Potentially Mistaking Enantiomers for Different Compounds Due to the Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon


Abstract
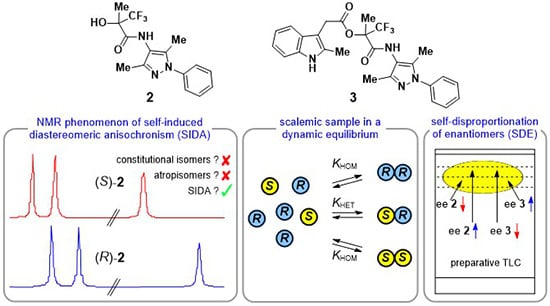
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Spectroscopy
2.2. Preparative TLC
3. Results and Discussion
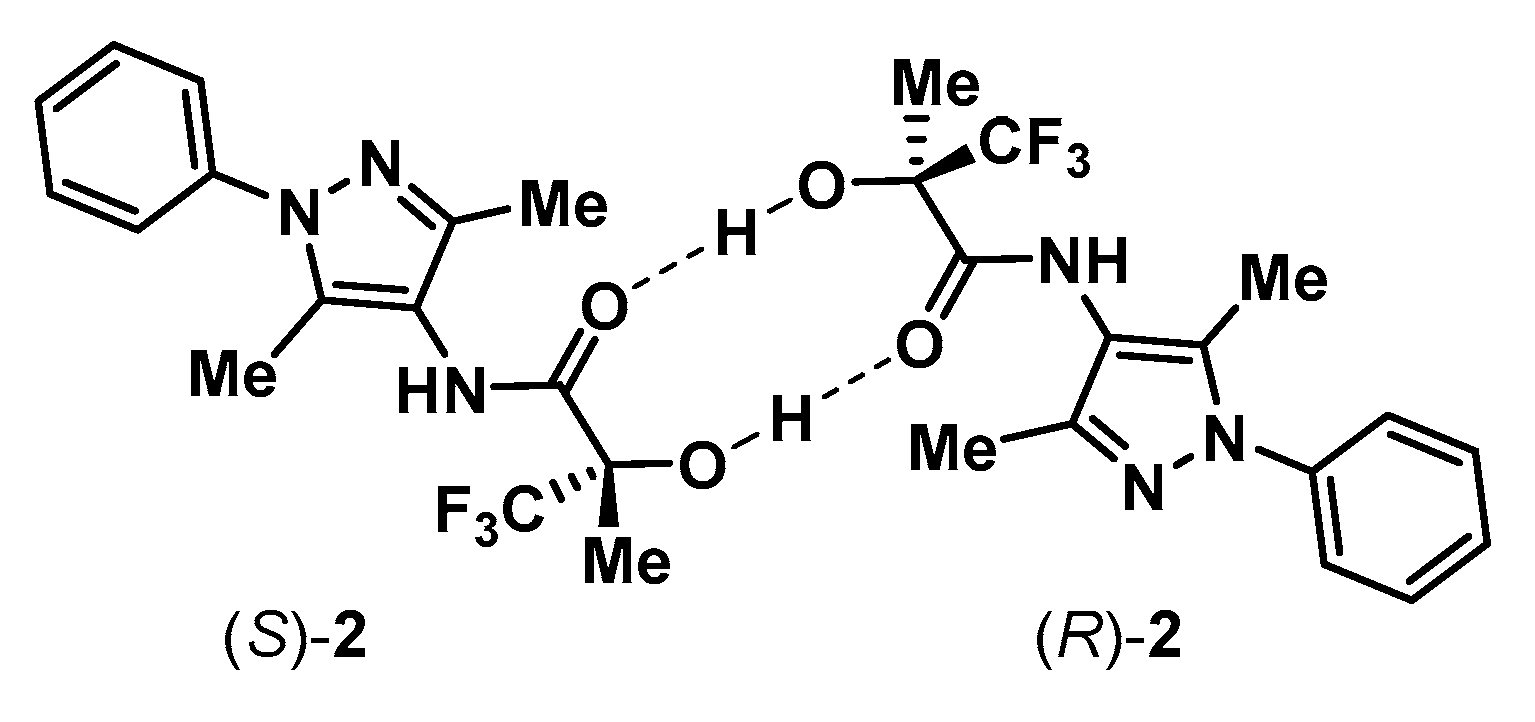
3.1. Analysis of Alcohols 2
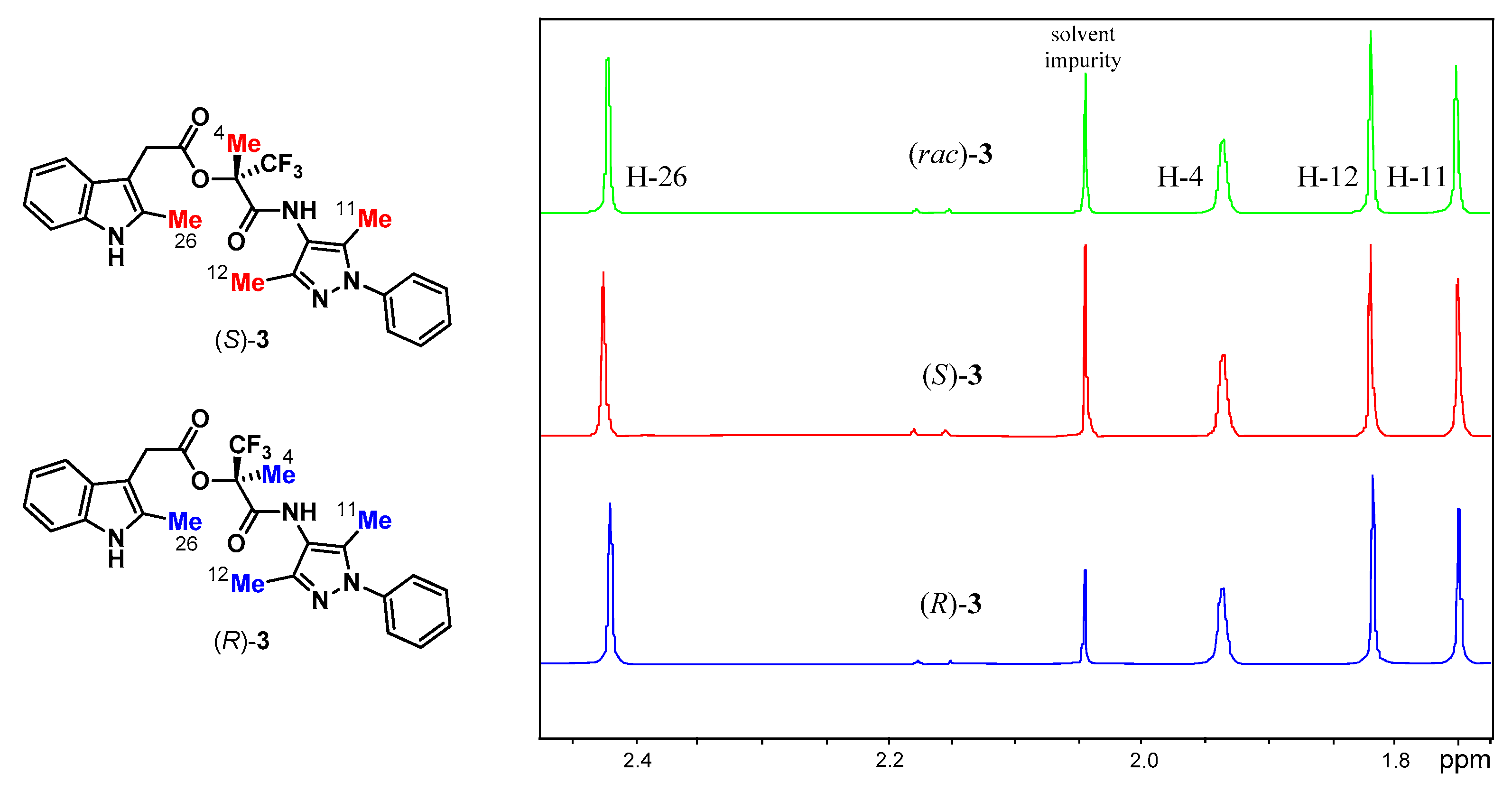
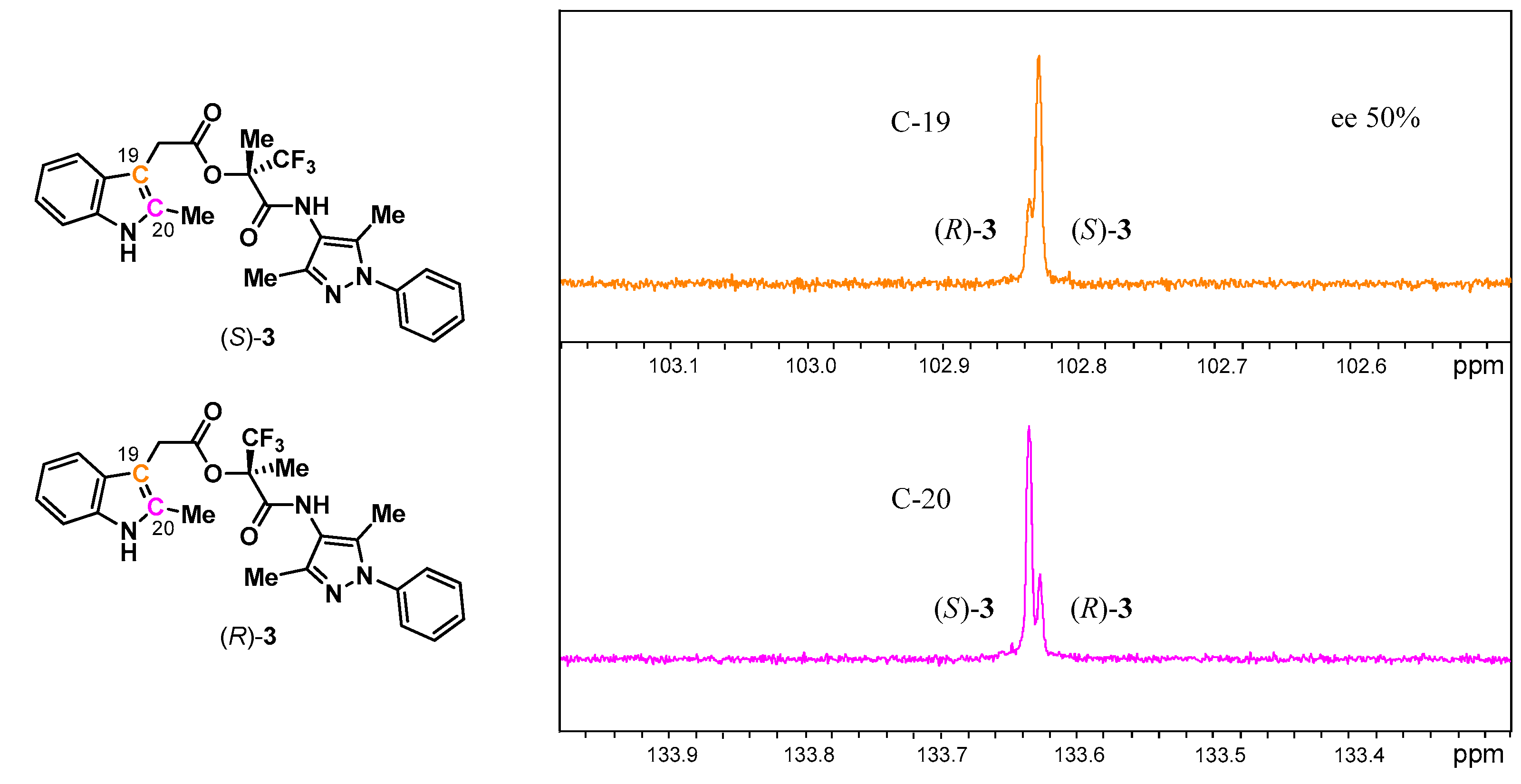
3.2. Analysis of Esters 3
4. Conclusions and Final Comments
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Background to SIDA and SDE
Appendix A.1. SIDA
Appendix A.1.1. The Origin of SIDA

Appendix A.1.2. Potential Sources of Error in the Quantitation of Ee by SIDA
Appendix A.1.3. Identification of Associate Preference by Diffusion
Appendix A.2. SDE
Appendix A.3. Some Notes on Terminology
References
- Szakács, Z.; Sánta, Z.; Lomoschitz, A.; Szántay, C., Jr. Self-induced recognition of enantiomers (SIRE) and its application in chiral NMR analysis. Trends Anal. Chem. 2018, 109, 180–197. [Google Scholar] [CrossRef]
- Nieminen, V.; Murzin, D.Y.; Klika, K.D. NMR and molecular modeling of the dimeric self-association of the enantiomers of 1,1’-bi-2-naphthol and 1-phenyl-2,2,2-trifluoroethanol in the solution state and their relevance to enantiomer self-disproportionation on achiral-phase chromatography (ESDAC). Org. Biomol. Chem. 2009, 7, 537–542. [Google Scholar] [CrossRef]
- Storch, G.; Haas, M.; Trapp, O. Attracting Enantiomers: Chiral Analytes That Are Simultaneously Shift Reagents Allow Rapid Screening of Enantiomeric Ratios by NMR Spectroscopy. Chem. Eur. J. 2017, 23, 5414–5418. [Google Scholar] [CrossRef]
- Klika, K.D.; Budovská, M.; Kutschy, P. Enantiodifferentiation of phytoalexin spirobrassinin derivatives using the chiral solvating agent (R)-(+)-1,1’-bi-2-naphthol in conjunction with molecular modeling. Tetrahedron Asymmetry 2010, 21, 647–658. [Google Scholar] [CrossRef]
