds via oxidative coupling at first, followed by intramolecular cyclization. This results in the one-step enantioselective synthesis of oxa[9]helicenes in good yields with up to 94% ee [111] (Scheme 36).
In 2015, a new general asymmetric synthetic methodology was successfully developed for [5]-, [6]-, and [7]helicenes with ultimate enantioselectivity (ee >99) based on the controlled transfer of reactant point chirality to the product with unidirectional helicity [112]. At first, triyne207, having inbuilt point chirality, was converted into tetrahydrohelicene diastereomers 208 and 209, being in the thermodynamic equilibrium, by cobalt-catalyzed tandem [2 + 2 + 2] cycloisomerization. The (M)-conformational helicity results in the 1,3-allylic strain, hence making 208 a highly energetic diastereomer in comparison to 209, the (P)-conformational helicity of which gives relief from this strain, when the point chirality is of (R)-configuration at the 9-position. Subsequently, the favored diastereomer under acidic conditions yielded fully aromatic helicenes 210 with greater than 99% ee (Scheme 37). Thus, (P)- and (M)-helicenes can be readily prepared starting from the (R)- and (S)- absolute configurations at the 9-position of the precursor, correspondingly.
One of the most important points of chiral helicenes is their configurational stability, which can be analyzed by using CD spectroscopy and chiral HPLC in combination with other techniques. Evidently, understanding the mechanism of racemization helps in designing more stable helicene structures. Recently, detailed theoretical racemization studies have been performed on [n]helicenes where n = 4–24 [113]. Consequently, it was found that n = 4–7 involves the one-step concerted mechanism of racemization. The smallest member of the carbohelicene family, [4]helicene has a C2V symmetric transition state (TS) with an energy barrier of 4.0 kcal mol-1. However, the TS of n = 5–7 possesses a Cs-geometry with the energy barriers of 24.4, 36.9, and 42.0 kcal mol−1, respectively. This is a result of the steric hindrance, which accounts for the high TS energies of racemization. For n ≥ 8, besides the steric hindrance, an additional factor of the π-π interaction starts to contribute to the overall structural stability, making the racemization process a multistep process and thus further increasing the energy barrier. For example, when n = 8,9, or 10, helicenes displayed 2, 4, and 6 intermediates for the racemization process, respectively. Thus, in general, when n = 8–24, helicenes involve (2n-14) consecutive displacements, traversing intermediates in the molecule during the racemization process for corresponding carbohelicenes. These theoretical results should be taken into account to control the desired properties of helicene structures in future research.
9. Conclusions
Helicenes are versatile compounds existing in achiral, stereodynamic, meso, and chiral forms. Additionally, they could be neutral, charged, carbocyclic, heterocyclic, or polymeric in nature, with electron-rich or -deficient characteristics. These conjugated molecules exhibit unique physical properties such as fluorescence and strong chiroptical features, making them exclusively useful for the chirogenesis processes, including chiral recognition using both fluorescence and circular dichroism spectroscopies. Their polyaromatic helical structures are also suitable for providing shielding and deshielding effects, making them potentially applicable as chiral NMR sensors.
The high configurational stability and facile functionalization of the helicene structures open intriguing prospects in the fields of chiral catalysts and auxiliaries for various asymmetric reactions. In the last three decades, there has been much research effort focusing on newer, simpler, and multigram synthetic procedures including different asymmetric methodologies for obtaining sufficient quantities of enantiopure helicenes for various practical applications, including chirogenesis. It has been established that modified helicene molecules with important properties can be developed by merging the helical structure with other (central, axial, and planar) chirality elements for specific applications. The helicity control may be conveniently achieved by means of the intramolecular cyclization of terminal rings, resulting in a decrease in the helicity. Alternatively, the introduction of functional groups on the terminal rings or presence of saturated bond(s) inside the molecule (helicenoid) increases the helicity. These modifications also govern the helicene chiral cavity space properties, and may thus be adopted for desired applications.
Besides chiral auxiliaries and chirogenesis, the helicene structures are good candidates for material chemistry and nanotechnology. We believe that there is still a large and diverse scope for helicene chemistry and application to be studied in the upcoming future, which will result in novel and interesting discoveries. We are yet to fully explore the real potential of helicene and related molecules.
Acknowledgments
M.H. and V.B. acknowledge funding from the European Union’s Seventh Framework Programme for Research, Technological Development, and Demonstration under Grant Agreement No. 621364 (TUTIC-Green).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hembury, G.A.; Borovkov, V.V.; Inoue, Y. Chirality sensing supramolecular systems. Chem. Rev. 2008, 108, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Chen, C.F. Helicenes: Synthesis and applications. Chem. Rev. 2012, 112, 1463–1535. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M. One hundred years of helicene chemistry. Part 1: Non-stereoselective syntheses of carbohelicenes. Chem. Soc. Rev. 2013, 42, 968–1006. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M.; Félix, G.; Peresuttiab, R. One hundred years of helicene chemistry. Part 2: Stereoselective syntheses and chiral separations of carbohelicenes. Chem. Soc. Rev. 2013, 42, 1007–1050. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M. One hundred years of helicene chemistry. Part 3: Applications and properties of carbohelicenes. Chem. Soc. Rev. 2013, 42, 1051–1095. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, N. Photochemical reactions applied to the synthesis of helicenes and helicene-like compounds. J. Photochem. Photobiol. C Photochem. Rev. 2014, 19, 1–19. [Google Scholar] [CrossRef]
