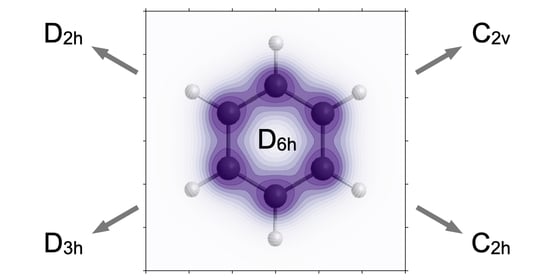
3.1. Electron Symmetry Breaking and Charge Migration Induced by Linearly Polarized Laser Pulses in Oriented Benzene with D6h Nuclear Scaffold
This Section presents the results of quantum dynamics simulations (cf.
Section 2.6) of eight different examples of electron symmetry breaking in the oriented model benzene (cf.
Section 2.2) induced by eight different linearly polarized laser pulses labeled
p = 1, 2, 3, …, or two sequential laser pulses (cf.
Section 2.1). The symmetry breakings G = D
6h → S(ρ
p(
r,t)) yield eight different point subgroups S(ρ
p(
r,t)) of G for the one-electron densities ρ
p(
r,t) (cf.
Section 2.8) which present eight different types of attosecond charge migration, right after the end of the laser pulse(s). The durations of the laser pulses, or two sequential laser pulses, are restricted to 10 fs, such that the nuclear scaffolds may be considered as frozen [
7,
8], implying conservation of nuclear D
6h symmetry.
The first two examples (labeled p = 1, 2) are motivated by the results of Ref. [
10] which discovered quantum control of electronic fluxes during attosecond charge migration in superposition states of oriented benzene. The charge migration is illustrated in Ref. [
10] by means of snapshots of the one-electron density. As a byproduct, but without explicit observation (even without mentioning the word “symmetry”!), Ref. [
10] demonstrates two different C
