The general method used by
TROVE is thoroughly explained in [
3,
39,
40]. The basic idea is to take advantage of the useful fact that in a symmetry-adapted basis set
, the ro-vibrational Hamiltonian matrix is block diagonal with the form
where the indices
and
label the basis functions,
and
denote irreducible representations of the symmetry group, and
(
) labels the components of the irrep
(
). To construct a symmetrised basis set, one can diagonalise an operator which commutes with all operations of the MS group. Of course, the ro-vibrational Hamiltonian is one such operator, by definition, so
TROVE utilises this by constructing, from the full ro-vibrational Hamiltonian, reduced Hamiltonians that also commute with the group operations, each reduced Hamiltonian depending on fewer coordinates than the full Hamiltonian. The reduced Hamiltonians are diagonalised separately and products of their—by necessity symmetrised—eigenfunctions are again symmetrised. The result is a symmetry-adapted basis set appropriate for the complete ro-vibrational coordinate space. We describe this symmetrisation procedure for ethane in the following sections.
6.1. Definition of the Internal Coordinates Used for Ethane
To construct the reduced Hamiltonians mentioned in the preceding section, one first separates the internal coordinates into subsets which transform into each other under the symmetry group operations. We use the convention in Section 1.2 of [
1] for transforming functions of Cartesian coordinates. The basis set associated with each coordinate subset is obtained in terms of one-dimensional primitive functions
, where
is the number of quanta for coordinate
. By averaging the Hamiltonian over the “ground state” (
;
) primitives associated with the coordinates in all subsets not under consideration, i.e.
where
are the coordinates in the subset of interest, one obtains the reduced Hamiltonian which depends only on these coordinates; it commutes with the operations in the MS group. Product functions of the form
are then used as a basis set for diagonalising this reduced Hamiltonian.
In the case of ethane, there are
small-amplitude vibrational coordinates, one torsional coordinate (describing the independent rotations of the CH
3 groups), and three rotational coordinates.
Figure 3 shows representative members of three of the vibrational coordinate subsets: the C−C bond length denoted by
R, one of six C−H
k bonds denoted by
r, and one of six bond angles ∠(H
–C–C) denoted by
. The small-amplitude vibrational coordinates
R,
, and
(
) measure the displacements of the respective internal coordinates from their equilibrium values, that is the coordinates actually used are
,
(
) and
(
). The
(
) coordinates are equivalent and so they have a common equilibrium value
(
).
The last vibrational subset is obtained from six dihedral angles
,
,
,
,
,
, one of which is labelled
in
Figure 4.
is the angle between the H
i−C−C and H
j−C−C planes, where protons
i and
j belong to the same CH
3 group. Only four of the six angles are linearly independent due to the constraints
=
=
. The positive directions of rotation for the
angles are from proton 1 → 2 → 3 for the one CH
3 group, and from proton 4 → 5 → 6 for the other. The independent coordinates constructed from the six
angles are
which transform as the
G representation of
G.
To describe the orientation of the ethane molecule with Euler angles, one approach is to attach coordinate axes on each CH
3 group. Here, the
z axis is the same for both and points from C
b to C
a, while the
and
axes point in the direction of C
a-H
1 and C
b-H
4, respectively, when the molecule is viewed in the Newman projection of
Figure 5. The
y axes ensure that the Cartesian axes are right handed. With this construction, the
and
Euler angles describing the direction of the
z axis for CH
3 group are the same while the
angles describing the rotation about the
z axis are different and are denoted by
and
. These increase in the counterclockwise direction due to the right hand rule. This essentially follows Section 15.4.4 of [
1] in the definition of the Euler angles according to the convention described in Section 10.1.1 of [
1].
As described in [
1], to achieve maximum separation of the torsional and rotational motion, it is expedient to define two new coordinates from
and
. We set the rotational coordinate
to
and hence our
x axis shown in
Figure 4 halves the angle between H
1−C−C and H
4−C−C and increases in the counterclockwise direction. The torsional angle
could be defined as
and hence, as indicated in
Figure 4, is the angle from H
4−C−C to H
1−C−C in the counterclockwise direction. In the TROVE calculations, we use a different choice and define
as the average of three dihedral angles. To define these, we form three pairs of protons
,
, and
with the two members belonging to different CH
3 groups. The pairs are chosen such that the protons
i and
j in each
pair form a dihedral angle
of
in the staggered equilibrium geometry of
Figure 1 and
for the eclipsed geometry (see
Figure 6), using the labelling of
Figure 2. In a general instantaneous geometry, each
pair defines a dihedral angle
(where the positive direction of rotation for the
angles is from proton 1 → 2 → 3) and the torsional angle is then given by a symmetric combination
With this definition,
,
and
correspond to eclipsed configurations, while, at
,
, and
, the molecule is in one of its three equilibrium geometries. The torsional angle
has definite transformation properties under the operations of
G (see also
Appendix D). As discussed in
Section 6.3, the two coordinate-pair values (
,
) and (
,
) describe the same physical situation. However, the coordinate which
is based on,
, has a range of
. Although a given geometry can be described by a value of
in the interval
, we must allow
to range over
to obtain a correct correlation with
(see
Section 6.3 below).