- Saleh, N.; Shena, C.; Crassous, J. Helicene-based transition metal complexes: Synthesis, properties and applications. Chem. Sci. 2014, 5, 3680–3694. [Google Scholar] [CrossRef]
- Aillard, P.; Voituriez, A.; Marinetti, A. Helicene-like chiral auxiliaries in asymmetric catalysis. Dalton Trans. 2014, 43, 15263–15278. [Google Scholar] [CrossRef] [PubMed]
- Virieux, D.; Sevrain, N.; Ayad, T.; Pirat, J.-L. Helical phosphorus derivatives. In Advances in Heterocyclic Chemistry; Eric, F.V.S., Christopher, A.R., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 116, pp. 37–83. [Google Scholar]
- Rajca, A.; Miyasaka, M. Functional Organic Materials; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; Chapter 15; pp. 547–558. [Google Scholar]
- Chen, C.-F.; Shen, Y. Helicene Chemistry—From Synthesis to Applications; Springer: Berlin/Heidelberg, Germany, 2017; ISBN 978-3-662-53168-6. [Google Scholar]
- Weitzenbock, R.; Lieb, H. New synthesis of chrysene. Monatshefte Chem. 1913, 33, 549–565. [Google Scholar] [CrossRef]
- Weitzenbock, R.; Klingler, A. Synthesis of the isomeric hydrocarbons 1,2,5,6-dibenzanthracene and 3,4,5,6-dibenzophenanthrene. J. Chem. Soc. 1918, 114, 494. [Google Scholar]
- Cook, J.W. Polycyclic aromatic hydrocarbons. Part XII. The orientation of derivatives of 1:2-benzanthracene, with notes on the preparation of some new homologues, and on the isolation of 3:4:5:6-dibenzphenanthrene. J. Chem. Soc. 1933, 0, 1592–1597. [Google Scholar] [CrossRef]
- Newman, M.S.; Lutz, W.B.; Lednicer, D. A new reagent for resolution by complex formation; the resolution of phenanthro-[3,4-c]phenanthrene. J. Am. Chem. Soc. 1955, 77, 3420–3421. [Google Scholar] [CrossRef]
- Newman, M.S.; Lednicer, D. The synthesis and resolution of hexahelicene. J. Am. Chem. Soc. 1956, 78, 4765–4770. [Google Scholar] [CrossRef]
- Morrison, D.J.; Trefz, T.K.; Piers, W.E.; McDonald, R.; Parvez, M. 7:8,9:10-Dibenzo-1,2,3,4-tetrafluoro-triphenylene: Synthesis, structure, and photophysical properties of a novel [5]helicene. J. Org. Chem. 2005, 70, 5309–5312. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Scott, L.T. Thermal cyclodehydrogenations to form 6-membered rings: Cyclizations of [5]helicenes. Org. Lett. 2007, 9, 3937–3940. [Google Scholar] [CrossRef] [PubMed]
- Minuti, L.; Taticchi, A.; Marrocchi, A.; Gacs-Baitz, E. Diels-Alder reaction of 3,3′,4,4′-tetrahydro-1,1′-binaphthalene. One-pot synthesis of a pentahelicenebenzoquinone. Tetrahedron 1997, 53, 6873–6878. [Google Scholar] [CrossRef]
- Ogawa, Y.; Ueno, T.; Tarikomi, M.; Seki, K.; Haga, K.; Uyehara, T. Synthesis of 2-acetoxy[5]helicene by sequential double aromatic oxy-Cope rearrangement. Tetrahedron Lett. 2002, 43, 7827–7829. [Google Scholar] [CrossRef]
- Pieters, G.; Gaucher, A.; Prim, D.; Marrot, J. First expeditious synthesis of 6,11-diamino-[6]carbohelicenes. Chem. Commun. 2009, 32, 4827–4828. [Google Scholar] [CrossRef] [PubMed]
- El Abed, R.; Aloui, F.; Genet, J.-P.; Ben Hassine, B.; Marinetti, A. Synthesis and resolution of 2-(diphenylphosphino)heptahelicene. J. Organomet. Chem. 2007, 692, 1156–1160. [Google Scholar] [CrossRef]
- Abbate, S.; Bazzini, C.; Caronna, T.; Fontana, F.; Gambarotti, C.; Gangemi, F.; Longhi, G.; Mele, A.; Sora, I.N.; Panzeri, W. Monoaza[5]helicenes. Part 2: Synthesis, characterisation and theoretical calculations. Tetrahedron 2006, 62, 139–148. [Google Scholar] [CrossRef]
- Meisenheimer, J.; Witte, K. Reduction of 2-nitronaphthalene. Chem. Ber. 1903, 36, 4153–4164. [Google Scholar] [CrossRef]
- Sundar, M.S.; Sahoo, S.; Bedekar, A.V. Synthesis and study of the structural properties of oxa[5]helicene derivatives. Tetrahedron Asymmetry 2016, 27, 777–781. [Google Scholar] [CrossRef]
- Dreher, S.D.; Weix, D.J.; Katz, T.J. Easy synthesis of functionalized hetero[7]helicenes. J. Org. Chem. 1999, 64, 3671–3678. [Google Scholar] [CrossRef] [PubMed]
- Eskildsen, J.; Krebs, F.C.; Faldt, A.; Sommer-Larsen, P.; Bechgaard, K. Preparation and structural properties of 7,8-dioxa[6]helicenes and 7a,14c-dihydro-7,8-dioxa[6]helicenes. J. Org. Chem. 2001, 66, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Oyama, H.; Nishimura, Y.; Nakasako, S.; Nozaki, K. k5-Phospha[7]helicenes: Synthesis, properties, and columnar aggregation with one-way chirality. Angew. Chem. Int. Ed. 2012, 51, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Oyama, H.; Nakano, K.; Harada, T.; Kuroda, R.; Naito, M.; Nobusawa, K.; Nozaki, K. Facile synthetic route to highly luminescent sila[7]helicene. Org. Lett. 2013, 15, 2104–2107. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, T.; Hashimoto, S.; Oba, T.; Nakamura, M. Azaboradibenzo[6]helicene: Carrier inversion induced by helical homochirality. J. Am. Chem. Soc. 2012, 134, 19600–19603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Edward, L.; Clennan, E.L.; Arulsamy, N. Photophysical and electrochemical characterization of a helical Viologen, N,N′-dimethyl-5,10-diaza[5]helicene. Org. Lett. 2014, 16, 4610–4613. [Google Scholar] [CrossRef] [PubMed]
- Guin, J.; Besnard, C.; Lacour, J. Synthesis, resolution, and stabilities of a cationic chromenoxanthene [4]helicene. Org. Lett. 2010, 12, 1748–1751. [Google Scholar]
- Rozen, S.; Dayan, S. At Last, 1,10-Phenanthroline-N,N′-dioxide, A new type of helicene, has been synthesized using HOF small middle dotCH(3)CN. Angew. Chem. Int. Ed. 1999, 38, 3471–3473. [Google Scholar] [CrossRef]
- Talele, H.R.; Sahoo, S.; Bedekar, A.V. Synthesis of chiral helical 1,3-oxazines. Org. Lett. 2012, 14, 3166–3169. [Google Scholar] [CrossRef] [PubMed]