2v-type symmetries of the time-dependent one-electron densities which are generated by two different linearly
ex and
ey polarized π/2 laser pulses with Gaussian shapes. Here, we employ two similar re-optimized π/2 laser pulses with sin
2-shapes (cf.
Section 2.1 and
Section 2.9), labeled p = 1 and 2, which are tailored to selective transitions from the initial (t = 0) electronic ground state |1A
1g> to the superposition states (1/√2)*(|1A
1g> + |1E
1ux>) and (1/√2)*(|1A
1g> + |1E
1uy>), respectively. The notation {|1E
1ux>, |1E
1uy>} denotes the doublet of two orthogonal degenerate states with IRREP E
1u and with the lowest energy E
1E1u which transform as the functions x and y (in brief: as x and y), respectively [
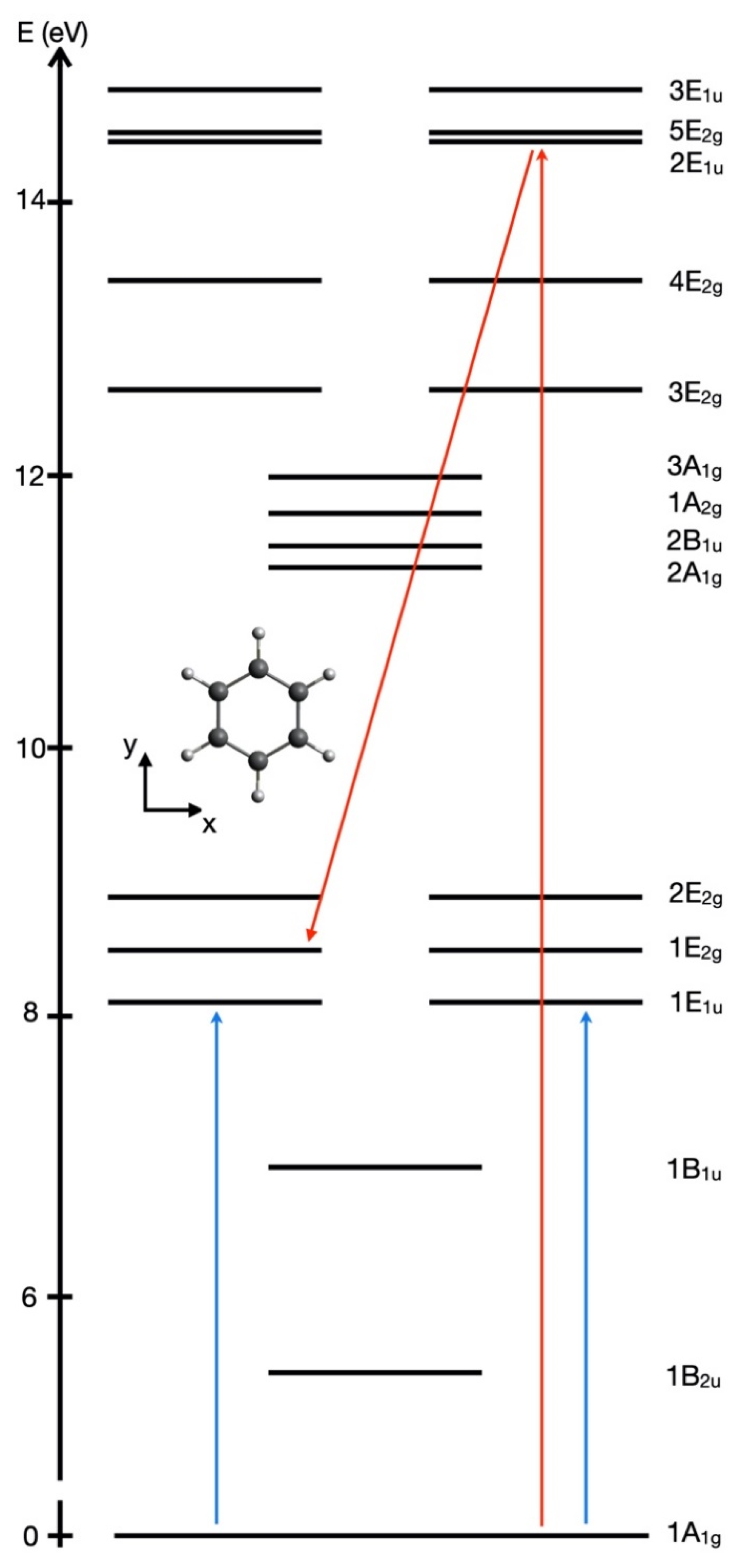
13]. The target transitions are illustrated schematically by vertical arrows from the energy level E
1A1g to E
1E1u in
Figure 3. The energy gap between their eigenenergies is ΔE
1A1g,1E1u = E
1E1u − E
1A1g = 8.17 eV with corresponding transition frequency ω
1A1g,1E1u = ΔE
1A1g,1E1u /ℏ and period T = T
1A1g,1E1u = 2π/ω
1A1g,1E1u = h/ ΔE
1A1g,1E1u = 506 as, cf. Equations (42) and (43). The pulses have the same electric field amplitudes
1(t) =
2(t) ≡
(t) which is shown in
Figure 5, but different polarization vectors
e1 =
ex =
e”
1 and
e2 =
ey =
e’
1. The identical laser parameters are specified in
Figure 5; the frequencies ω
1 = ω
2 are resonant with the transition frequency ω
1A1g,1E1u.