- Soloshonok, V.A. Remarkable amplification of the self-disproportionation of enantiomers on achiral-phase chromatography columns. Angew. Chem. Int. Ed. 2006, 45, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Roussel, C.; Kitagawa, O.; Sorochinsky, A.E. Self-disproportionation of enantiomers via achiral chromatography: A warning and an extra dimension in optical purifications. Chem. Soc. Rev. 2012, 41, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Klika, K.D. Terminology related to the phenomenon ‘self-disproportionation of enantiomers’ (SDE). Helv. Chem. Acta 2014, 97, 1583–1589. [Google Scholar] [CrossRef]
- Han, J.; Kitagawa, O.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. The self-disproportionation of enantiomers (SDE): A menace or an opportunity? Chem. Sci. 2018, 9, 1718–1739. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Katagiri, T.; Soloshonok, V.A. Unconventional preparation of racemic crystals of isopropyl 3,3,3-trifluoro-2-hydroxypropanoate and their unusual crystallographic structure: The ultimate preference for homochiral intermolecular interactions. Chem. Commun. 2013, 49, 373–375. [Google Scholar] [CrossRef]
- Wu, D.H.; Chen, A.D.; Johnson, C.S., Jr. An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses. J. Magn. Reson. Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- De Vita, E.; Schüler, P.; Lovell, S.; Lohbeck, J.; Kullmann, S.; Rabinovich, E.; Sananes, A.; Hessling, B.; Hamon, V.; Papo, N. Depsipeptides Featuring a Neutral P1 Are Potent Inhibitors of Kallikrein-Related Peptidase 6 with On-Target Cellular Activity. J. Med. Chem. 2018, 61, 8859–8874. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Katagiri, T.; Ono, T.; Wzorek, A.; Aceña, J.L.; Soloshonok, V.A. Optical purifications via self-disproportionation of enantiomers by achiral chromatography: Case study of a series of α-CF3-containing secondary alcohols. Chirality 2013, 25, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Aceña, J.L.; Soloshonok, V.A. Self-disproportionation of enantiomers of chiral, non-racemic fluoroorganic compounds: Role of fluorine as enabling element. Synthesis 2013, 45, 141–152. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ueki, H.; Yasumoto, M.; Mekala, S.; Hirschi, J.S.; Singleton, D.A. Phenomenon of Optical Self-Purification of Chiral Non-Racemic Compounds. J. Am. Chem. Soc. 2007, 129, 12112–12113. [Google Scholar] [CrossRef]
- Yasumoto, M.; Ueki, H.; Soloshonok, V.A. Self-disproportionation of enantiomers of 3,3,3-trifluorolactic acid amides via sublimation. J. Fluor. Chem. 2010, 131, 266–269. [Google Scholar] [CrossRef]
- Yasumoto, M.; Ueki, H.; Soloshonok, V.A. Self-disproportionation of enantiomers of α-trifluoromethyl lactic acid amides via sublimation. J. Fluor. Chem. 2010, 131, 540–544. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Orita, H.; Ueki, H.; Soloshonok, V.A. First principle lattice energy calculations for enantiopure and racemic crystals of α-(trifluoromethyl)lactic acid: Is self-disproportionation of enantiomers controlled by thermodynamic stability of crystals? J. Fluor. Chem. 2010, 131, 461–466. [Google Scholar] [CrossRef]
- Albrecht, M.; Soloshonok, V.A.; Schrader, L.; Yasumoto, M.; Suhm, M.A. Chirality-dependent sublimation of α-(trifluoromethyl)-lactic acid: Relative vapor pressures of racemic, eutectic, and enantiomerically pure forms, and vibrational spectroscopy of isolated (S,S) and (S,R) dimers. J. Fluor. Chem. 2010, 131, 495–504. [Google Scholar] [CrossRef]