- Jierry, L.; Harthong, S.; Aronica, C.; Mulatier, J.-C.; Guy, L.; Guy, S. Efficient dibenzo[c]acridine helicene-like synthesis and resolution: Scaleup, structural control, and high chiroptical properties. Org. Lett. 2012, 14, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Wynberg, H. Some observations on the chemical, photochemical, and spectral properties of thiophenes. Acc. Chem. Res. 1971, 4, 65–73. [Google Scholar] [CrossRef]
- Janke, R.H.; Haufe, G.; Würthwein, E.-U.; Borken, J.H. Racemization barriers of helicenes: A computational study. J. Am. Chem. Soc. 1996, 118, 6031–6035. [Google Scholar] [CrossRef]
- Groen, M.B.; Wynberg, H. Optical properties of some heterohelicenes. Absolute configuration. J. Am. Chem. Soc. 1971, 93, 2968–2974. [Google Scholar] [CrossRef]
- Fujikawa, T.; Segawa, Y.; Itami, K. Synthesis, structures, and properties of π-extended double helicene: A combination of planar and nonplanar π-systems. J. Am. Chem. Soc. 2015, 137, 7763–7768. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, P.; Xu, L.; Yang, J.; Shi, J.; Wang, Z.; Cheng, Y.; Wang, H. Synthesis for the mesomer and racemate of thiophene-based double helicene under irradiation. J. Org. Chem. 2013, 78, 6316–6321. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.S.; Wise, R.M. The synthesis and resolution of 1,12-dimethylbenzo[c]phenanthrene-5-acetic acid. J. Am. Chem. Soc. 1956, 78, 450–454. [Google Scholar] [CrossRef]
- Martin, R.H. The Helicenes. Angew. Chem. Int. Ed. 1974, 13, 649–660. [Google Scholar] [CrossRef]
- McCarthy, M.; Guiry, P.J. Axially chiral bidentate ligands in asymmetric catalysis. Tetrahedron 2001, 57, 3809–3844. [Google Scholar] [CrossRef]
- Chen, Y.; Yekta, S.; YudiA, K. Modified BINOL ligands in asymmetric catalysis. Chem. Rev. 2003, 103, 3155–3212. [Google Scholar] [CrossRef] [PubMed]
- Berthod, M.; Mignani, G.; Woodward, G.; Marc Lemaire, M. Modified BINAP: The how and the why. Chem. Rev. 2005, 105, 1801–1836. [Google Scholar] [CrossRef] [PubMed]
- Hassine, B.B.; Gorsane, M.; Pecher, J.; Martin, R.H. Diastereoselective sodium borohydride reductions of (dl) α-keto esters. Bull. Soc. Chim. Belg. 1985, 94, 597–603. [Google Scholar] [CrossRef]
- Hassine, B.B.; Gorsane, M.; Geerts-Evrard, F.; Pecher, J.; Martin, R.H.; Castelet, D. Payne syntheses and enantioselective syntheses of epoxides from (E)-stilbene and α-methylstyrene. Bull. Soc. Chim. Belg. 1986, 95, 557–566. [Google Scholar] [CrossRef]
- Hassine, B.B.; Gorsane, M.; Pecher, J.; Martin, R.H. Potential asymmetric syntheses via the ene reaction. Bull. Soc. Chim. Belg. 1987, 96, 801–808. [Google Scholar]
- Hassine, B.B.; Gorsane, M.; Pecher, J.; Martin, R.H. Asymmetric syntheses and potential asymmetric synthesis of α-amino alcohols: Hydroxyamination of olefins by the Sharpless method. Bull. Soc. Chim. Belg. 1985, 94, 759–769. [Google Scholar] [CrossRef]
- Hassine, B.B.; Gorsane, M.; Pecher, J.; Martin, R.H. Atrolactic synthesis in the evaluation of the efficiency of inducers of asymmetric synthesis. Bull. Soc. Chim. Belg. 1986, 95, 547–556. [Google Scholar]
- Yamamoto, K.; Shimizu, T.; Igawa, K.; Tomooka, K.; Hirai, G.; Suemune, H.; Usui, K. Rational design and synthesis of [5]helicene-derived phosphine ligands and their application in Pd-catalyzed asymmetric reactions. Sci. Rep. 2016, 6, 36211. [Google Scholar] [CrossRef] [PubMed]
- Tsujihara, T.; Inada-Nozaki, N.; Takehara, T.; Zhou, D.-Y.; Suzuki, T.; Kawano, T. Nickel-catalyzed construction of chiral 1-[6]helicenols and application in the synthesis of [6]helicene-based phosphinite ligands. Eur. J. Org. Chem. 2016, 2016, 4948–4952. [Google Scholar] [CrossRef]
- Reetz, M.T.; Sostmann, S. Kinetic resolution in Pd-catalyzed allylic substitution using the helical PHelix ligand. J. Organomet. Chem. 2000, 603, 105–109. [Google Scholar] [CrossRef]
- Reetz, M.T.; Sostmann, S. First enantioselective catalysis using a helical diphosphane. Tetrahedron Lett. 1997, 38, 3211–3214. [Google Scholar] [CrossRef]
- Nakano, D.; Yamaguchi, M. Enantioselective hydrogenation of itaconate using rhodium bihelicenol phosphite complex. Matched/mismatched phenomena between helical and axial chirality. Tetrahedron Lett. 2003, 44, 4969–4971. [Google Scholar] [CrossRef]
- Yavari, K.; Aillard, P.; Zhang, Y.; Nuter, F.; Retailleau, P.; Voituriez, A.; Marinetti, A. Helicenes with embedded phosphole units in enantioselective gold catalysis. Angew. Chem. Int. Ed. 2014, 53, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Aillard, P.; Voituriez, A.; Dova, D.; Cauteruccio, S.; Licandro, E.; Marinetti, A. Phosphathiahelicenes: Synthesis and uses in enantioselective gold catalysis. Chem. Eur. J. 2014, 20, 12373–12376. [Google Scholar] [CrossRef] [PubMed]
- Cauteruccio, S.; Dova, D.; Benaglia, M.; Genoni, A.; Orlandi, M.; Licandro, E. Synthesis, characterisation, and organocatalytic activity of chiral tetrathiahelicene diphosphine oxides. Eur. J. Org. Chem. 2014, 2014, 2694–2702. [Google Scholar] [CrossRef]
- Šamal, M.; Misek, J.; Stara, I.G.; Stary, I. Organocatalysis with azahelicenes: The first use of helically chiral pyridine-based catalysts in the asymmetric acyl transfer reaction. Collect. Czechoslov. Chem. Commun. 2009, 74, 1151–1159. [Google Scholar] [CrossRef]
- Misek, J.; Teply, F.; Stara, I.G.; Tichy, M.; Saman, D.; Cisarova, I.; Vojtisek, P.; Stary, I. A straightforward route to helically chiral N-heteroaromatic compounds: Practical synthesis of racemic 1,14-diaza[5]helicene and optically pure 1- and 2-aza[6]helicenes. Angew. Chem. Int. Ed. 2008, 47, 3188–3191. [Google Scholar] [CrossRef] [PubMed]
- Crittall, M.R.; Rzepa, H.S.; Carbery, D.R. Design, synthesis, and evaluation of a helicenoidal DMAP Lewis base catalyst. Org. Lett. 2011, 13, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Ruble, J.C.; Latham, H.A.; Fu, G.C. Effective kinetic resolution of secondary alcohols with a planar−chiral analogue of 4-(Dimethylamino)pyridine. Use of the Fe(C5Ph5) group in asymmetric catalysis. J. Am. Chem. Soc. 1997, 119, 1492–1493. [Google Scholar] [CrossRef]
- Ruble, J.C.; Tweddell, J.; Fu, G.C. Kinetic resolution of arylalkylcarbinols catalyzed by a planar-chiral derivative of DMAP: A new benchmark for nonenzymatic acylation. J. Org. Chem. 1998, 63, 2794–2795. [Google Scholar] [CrossRef]