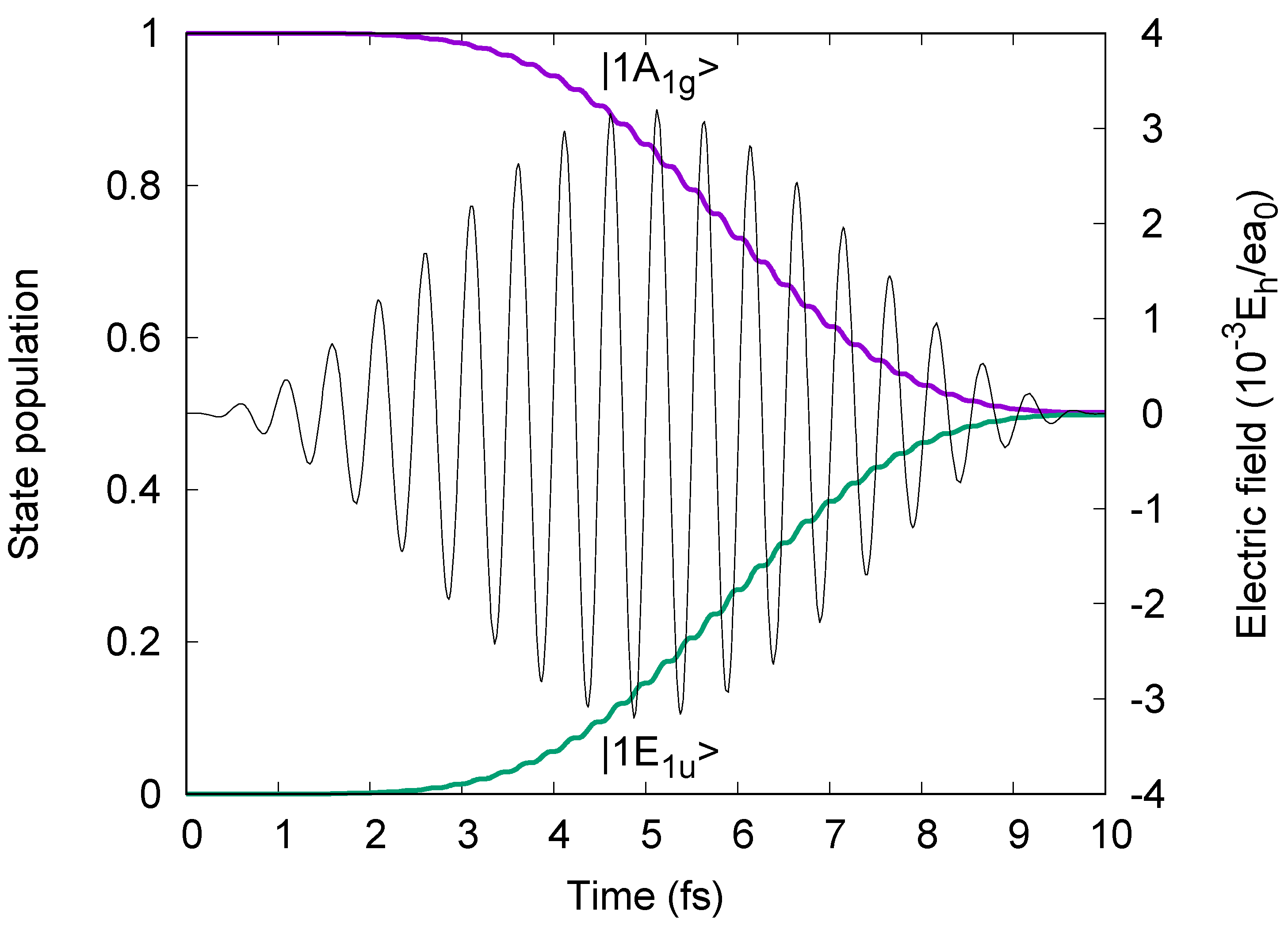
Figure 5 also shows the resulting two-state population dynamics P
1A1g(t) and P
1E1ux(t) = P
1E1uy(t) ≡ P
1E1u(t), documenting perfect preparation of the target superposition states, with the same amplitudes (= 1/√2) of the ground and degenerate excited states at the end (t= τ
1 = τ
2 ≡ τ) of the laser pulses. The populations of all other states are entirely negligible.