In conclusion, the coordinate subsets, for which we initially diagonalise reduced Hamiltonians, are
the C−C bond length R;
six C−H bond lengths , k = 1, 2, …, 6;
six bond angles ∠(H-C-C) = , k = 1, 2, …, 6;
four dihedral-angle coordinates , , , and ,
the torsional angle ; and
the three rotational angles .
The primitive basis functions for subsets 1, 2, 3, and 5 are obtained using the Numerov–Cooley approach [
3,
41,
42], while we use harmonic-oscillator eigenfunctions [
1] for Subset 4. The primitive basis set 5 is obtained by solving the 1D torsional Schrödinger equation
using the basis set constructed from the normalised,
-periodic Fourier series functions
and
. Here,
is the purely torsional element of the TROVE kinetic-energy
g-matrix (see [
3]) and
is the one-dimensional,
-periodic torsional part of the 18D potential energy function of ethane with the remaining 17 vibrational coordinates set to their equilibrium values. For both the kinetic-energy function
and the potential function
, we must consider
-values in the extended interval
as detailed in
Section 6.3 below.
6.2. Transformation of the Vibrational Coordinates under G36
As described in
Section 3, one can construct every operation of
G with only four generating operations. In the
TROVE calculations, we use the generators
,
,
and
. Then, to describe the transformation properties of a coordinate for all elements in
G, we need only determine these properties for the four generating operations. We have implemented in
TROVE the procedure to generate the irreducible representations of
G based on these four group generators and the multiplication rules in
Table 1. The latter can be conveniently represented as the following recursive rule:
where the operations
,
and
(
) are as organised in
Table 3.
Each of the vibrational coordinates and the torsional coordinate can be expressed as a function of the nuclear Cartesian coordinates. In the following, and in
Appendix C, we describe the procedure in determining the transformation of these coordinates under the
G operations in a systematic way, although in the majority of cases the result is intuitive.
The C−C bond length R is invariant under all G operations. We label the six C−Hk bond lengths , and the six bond angles ∠H-C-C = , by the generic labels , as the two subsets transform identically. The transformation properties are determined by recognising that after the operation ∈ G has been carried out, proton 1, e.g., occupies the position previously occupied by the proton k (and the C nucleus to which it is bound has moved with it), and therefore the transformed value of the C−H1 bond length is = , the original value of the C−Hk bond length, with analogous considerations for the bond lengths , …, , and the angles.
For (, , ), we obtain the following transformation properties under the generating operations:
for (123)(456), for
for , for
while for they are given by
for , for
for , for .
The dihedral-angle coordinates
are mixed by the
G operations. The transformation properties are most easily determined by using the correspondence with the transformation of the
and
coordinates. For
, we write
with
where we have taken into account the constraint
=
in the third row of
.
After we have carried out the operation
, protons 1, 2, and 3 are found at the positions initially occupied by protons 3, 1, and 2, respectively, and so the angles
,
,
are permuted as follows
with
and we can finally calculate the transformed values of
as
where
With the upper 2 × 2 corner of this matrix, we can express the transformed values in terms of . The transformation matrix for for , and the ones for the other generating operations, obtained in a similar manner, are
for , for
for , for
and those for (, ) are given by
for , for
for , for
6.3. The Extended Molecular Symmetry Group G36(EM) and the Transformation of the Torsional Coordinate
As explained in Section 15.4.4 of [
1], our separation of the rotational and torsional degrees of freedom has led
and
being double-valued. That is, there are two sets of
values associated with the same physical situation. This is most straightforwardly seen by considering Equation (
18) and the three angles
,
, and
appearing in it. The angle
is determined entirely by the positions in space of protons 1, 2, 3 and their carbon nucleus C
;
is determined by the positions of protons 4, 5, 6 and their carbon nucleus C
; and
is the average of the two. Due to the
periodicity of
, increasing it by
does not change the positions in space of the nuclei in C
aH
3, however, in this case,
→
and
. That is, the two coordinate pairs (
,
) and (
,
) describe identical physical situations.