- Spivey, A.C.; Fekner, T.; Spey, S.E. Axially chiral analogues of 4-(Dimethylamino)pyridine: Novel catalysts for nonenzymatic enantioselective acylations. J. Org. Chem. 2000, 65, 3154–3159. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Takenaka, N. Helical chiral pyridine N-oxides: A new family of asymmetric catalysts. Chem. Eur. J. 2009, 15, 7268–7276. [Google Scholar] [CrossRef] [PubMed]
- Narcis, M.J.; Takenaka, N. Helical-chiral small molecules in asymmetric catalysis. Eur. J. Org. Chem. 2014, 2014, 21–34. [Google Scholar] [CrossRef]
- Takenaka, N.; Sarangthem, R.S.; Captain, B. Helical chiral pyridine N-oxides: A new family of asymmetric catalysts. Angew. Chem. Int. Ed. 2008, 47, 9708–9710. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Captain, B.; Takenaka, N. Helical chiral 2,20-bipyridine N-monoxides as catalysts in the enantioselective propargylation of aldehydes with allenyltrichlorosilane. Org. Lett. 2011, 13, 1654–1657. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Zhu, R.; An, Y.; Wheeler, S.E. Origin of enantioselectivity in the propargylation of aromatic aldehydes catalyzed by helical N-oxides. J. Am. Chem. Soc. 2012, 134, 3095–3102. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, N.; Chen, J.; Captain, B.; Sarangthem, R.S.; Chandrakumar, A. Helical chiral 2-aminopyridinium ions: A new class of hydrogen bond donor catalysts. J. Am. Chem. Soc. 2010, 132, 4536–4537. [Google Scholar] [CrossRef] [PubMed]
- Narcis, M.J.; Sprague, D.J.; Captain, B.; Takenaka, N. Enantio- and periselective nitroalkene Diels–Alder reaction. Org. Biomol. Chem. 2012, 10, 9134–9136. [Google Scholar] [CrossRef] [PubMed]
- Sato, I.; Yamashima, R.; Kadowaki, K.; Yamamoto, J.; Shibata, T.; Soai, K. Asymmetric induction by helical hydrocarbons: [6]- and [5]helicenes. Angew. Chem. Int. Ed. 2001, 40, 1096–1098. [Google Scholar] [CrossRef]
- Kawasaki, T.; Suzuki, K.; Licandro, E.; Bossi, A.; Maiorana, S.; Soai, K. Enantioselective synthesis induced by tetrathia-[7]-helicenes in conjunction with asymmetric autocatalysis. Tetrahedron Asymmetry 2006, 17, 2050–2053. [Google Scholar] [CrossRef]
- Dreher, S.D.; Katz, T.J.; Lam, K.-C.; Rheingold, A.L. Application of the Russig-Laatsch reaction to synthesize a bis[5]helicene chiral pocket for asymmetric catalysis. J. Org. Chem. 2000, 65, 815–822. [Google Scholar] [CrossRef]
- Amemiya, R.; Yamaguchi, M. Chiral recognition in noncovalent bonding interactions between helicenes: Right-handed helix favors right-handed helix over left-handed helix. Org. Biomol. Chem. 2008, 6, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Okubo, H.; Yamaguchi, M.; Kabuto, C. Macrocyclic amides consisting of helical chiral 1,12-dimethylbenzo[c]phenanthrene-5,8-dicarboxylate. J. Org. Chem. 1998, 63, 9500–9509. [Google Scholar] [CrossRef]
- Reetz, M.T.; Sostmann, S. 2,15-Dihydroxy-hexahelicene (HELIXOL): Synthesis and use as an enantioselective fluorescent sensor. Tetrahedron 2001, 57, 2515–2520. [Google Scholar] [CrossRef]
- Nakazaki, M.; Yamamoto, K.; Ikeda, T.; Kitsuki, T.; Okamoto, Y. Synthesis and chiral recognition of novel crown ethers incorporating helicene chiral centres. J. Chem. Soc. Chem. Commun. 1983, 0, 787–788. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ikeda, T.; Kitsuki, T.; Okamoto, Y.; Chikamatsu, H.; Nakazaki, M. Synthesis and chiral recognition of optically active crown ethers incorporating a helicene moiety as the chiral centre. J. Chem. Soc. Perkin Trans. 1 1990, 0, 271–276. [Google Scholar] [CrossRef]
- Pandey, A.D.; Mohammed, H.; Pissurlenkar, R.R.S.; Karnik, A.V. Size-induced chiral discrimination switching by (S)-(-)-2(α-hydroxyethyl)benzimidazole-derived azacrowns. ChemPlusChem 2015, 80, 475–479. [Google Scholar] [CrossRef]
- Weix, D.J.; Dreher, S.D.; Katz, T.J. [5]HELOL phosphite: A helically grooved sensor of remote chirality. J. Am. Chem. Soc. 2000, 122, 10027–10032. [Google Scholar] [CrossRef]
- Wang, D.Z.; Katz, T.J. A [5]HELOL analogue that senses remote chirality in alcohols, phenols, amines, and carboxylic acids. J. Org. Chem. 2005, 70, 8497–8502. [Google Scholar] [CrossRef] [PubMed]
- Schweinfurth, D.; Zalibera, M.; Kathan, M.; Shen, C.; Mazzolini, M.; Trapp, N.; Crassous, J.; Gescheidt, G.; Diederich, F. Helicene quinones: Redox-triggered chiroptical switching and chiral recognition of the semiquinone radical anion lithium salt by electron nuclear double resonance spectroscopy. J. Am. Chem. Soc. 2014, 136, 13045–13052. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Khose, V.N.; Mori, T.; Borovkov, V.; Karnik, A.V. Sui generis helicene-based supramolecular chirogenic system: Enantioselective sensing, solvent control, and application in chiral group transfer reaction. ACS Omega 2017, 2, 592–598. [Google Scholar] [CrossRef]
- Hasan, M.; Pandey, A.D.; Khose, V.N.; Mirgane, N.A.; Karnik, A.V. Sterically congested chiral 7,8-dioxa[6]helicene and Its dihydro analogues: Synthesis, regioselective functionalization, and unexpected domino Prins reaction. Eur. J. Org. Chem. 2015, 2015, 3702–3712. [Google Scholar] [CrossRef]
- Hasan, M.; Khose, V.N.; Pandey, A.D.; Borovkov, V.; Karnik, A.V. Tailor-made supramolecular chirogenic system based on Cs-symmetric rigid organophosphoric acid host and amino alcohols: Mechanistic studies, bulkiness effect, and chirality sensing. Org. Lett. 2016, 18, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Anger, E.; Iida, H.; Yamaguchi, T.; Hayashi, K.; Kumano, D.; Crassous, J.; Vanthuyne, N.; Roussel, C.; Yashima, E. Synthesis and chiral recognition ability of helical polyacetylenes bearing helicene pendants. Polym. Chem. 2014, 5, 4909–4914. [Google Scholar] [CrossRef]