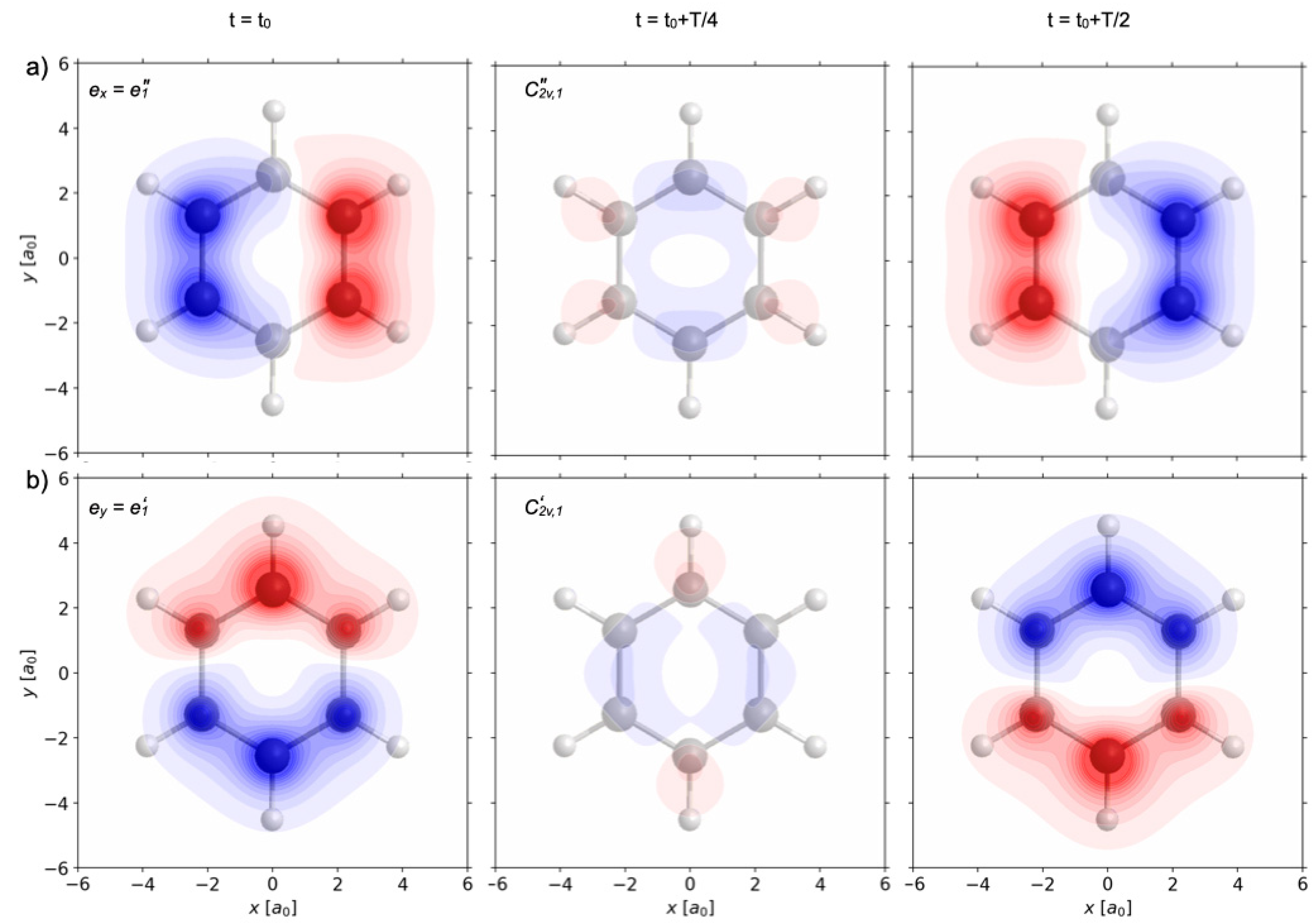
Figure 6a,b illustrate the field-free time evolutions of the one-electron densities ρ
p(
r,t) after the end of the re-optimized π/2 laser pulses labeled p=1 and 2, respectively. They are represented by three snapshots of ρ
p(
r,t) − ρ
p(
r,t=0) = ρ
p(
r,t) − ρ
1A1g(
r) integrated over z, i.e., ∫ [ρ
p(
r,t) − ρ
1A1g(
r)]dz. The snapshots are taken at t= t
0, t
0 + T/4 and t
0 + T/2, with period T = T
1A1g,1E1u. Here, t
0 is the first time after the end of the laser pulse when the phase factor exp[….] of c
e,1E1u(t) is equal to 1, cf. Equation (26). This equality is obtained during the first period T after the end of the laser pulse, t
f = τ ≤ t
0 ≤ τ + T, such that the nuclei can still be considered as frozen, conserving nuclear symmetry. Sequel snapshots at t = t
0 + (3/4)T and at t
0 + T look the same as those at t = t
0 + T/4 and at t
0, respectively. The subtraction of the initial one-electron density ρ
p(
r,t = 0) = ρ
1A1g(
r) from ρ
p(
r,t) yields what may be called the “swapping one-electron density”, i.e., that part of the one-electron density which is driven by the laser pulse away from the original reference ρ
1A1g(
r). It facilitates recognition of the time evolution and the symmetry of the one-electron density after the laser pulse; in contrast, the temporal changes of ρ
p(
r,t) would be hardly recognizable.