- Yasumoto, M.; Ueki, H.; Ono, T.; Katagiri, T.; Soloshonok, V.A. Self-disproportionation of enantiomers of isopropyl 3,3,3-(trifluoro)lactate via sublimation: Sublimation rates vs. enantiomeric composition. J. Fluor. Chem. 2010, 131, 535–539. [Google Scholar] [CrossRef]
- Katagiri, T.; Takahashi, S.; Tsuboi, A.; Suzaki, M.; Uneyama, K. Discrimination of enantiomeric excess of optically active trifluorolactate by distillation: Evidence for a multi-center hydrogen bonding network in the liquid state. J. Fluor. Chem. 2010, 131, 517–520. [Google Scholar] [CrossRef]
- Katagiri, T.; Uneyama, K. Chiral Recognition by Multicenter Single Proton Hydrogen Bonding of Trifluorolactates. Chem. Lett. 2001, 30, 1330–1331. [Google Scholar] [CrossRef]
- Katagiri, T.; Yoda, C.; Furuhashi, K.; Ueki, K.; Kubota, T. Separation of an Enantiomorph and Its Racemate by Distillation: Strong Chiral Recognizing Ability of Trifluorolactates. Chem. Lett. 1996, 25, 115–116. [Google Scholar] [CrossRef]
- Han, J.; Nelson, D.J.; Sorochinsky, A.E.; Soloshonok, V.A. Self-disproportionation of enantiomers via sublimation; new and truly green dimension in optical purification. Curr. Org. Synth. 2011, 8, 310–317. [Google Scholar] [CrossRef]
- Wallach, O. Zur Kenntniss der Terpene und der ätherischen Oele. Ueber gebromte Derivate der Carvonreihe. Justus Liebigs Ann. Chem. 1895, 286, 119–143. [Google Scholar] [CrossRef]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates, and Resolutions; J. Wiley & Sons, Inc.: New York, NY, USA, 1981. [Google Scholar]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A.; Klika, K.D. Enantiomeric Enrichments via the Self-Disproportionation of Enantiomers (SDE) by Achiral, Gravity-Driven Column Chromatography: A Case Study Using N-(1-Phenylethyl)acetamide for Optimizing the Enantiomerically Pure Yield and Magnitude of the SDE. Helv. Chim. Acta 2015, 98, 1147–1159. [Google Scholar] [CrossRef]