- An, Z.; Yamaguchi, M. Chiral recognition in aggregation of gold nanoparticles grafted with helicenes. Chem. Commun. 2012, 48, 7383–7385. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, W.; Miyagawa, M.; An, Z.; Yamaguchi, M. Optical resolution of aromatic alcohols using silica nanoparticles grafted with helicene. Org. Lett. 2012, 14, 3123–3125. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Ichinose, W.; Yamaguchi, M. Equilibrium shift in solution: Molecular shape recognition and precipitation of a synthetic double helix using helicene-grafted silica nanoparticles. Chem. Eur. J. 2014, 20, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Ishii, R.; Nakagawa, H.; Kawazura, H. Inclusion of Trithia[5]heterohelicene by various serum albumins. An effective probe for chiral discrimination. Chem. Pharm. Bull. 1997, 45, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Kobori, Y.; Yamada, K. Spontaneous chirality conversion of [5]thiaheterohelicene in charge-transfer complexes in SDS micelles. Chirality 2001, 13, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Gomi, K.; Yamada, K. Chiral recognition of thiaheterohelicenes by alkyl b-D-pyranoside micelles. Influence of extension of helix. Chem. Pharm. Bull. 2001, 49, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Kobori, Y.; Yoshida, M.; Yamada, K. Chiral recognition by single bilayered phosphatidylcholine vesicles using [5]thiaheterohelicene as a probe. Chem. Commun. 2001, 0, 2692–2693. [Google Scholar] [CrossRef]
- Nakagawa, H.; Yoshida, M.; Kobori, Y.; Yamada, K. Study of chiral recognition of bilayered phosphatidylcholine vesicles using a helicene probe: Characteristic function of cholesterol. Chirality 2003, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Yamada, K. Difference in chiral recognition of gel and liquid-crystalline phases of phosphatidylcholine vesicles as determined by circular dichroism spectroscopy. Chem. Pharm. Bull. 2005, 53, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Osuga, H.; Suzuki, H.; Shogase, Y.; Kitahara, Y. Synthesis, enzymic resolution and enantiomeric enhancement of bis(hydroxymethyl)[7]thiaheterohelicenes. J. Chem. Soc. Perkin Trans. 1 1998, 0, 935–940. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.X.; Sugiyama, H.; Umano, T.; Osuga, H.; Tanaka, K. (P)-Helicene displays chiral selection in binding to Z-DNA. J. Am. Chem. Soc. 2004, 126, 6566–6567. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Osuga, H.; Kitahara, Y. Elongation and contraction of molecular springs. Synthesis, structures, and properties of bridged [7]thiaheterohelicenes. J. Org. Chem. 2002, 67, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, K.; Sannohe, Y.; Kaieda, S.; Tanaka, K.; Osuga, H.; Tahara, H.; Xu, Y.; Kawase, T.; Bando, T.; Sugiyama, H. A chiral wedge molecule inhibits telomerase activity. J. Am. Chem. Soc. 2010, 132, 3778–3782. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, G.; Kawakami, K.; Sasaki, S. Enantioselective binding of chiral 1,14-dimethyl[5]helicene–spermine ligands with B- and Z-DNA. Bioorg. Med. Chem. 2013, 21, 6063–6068. [Google Scholar] [CrossRef] [PubMed]
- Kel, O.; Frstenberg, A.; Mehanna, N.; Nicolas, C.; Laleu, B.; Hammarson, M.; Albinsson, B.; Lacour, J.; Vauthey, E. Chiral selectivity in the binding of [4]helicene derivatives to double-stranded DNA. Chem. Eur. J. 2013, 19, 7173–7180. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Jiang, L.; Liang, W.; Gui, J.; Xu, D.; Wu, W.; Nakai, Y.; Nishijima, M.; Fukuhara, G.; Mori, T.; et al. Inherently chiral azonia[6]helicene-modified β-cyclodextrin: Synthesis, characterization, and chirality sensing of underivatized amino acids in water. J. Org. Chem. 2016, 81, 3430–3434. [Google Scholar] [CrossRef] [PubMed]
- Okubo, H.; Nakano, D.; Anzai, S.; Yamaguchi, M. Synthesis of symmetrical polynitrohelicenes and their chiral recognition in the charge transfer complexation. J. Org. Chem. 2001, 66, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Murguly, E.; McDonald, R.; Branda, N.R. Chiral discrimination in hydrogen-bonded [7]helicenes. Org. Lett. 2000, 2, 3169–3172. [Google Scholar] [CrossRef] [PubMed]
- Mikes, F.; Boshart, G.; Gil-Av, E. Resolution of optical isomers by high-performance liquid chromatography, using coated and bonded chiral charge-transfer complexing agents as stationary phases. J. Chromatogr. A 1976, 122, 205–221. [Google Scholar] [CrossRef]
- Thongpanchang, T.; Paruch, K.; Katz, T.J.; Rheingold, A.R.; Lam, K.-C.; Liable-Sands, L. Why (1S)-camphanates are excellent resolving agents for helicen-1-ols and why they can be used to analyze absolute configurations. J. Org. Chem. 2000, 65, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- Bucinskas, A.; Waghray, D.; Bagdziunas, G.; Thomas, J.; Grazulevicius, J.V.; Dehaen, W. Synthesis, functionalization, and optical properties of chiral carbazole-based diaza[6]helicenes. J. Org. Chem. 2015, 80, 2521–2528. [Google Scholar] [CrossRef] [PubMed]
- Waghray, D.; Bagdziunas, G.; Jacobs, J.; Meervelt, L.V.; Grazulevicius, J.V.; Dehaen, W. Diastereoselective strategies towards thia[n]helicenes. Chem. Eur. J. 2015, 21, 18791–18798. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Hidehira, Y.; Takahashi, K.; Hiyama, T.; Nozaki, K. Stereospecific synthesis of hetero[7]helicenes by Pd-catalyzed double N-arylation and intramolecular O-arylation. Angew. Chem. Int. Ed. 2005, 44, 7136–7138. [Google Scholar] [CrossRef] [PubMed]
- Sako, M.; Takeuchi, Y.; Tsujihara, T.; Kodera, J.; Kawano, T.; Takizawa, S.; Sasai, T. Efficient enantioselective synthesis of oxahelicenes using redox/acid cooperative catalysts. J. Am. Chem. Soc. 2016, 138, 11481–11484. [Google Scholar] [CrossRef] [PubMed]
- Šámal, M.; Chercheja, S.; Rybáček, J.; Chocholoušová, J.V.; Vacek, J.; Bednárová, L.; Šaman, D.; Stará, I.G.; Starý, I. An ultimate stereocontrol in ssymmetric synthesis of optically pure fully aromatic helicenes. J. Am. Chem. Soc. 2015, 137, 8469–8474. [Google Scholar] [CrossRef] [PubMed]
- Barroso, J.; Cabellos, J.L.; Pan, S.; Murillo, F.; Zarate, X.; Fernandez-Herrera, M.A.; Merino, G. Revisiting the racemization mechanism of helicenes. Chem. Commun. 2018, 54, 188–191. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Conventional aromatic compounds, 1–3, and helicene compounds, 4–6 (numbers in bracket represent ortho-fused aromatic rings).
Figure 1.
Conventional aromatic compounds, 1–3, and helicene compounds, 4–6 (numbers in bracket represent ortho-fused aromatic rings).