One way of avoiding the ambiguity described above would be to use a molecule-fixed axis system with, for example,
=
. This molecule-axis system is attached to the CH
3 group with protons 1, 2, 3, and not influenced by the other CH
3 group. A
coordinate chosen in this manner has no ambiguity. However, as already mentioned it is advantageous to choose
according to Equation (
18) since this choice (called the Internal Axis Method (IAM), see Section 15.2.2 of [
1]) yields a particularly useful form of the rotation-torsion-vibration Hamiltonian with minimised coupling between rotation and torsion. It is desirable to keep the definition of
from Equation (
18) and find a way of treating the ambiguity. We use here the procedure first proposed by Hougen [
43] in 1964, primarily for dimethylacetylene H
CCCCH
, whose internal rotation is essentially free, but generally applicable to other molecules such as ethane H
CCH
, hydrogen peroxide HOOH, and disulfane HSSH with two identical moieties carrying out internal rotation. Recently, these ideas have been extended to molecules with two different moieties and applied to oxadisulfane (also known as hydrogen thio-peroxide) HSOH [
44,
45], and more recently they have been applied to computation of the rotation-torsion-vibrational spectra of hydrogen peroxide HOOH in Ref. [
21,
26], where the HOOH kinetic energy operator was built using the
x-axis chosen to halve the dihedral angle between the H
1−O−O and H
4−O−O planes, as well as in Ref. [
46].
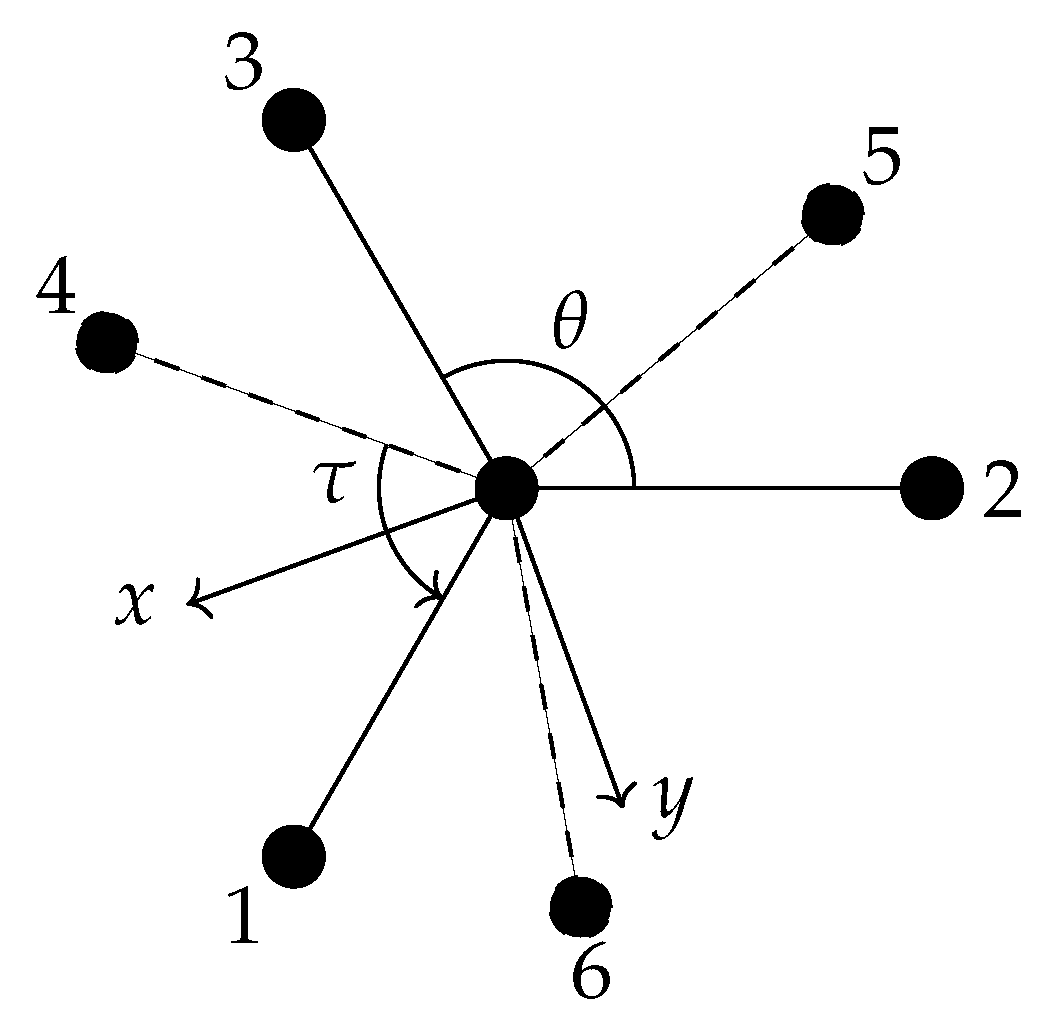
We consider the effect of
G operations on
, described thoroughly in Section 12.1.1 of [
1], where the axes are rotated in such a way that the coordinates of the nuclei—measured in a space-fixed axis system—remain the same after the operation, assuming the C
aH
3 group is in the equilibrium configuration. Considering the operation (123), we see that, using our convention,
becomes
or, equivalently,
. This would correspond to the pair
becoming
or
, which are no longer equivalent but correspond to the same physical situation as noted before. For
, the changes become
or
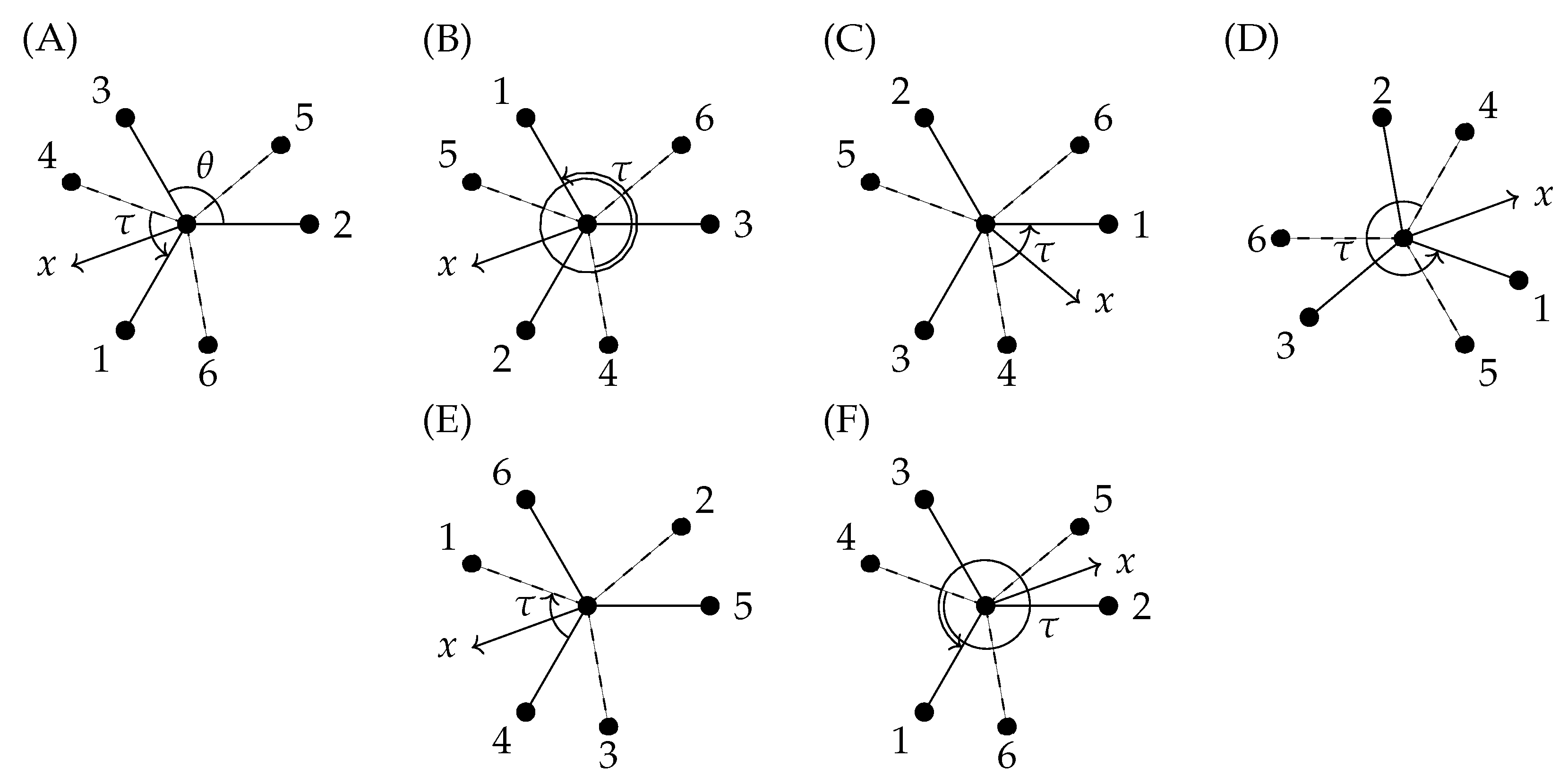
. These are illustrated in
Figure 7A,F, respectively.