Appendix A proves that the electron symmetry of ρ
p(
r,t) is the same as for the swapping one-electron density, ρ
p(
r,t) − ρ
1A1g(
r).
Altogether, the time evolution of the laser-driven one-electron densities illustrated in
Figure 6a,b demonstrate two different types of periodic charge migrations, essentially from the left to the right CC bonds (and back, p = 1), or from the domains of the bottom to the top C-atoms (and back, p = 2), respectively, with period T = T
1A1g,1E1u. At time t = t
0, the swapping one-electron densities are accumulated in the left CC bond (p = 1), or in the domain of the bottom C-atom (p = 2). At t = t
0 + T/4, they “tunnel” [
34] between the sites, and at t
0 + T/2, they arrive on the opposite sites. At t = t
0 + (3/4) T, they tunnel back (snapshot identical to the one for t = t
0 + T/4), and at t = t
0 + T, they are back to the densities at t= t
0, completing the first cycle of periodic charge migration.
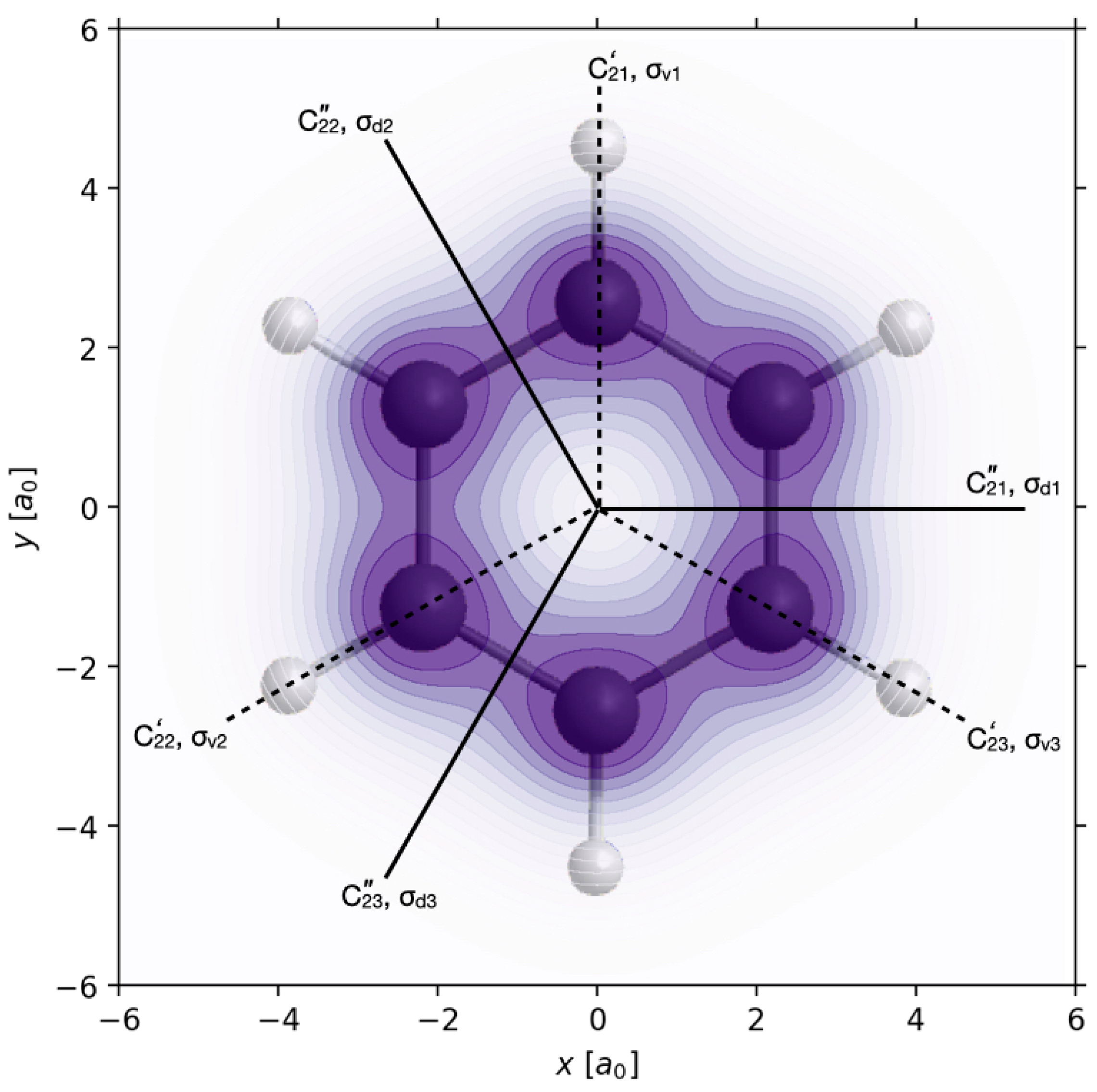