Figure 2.
Examples of carbohelicenes: [5]carbohelicenes 7–9 with additional fused rings and functionalized [5]-10, [6]-11, and [7]-12 carbohelicenes.
Figure 2.
Examples of carbohelicenes: [5]carbohelicenes 7–9 with additional fused rings and functionalized [5]-10, [6]-11, and [7]-12 carbohelicenes.

Figure 3.
Examples of heterohelicenes, 13–20.

Figure 4.
Charged helicenes, 21–22.

Figure 5.
Special structural types of helicene 23 and helicene-like molecules 24–25.

Figure 6.
Schematic representation of (a) in-plane turn angle and (b) helicity and enantiomers of [6]helicene.
Figure 6.
Schematic representation of (a) in-plane turn angle and (b) helicity and enantiomers of [6]helicene.

Figure 7.
Examples of meso double helicenes 26–28.

Figure 8.
(a) General explanation of terms used for helicene structures and chiral cavity (shaded area); (b) representative [6]- and [7]helicenes (R = R’ or R ≠ R’) with mono- or di-functionalized peripheral rings.
Figure 8.
(a) General explanation of terms used for helicene structures and chiral cavity (shaded area); (b) representative [6]- and [7]helicenes (R = R’ or R ≠ R’) with mono- or di-functionalized peripheral rings.

Scheme 1.
Diastereoselective reaction using racemic [7]helicene 29 as a chiral auxiliary.

Scheme 2.
Diastereoselective ene reaction using racemic [7]helicene 32 as a chiral auxiliary.

Scheme 3.
Diastereoselective hydroxyamination of stilbene reaction using racemic [7]helicene 33 as a chiral auxiliary.
Scheme 3.
Diastereoselective hydroxyamination of stilbene reaction using racemic [7]helicene 33 as a chiral auxiliary.

Scheme 4.
Enantioselective epoxidation of alkenes 35 and 39 using enantioenriched [7]helicene 38 as a chiral additive.
Scheme 4.
Enantioselective epoxidation of alkenes 35 and 39 using enantioenriched [7]helicene 38 as a chiral additive.

Scheme 5.
Synthesis and resolution of enantiopure phosphine ligands 43 and 44.

Scheme 6.
Pd-catalyzed asymmetric alkylation of rac-1,3-diphenylallyl acetate 50 using helical phosphine ligands 43 and 44.
Scheme 6.
Pd-catalyzed asymmetric alkylation of rac-1,3-diphenylallyl acetate 50 using helical phosphine ligands 43 and 44.

Figure 9.
Other helicene-based phosphorus-containing ligands 53–55 used for Pd-catalyzed allylic substitution reaction of 50 and subsequent kinetic resolution.
Figure 9.
Other helicene-based phosphorus-containing ligands 53–55 used for Pd-catalyzed allylic substitution reaction of 50 and subsequent kinetic resolution.

| (P)-53 | [(η3-C3H5)-PdCl]2 (2.5 mol%), (P)-53 (10 mol%), BSA, LiOAc, CH2Cl2, rt, 24 h | 90 | 96 | [52] |
| (P)-54 | [(η3-C3H5)-PdCl]2 (2.5 mol%), (P)-54 (10 mol%), BSA, LiOAc, CH2Cl2, rt, 24 h | 97 | 99 | [52] |
Scheme 7.
Synthesis and optical resolution of 53 and 54 by using the Ni-catalyzed [2 + 2 + 2] cycloaddition reaction as a key step.
Scheme 7.
Synthesis and optical resolution of 53 and 54 by using the Ni-catalyzed [2 + 2 + 2] cycloaddition reaction as a key step.

Scheme 8.
Pd-catalyzed asymmetric allylic substitution reactions of 50 by using (M)-helicenoid phosphine ligand 43 (a) with indole 63 and (b) with alcohols 65.
Scheme 8.
Pd-catalyzed asymmetric allylic substitution reactions of 50 by using (M)-helicenoid phosphine ligand 43 (a) with indole 63 and (b) with alcohols 65.

Scheme 9.
Pd-catalyzed asymmetric Suzuki–Miyaura coupling between 67 and 68 using (P)-44 as the helicene phosphine ligand.
Scheme 9.
Pd-catalyzed asymmetric Suzuki–Miyaura coupling between 67 and 68 using (P)-44 as the helicene phosphine ligand.

Scheme 10.
Comparative Rh-catalyzed asymmetric hydrogenation of itaconate ester 70 using helicene-based ligands 55 and 72.
Scheme 10.
Comparative Rh-catalyzed asymmetric hydrogenation of itaconate ester 70 using helicene-based ligands 55 and 72.

Scheme 11.
Photochemical diastereoselective synthesis of [6]helicene phosphine oxide 74 and phospha[7]helicene 75.
Scheme 11.
Photochemical diastereoselective synthesis of [6]helicene phosphine oxide 74 and phospha[7]helicene 75.


Scheme 12.
(a) Synthesis of endo (SP,P)-76 and (b) its application as a catalyst for the enantioselective cycloisomerization of N-tethered enyne 78.
Scheme 12.
(a) Synthesis of endo (SP,P)-76 and (b) its application as a catalyst for the enantioselective cycloisomerization of N-tethered enyne 78.