To deal with the double-valuedness of (
,
), we extend the symmetry description in the manner first introduced by Hougen [
43], by extending
G to the extended molecular symmetry group
G(EM) as explained in Section 15.4.4 of [
1]. To appreciate the definition of
G(EM), we note that the internal coordinates
R,
and
(
k = 1, 2, …, 6),
,
,
,
,
, and
used for ethane in the present work are all “space-fixed” in the sense of Section 15.4.4 in [
1]; the instantaneous values of these coordinates can be unambiguously obtained from the instantaneous coordinate values of the nuclei in a Cartesian, space-fixed axis system [
1], e.g.,
.
Appendix C explains in detail how the internal coordinates values are determined from the Cartesian coordinate values.
We present here the ideas of Hougen [
43], using the more modern notation of Section 15.4.4 in [
1]. The extension of
G to
G(EM) involves the introduction of a fictitious operation
(taken to be different from the identity
E) which, for ethane, we can think of as letting the C
aH
3 do a full torsional revolution relative to the C
bH
3. That is,
has the effect of transforming
→
and
→
. After the application of
the molecule-fixed axis system is back where it started, and so we take
=
E.
does not affect the space-fixed nuclear coordinates; it has the same effect as the identity operation on the complete rotation-torsion-vibration wavefunction of ethane.
We now define four generating operations
a,
b,
c and
d that transform
and
unambiguously and have the same effect on the space-fixed coordinates [
R,
and
(
k = 1, 2, …, 6),
,
,
,
,
, and
] as the
G operations (123), (456), (14)(26)(35)(
)
and (23)(56)
, respectively. Tables A-28 and A-33 of [
1] are the character tables of
G and
G(EM), respectively. Comparison of the two tables shows that the four generating operations
,
,
and
chosen for
G in the present work correspond to
,
,
c, and
, respectively. Following Section 15.4.4 of [
1], we
define in
Table 4 the effect on
and
of the
G(EM) generators. The approach of Bunker and Jensen [
1] to simply postulate, by definition, the effect of the
G(EM) generators on
and
may appear arbitrary. It is not, however. We show in
Figure 7 how, for each of the four
G(EM) generators
,
,
c, and
, the effect on
and
can be explained as the effect of the
G partner of the
G(EM) generator in question (
Table 4).
The group generated by the five operations
a,
b,
c,
d, and
has 72 elements and, as mentioned above, it is denoted
G(EM), called an extended molecular symmetry (EMS) group, and its character table is given as Table A-33 of [
1]. This character table shows that we can think of
G(EM) as a direct product
where the group
is constructed from the generating operations
a,
b,
c and
d; it is isomorphic to
G, so that it has the irreducible representations given in
Table 2. The two-element group
is cyclic of order 2 and has two irreps
and
, both one-dimensional, with the representation matrices 1 or
, respectively, under
. The irreps of
G(EM) are straightforwardly constructed from those of
G. Each irrep
of
G in
Table 2 gives rise to two irreps of
G(EM),
=
and
=
as given in Table A-33 of [
1]. An irrep
has identical characters for the operations
E and
,
=
, whereas for the irrep
,
=
. As long as we pretend that
≠
E, we must also pretend that the coordinate values
and
=
describe different physical situations. As a consequence, we must allow
to be periodic with a period of
as already mentioned in connection with Equation (
21). The torsional potential energy function
is periodic with period
,
=
for
∈
, and this symmetry causes the torsional wavefunctions
from Equation (
21) to be either symmetric (of
symmetry) or antisymmetric (of
symmetry) under
.
We know that, in reality, = E, and so only functions and coordinates with symmetries occur in nature. Since we can form, for example, basis functions of an allowed symmetry as products of an even number of factors, each with a forbidden symmetry , say, we need to consider also the symmetries initially for the torsional and rotational basis functions. The final wavefunctions resulting from our theoretical calculations should be subjected to a “reality check”: They must necessarily have a symmetry in G(EM). In particular, torsional basis functions of d symmetry must be combined with rotational basis functions of d symmetry to produce a torsion-rotation basis function of an allowed s symmetry.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