Inspection of the swapping one-electron density ρ
p=1(
r,t) − ρ
1A1g(
r) ≡ ρ
e”1 (
r,t) − ρ
1A1g(
r) (
Figure 6a) which is prepared by the
e1 =
ex =
e”
1 polarized re-optimized π/2 laser pulse reveals that it conserves three and only three symmetry elements of the frozen nuclear symmetry point group G = D
6h, namely the binary rotation axis C
2”
1, the related dihedral plane σ
d1, and the horizontal plane σ
h. The corresponding symmetry operations constitute the subgroup S(ρ
p=1(
r,t)) = C
2v”
1 = {E, C
2”
1, σ
h, σ
d1}. Accordingly, the one-electron density ρ
e”1 (
r,t) evolves with C
2v”
1 symmetry. In conclusion, the
e1 =
ex =
e”
1 polarized re-optimized π/2 laser pulse yields electron symmetry breaking G=D
6h → S(ρ
p=1(
r,t)) = C
2v”
1.
By analogy, inspection of the swapping one-electron density ρ
p=2(
r,t) − ρ
1A1g(
r) (
Figure 6b) which is prepared by the
e2 =
ey =
e’
1 polarized re-optimized π/2 laser pulse reveals that it conserves a different set of three symmetry elements. The corresponding symmetry operations constitute the subgroup S(ρ
p=2(
r,t)) = C
2v’
1 = {E, C
2’
1, σ
h, σ
v1} of G = D
6h. In conclusion, the
e2 =
ey =
e’
1 polarized re-optimized π/2 laser pulse yields electron symmetry breaking G = D
6h → S(ρ
p=2(
r,t)) = C
2v’
1.
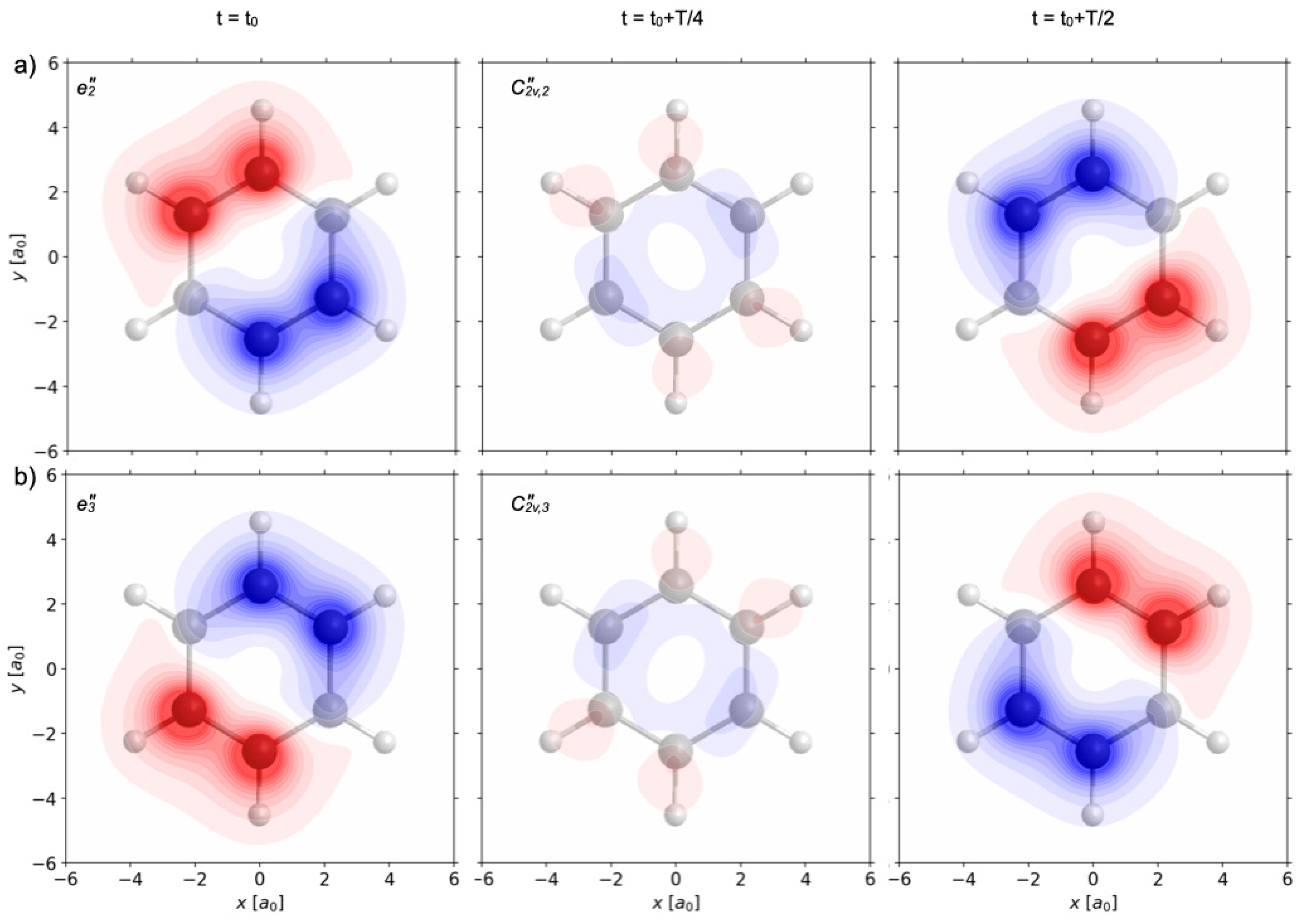
Figure 7a,b show the effects of two other re-optimized π/2 laser pulses labeled p = 3,4. They have the same electric field amplitudes
3(t) =
4(t) =
(t) with the same laser parameters as the previous ones (p = 1,2, cf.
Figure 5), but different polarizations,
e3 =
e”
2 = C
3 e”
1 and
e4 =
e”
3 = C
32 e”
1, respectively. As shown in
Section 2.7 (cf. the rule (35)), the rotations of the laser polarizations cause corresponding rotations of the laser-driven wave functions which may be expanded in terms of corresponding rotated eigenfunctions. The resulting population dynamics is the same as for the non-rotated wave functions, cf.