Scheme 13.
Synthesis of azahelicenes 81 and 82 via Co+1-catalyzed [2 + 2 + 2] cyclotrimerization reaction.
Scheme 13.
Synthesis of azahelicenes 81 and 82 via Co+1-catalyzed [2 + 2 + 2] cyclotrimerization reaction.

Scheme 14.
Kinetic resolution of rac-phenylethanol 80 by (M)-81 and (M)-82.

Figure 10.
Structures of simple dimethylaminopyridine (DMAP) and helically, axially, and planar chiral DMAP catalysts 88–90, respectively.
Figure 10.
Structures of simple dimethylaminopyridine (DMAP) and helically, axially, and planar chiral DMAP catalysts 88–90, respectively.

Scheme 15.
Representative general synthetic scheme for 1-azahelicene-N-oxide 93 using Stille–Kelly coupling reaction.
Scheme 15.
Representative general synthetic scheme for 1-azahelicene-N-oxide 93 using Stille–Kelly coupling reaction.

Scheme 16.
Desymmetrization of meso epoxide 97 using 1-azahelicene-N-oxide 91–93 as catalysts.

Scheme 17.
(a) Two-step synthesis of helicene (P)-99 from (P)-93 and (b) asymmetric synthesis of (S)-propargylic alcohols 102 from aldehyde 100 and allenyltrichlorosilane 101 by using (P)-99 as a catalyst.
Scheme 17.
(a) Two-step synthesis of helicene (P)-99 from (P)-93 and (b) asymmetric synthesis of (S)-propargylic alcohols 102 from aldehyde 100 and allenyltrichlorosilane 101 by using (P)-99 as a catalyst.

Figure 11.
Proposed transition state models showing preference for (a) favorable Si-face attack and (b) unfavorable Re-face attack, with (P)-99 catalyst. The bottom and top parts of the transition state models are marked in blue and black, respectively, whilst the coordinated SiCl2 group with the corresponding bonds are marked in magenta.
Figure 11.
Proposed transition state models showing preference for (a) favorable Si-face attack and (b) unfavorable Re-face attack, with (P)-99 catalyst. The bottom and top parts of the transition state models are marked in blue and black, respectively, whilst the coordinated SiCl2 group with the corresponding bonds are marked in magenta.

Scheme 18.
Asymmetric addition reaction between 4,7-dihydroindole 105 and nitrostyrene 106 using chiral (P)-helicenes 103 and 104 as hydrogen bond donor catalysts.
Scheme 18.
Asymmetric addition reaction between 4,7-dihydroindole 105 and nitrostyrene 106 using chiral (P)-helicenes 103 and 104 as hydrogen bond donor catalysts.

Scheme 19.
(M)-helicene 108 as a hydrogen bond catalyst for asymmetric Diels–Alder reaction between cyclopentadiene 109 and nitroethylene 110.
Scheme 19.
(M)-helicene 108 as a hydrogen bond catalyst for asymmetric Diels–Alder reaction between cyclopentadiene 109 and nitroethylene 110.

Scheme 20.
Asymmetric autocatalysis initiated by carbohelicenes (P)-5, (P)-6, and heterohelicene (P)-112.
Scheme 20.
Asymmetric autocatalysis initiated by carbohelicenes (P)-5, (P)-6, and heterohelicene (P)-112.

Scheme 21.
(a) Synthesis of (P,P)-HELOL 115 and (b) its application in asymmetric diethyl zinc addition to benzaldehydes 100.
Scheme 21.
(a) Synthesis of (P,P)-HELOL 115 and (b) its application in asymmetric diethyl zinc addition to benzaldehydes 100.

Scheme 22.
Synthesis of (P,P)-helicene cycloamides 120–122 from diketone 123.

Scheme 23.
(P,P)-helicene cycloamide dimer 120 catalyzed asymmetric diethyl zinc addition to aromatic aldehydes 100.
Scheme 23.
(P,P)-helicene cycloamide dimer 120 catalyzed asymmetric diethyl zinc addition to aromatic aldehydes 100.

Scheme 24.
(a) Synthesis and resolution of HELIXOL host, 127; (b) Structures of chiral guest amines 128–131 and amino alcohols 132–135 used for enantioselective recognition studies.
Scheme 24.
(a) Synthesis and resolution of HELIXOL host, 127; (b) Structures of chiral guest amines 128–131 and amino alcohols 132–135 used for enantioselective recognition studies.

Figure 12.
[5]helicene- and [6]helicene-bearing crown ethers 140 and 141 capable of enantioselective extraction of chiral amine salts 142–144 from aqueous to organic layer.
Figure 12.
[5]helicene- and [6]helicene-bearing crown ethers 140 and 141 capable of enantioselective extraction of chiral amine salts 142–144 from aqueous to organic layer.

Scheme 25.
(a) 31P based diastereomeric NMR sensing of alcohols and amines using (M,M)-HELOL 115 and (b) examples of remote chirality sensing with molecules 147 and 148.
Scheme 25.
(a) 31P based diastereomeric NMR sensing of alcohols and amines using (M,M)-HELOL 115 and (b) examples of remote chirality sensing with molecules 147 and 148.

Scheme 26.
Synthesis and resolution of helicene quinone 149 from triyne 150 using the nickel-catalyzed [2 + 2 + 2] cyclotrimerization reaction as a key step.
Scheme 26.
Synthesis and resolution of helicene quinone 149 from triyne 150 using the nickel-catalyzed [2 + 2 + 2] cyclotrimerization reaction as a key step.

Figure 13.
(a) Mechanism of reversible electrochemical switching of helicene quinone 149 to helicene semiquinone radical anions 149●−; (b) observed reversible chiroptical switching between 149 and 149●−; monitored by CD spectroscopy in acetonitrile. Reversible electrochemical switching for several cycles is shown for the (M)-(−) enantiomer at two different wavelengths (top right) along with the corresponding CD spectra. (Figure 13b) reprinted from [83]. © 2014, American Chemical Society.
Figure 13.
(a) Mechanism of reversible electrochemical switching of helicene quinone 149 to helicene semiquinone radical anions 149●−; (b) observed reversible chiroptical switching between 149 and 149●−; monitored by CD spectroscopy in acetonitrile. Reversible electrochemical switching for several cycles is shown for the (M)-(−) enantiomer at two different wavelengths (top right) along with the corresponding CD spectra. (Figure 13b) reprinted from [83]. © 2014, American Chemical Society.

Figure 14.
ENDOR spectra of diastereomeric species, 153 (black) and 154 (red) in THF at 210 K, only the low-frequency region is shown. Reprinted from [83]. © 2014, American Chemical Society.
Figure 14.
ENDOR spectra of diastereomeric species, 153 (black) and 154 (red) in THF at 210 K, only the low-frequency region is shown. Reprinted from [83]. © 2014, American Chemical Society.