- Kwiatkowska, M.; Marcinkowska, M.; Wzorek, A.; Pajkert, R.; Han, J.; Klika, K.D.; Soloshonok, V.A.; Röschenthaler, G.-V. The self-disproportionation of enantiomers (SDE) via column chromatography of β-amino-α,α-difluorophosphonic acid derivatives. Amino Acids 2019, 51, 1377–1385. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE): The effect of scaling down, potential problems versus prospective applications, possible new occurrences, and unrealized opportunities? Electrophoresis 2019, 40, 1869–1880. [Google Scholar] [CrossRef]
- Nakamura, T.; Tateishi, K.; Tsukagoshi, S.; Hashimoto, S.; Watanabe, S.; Soloshonok, V.A.; Aceña, J.L.; Kitagawa, O. Self-disproportionation of enantiomers of non-racemic chiral amine derivatives through achiral chromatography. Tetrahedron 2012, 68, 4013–4017. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumann, A.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D.; Miller, A.K. Potentially Mistaking Enantiomers for Different Compounds Due to the Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon. Symmetry 2020, 12, 1106. https://0-doi-org.brum.beds.ac.uk/10.3390/sym12071106
Baumann A, Wzorek A, Soloshonok VA, Klika KD, Miller AK. Potentially Mistaking Enantiomers for Different Compounds Due to the Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon. Symmetry. 2020; 12(7):1106. https://0-doi-org.brum.beds.ac.uk/10.3390/sym12071106
Chicago/Turabian StyleBaumann, Andreas, Alicja Wzorek, Vadim A. Soloshonok, Karel D. Klika, and Aubry K. Miller. 2020. "Potentially Mistaking Enantiomers for Different Compounds Due to the Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon" Symmetry 12, no. 7: 1106. https://0-doi-org.brum.beds.ac.uk/10.3390/sym12071106