Scheme 27.
Multistep synthesis of helicene camphanate 155 from 2,7-dihydroxynaphthalene 158.

Figure 15.
Fluorescence response of helicene camphanate 155 for trans-1,2-cyclohexanediamine 157 (a) in benzene and (b) in toluene solvent; (c) Schematic representation of the donor–acceptor hydrogen bonding between 155 and 157 in nonpolar solvent with turn-on fluorescence response; (d) Time-dependent changes observed in 1H NMR spectra of host 155 with 1:1 (R,R)-157 in CDCl3. Reprinted from [84]. Source: http://0-pubs-acs-org.brum.beds.ac.uk/doi/abs/10.1021/acsomega.6b00522.
Figure 15.
Fluorescence response of helicene camphanate 155 for trans-1,2-cyclohexanediamine 157 (a) in benzene and (b) in toluene solvent; (c) Schematic representation of the donor–acceptor hydrogen bonding between 155 and 157 in nonpolar solvent with turn-on fluorescence response; (d) Time-dependent changes observed in 1H NMR spectra of host 155 with 1:1 (R,R)-157 in CDCl3. Reprinted from [84]. Source: http://0-pubs-acs-org.brum.beds.ac.uk/doi/abs/10.1021/acsomega.6b00522.

Scheme 28.
(a) Chiral group transfer reaction between 155 and (R,R)-157 in chloroform and (b) proposed shift in conformational dynamic equilibrium of 156 in the presence of (R,R)-157 in chloroform.
Scheme 28.
(a) Chiral group transfer reaction between 155 and (R,R)-157 in chloroform and (b) proposed shift in conformational dynamic equilibrium of 156 in the presence of (R,R)-157 in chloroform.

Scheme 29.
Synthesis of 163 from dihyrofurofuran helicene 159.

Scheme 30.
(a) One step synthesis of polymeric helicene 166 and (b) its application against enantioselective adsorption of racemic guests 165, 167, and 168.
Scheme 30.
(a) One step synthesis of polymeric helicene 166 and (b) its application against enantioselective adsorption of racemic guests 165, 167, and 168.

Figure 16.
Enantiomeric structures of helicene thiol 169 and representations of the gold nanoparticles formed from 169. Modified and reprinted, with permission from [88] © 2012, Royal Society of Chemistry.
Figure 16.
Enantiomeric structures of helicene thiol 169 and representations of the gold nanoparticles formed from 169. Modified and reprinted, with permission from [88] © 2012, Royal Society of Chemistry.

Figure 17.
Helicene silica nanoparticle 170 and its ability towards chiral recognition and resolution of aromatic alcohol 80 and 171 and double helix oligomer 172.
Figure 17.
Helicene silica nanoparticle 170 and its ability towards chiral recognition and resolution of aromatic alcohol 80 and 171 and double helix oligomer 172.

Figure 18.
Stereodynamic hydroxymethyl trithia[5]helicene 173 and a principle of its use as an induced CD host.
Figure 18.
Stereodynamic hydroxymethyl trithia[5]helicene 173 and a principle of its use as an induced CD host.

Scheme 31.
Photochemical synthesis and enzymatic resolution of helicene 174; (a) hv, I2, propylene oxide, benzene, room temperature; (b) tetra-n-butylammonium fluoride, THF, 0 °C; (c) Pseudomonas cepacia, CH2Cl2, vinyl acetate, room temprature, 25 h; (d) Candida antarctica, CH2Cl2, vinyl acetate, 28–29 °C, 9.5 h.
Scheme 31.
Photochemical synthesis and enzymatic resolution of helicene 174; (a) hv, I2, propylene oxide, benzene, room temperature; (b) tetra-n-butylammonium fluoride, THF, 0 °C; (c) Pseudomonas cepacia, CH2Cl2, vinyl acetate, room temprature, 25 h; (d) Candida antarctica, CH2Cl2, vinyl acetate, 28–29 °C, 9.5 h.

Scheme 32.
Synthesis of functionalized helicene 180 and thiaethers 181–183 from [7]helicene 174.

Scheme 33.
Synthesis of helicene-spermine 184.

Figure 19.
Structure of (M)-enantiomers of 191.

Scheme 34.
Synthesis of 3-aza[6]helicene modified β-cyclodextrin 192 as a water-soluble amino acid sensor.
Scheme 34.
Synthesis of 3-aza[6]helicene modified β-cyclodextrin 192 as a water-soluble amino acid sensor.

Figure 20.
Helical discrimination upon supramolecular charge transfer complexation between electron-deficient helicene 195 and electron-rich helicene 196.
Figure 20.
Helical discrimination upon supramolecular charge transfer complexation between electron-deficient helicene 195 and electron-rich helicene 196.

Figure 21.
Hydrogen bonding-based self-assembly in pyridinone-containing helicenes 197–199 and 2-quinolinone 200, as a NMR refrence.
Figure 21.
Hydrogen bonding-based self-assembly in pyridinone-containing helicenes 197–199 and 2-quinolinone 200, as a NMR refrence.

Figure 22.
Selected examples of chiral reagents for helicene optical resolution.

Scheme 35.
Synthesis of chromatographically separable diastereomers 202 and 203 obtained by Pd-catalyzed reaction between rac-azahelicene 201 and (S)-phenylethylamine.
Scheme 35.
Synthesis of chromatographically separable diastereomers 202 and 203 obtained by Pd-catalyzed reaction between rac-azahelicene 201 and (S)-phenylethylamine.

Scheme 36.
Modified BINOL-based vanadium complex 204, catalyzed asymmetric synthesis of oxa[9]helicenes 205 from polycyclic phenols 206.
Scheme 36.
Modified BINOL-based vanadium complex 204, catalyzed asymmetric synthesis of oxa[9]helicenes 205 from polycyclic phenols 206.

Scheme 37.
Asymmetric synthesis of helicenes 210 based on the principle of point-to-helical chirality conversion.
Scheme 37.
Asymmetric synthesis of helicenes 210 based on the principle of point-to-helical chirality conversion.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hasan, M.; Borovkov, V. Helicene-Based Chiral Auxiliaries and Chirogenesis. Symmetry 2018, 10, 10. https://0-doi-org.brum.beds.ac.uk/10.3390/sym10010010
AMA Style
Hasan M, Borovkov V. Helicene-Based Chiral Auxiliaries and Chirogenesis. Symmetry. 2018; 10(1):10. https://0-doi-org.brum.beds.ac.uk/10.3390/sym10010010
Chicago/Turabian StyleHasan, Mohammed, and Victor Borovkov. 2018. "Helicene-Based Chiral Auxiliaries and Chirogenesis" Symmetry 10, no. 1: 10. https://0-doi-org.brum.beds.ac.uk/10.3390/sym10010010
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.