Figure 5. Accordingly, the resulting time evolutions of the one-electron densities appear as images of the one shown in
Figure 6a, rotated by 120° (C
3) and by 240° (C
32), respectively. By analogy with the previous analyses, inspection of the resulting swapping one-electron densities ρ
3(
r,t) − ρ
1A1g(
r) ≡ ρ
e”2 (
r,t) − ρ
1A1g(
r) and ρ
4(
r,t) − ρ
1A1g(
r) ≡ ρ
e”3 (
r,t) − ρ
1A1g(
r) show that the laser pulses labeled p = 3 and 4 yield electron symmetry breaking from G = D
6h to two subgroups S(ρ
p=3(
r,t)) = C
2v”
2 = {E, C
2”
2, σ
h, σ
d2} and S(ρ
p=4(
r,t)) = C
2v”
3 = {E, C
2”
3, σ
h, σ
d3}—these are different from the previous subgroups S(ρ
p=1(
r,t)) and S(ρ
p=2(
r,t))
. Likewise,
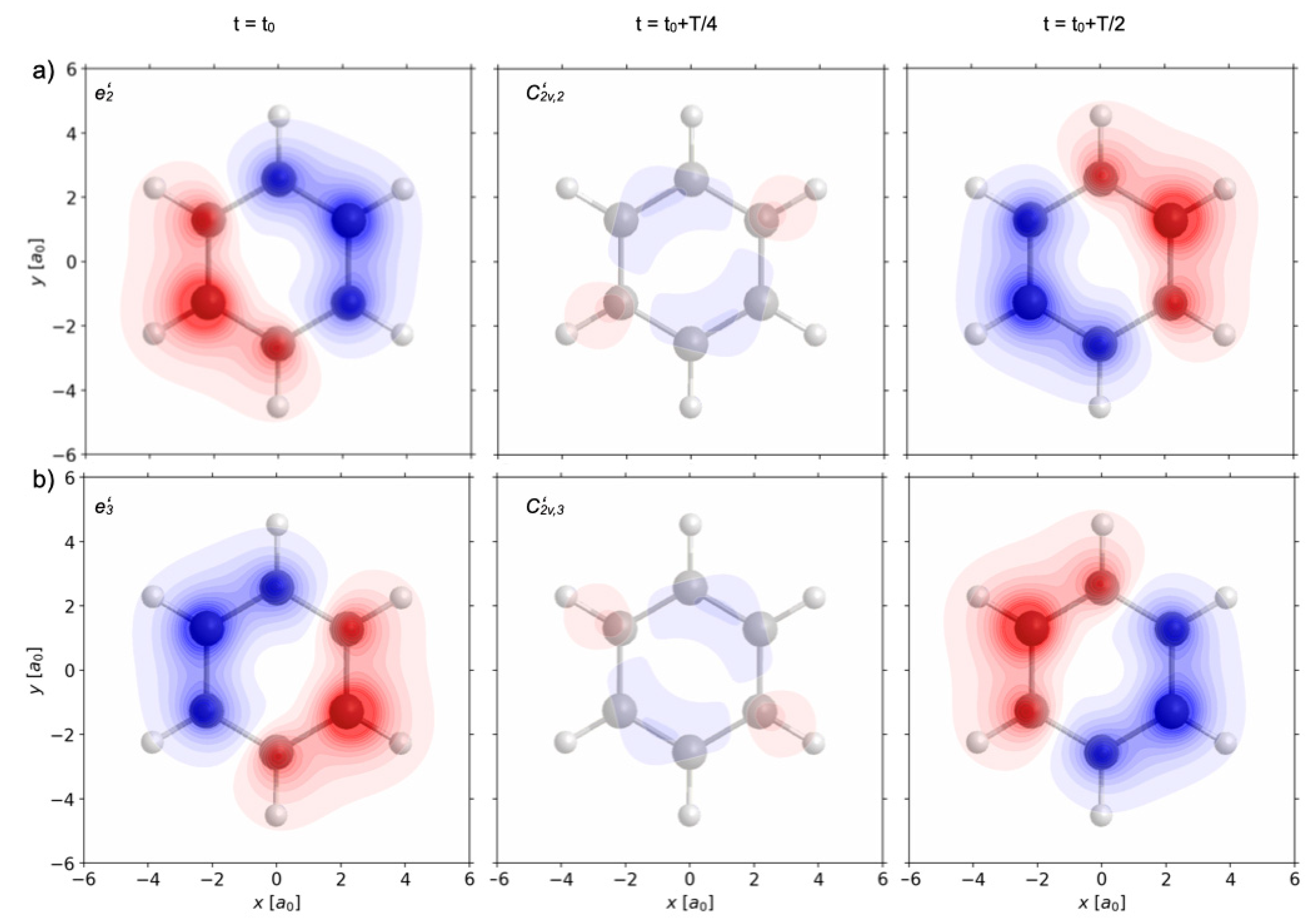
Figure 8a,b show the effects of two other re-optimized π/2 laser pulses labeled p = 5, 6. They have the same electric field amplitudes
5(t) =
6(t) =
(t) and the same laser parameters as the previous ones (p = 1, 2, 3, 4, cf.
Figure 5), but different polarizations,
e5 =
e’
2 = C
3 e’
1 and
e6 =
e’
3 = C
32 e’
1, respectively. The resulting time evolutions of the swapping one-electron densities appear as images of the one shown in
Figure 6b, rotated by 120° (C
3) and by 240° (C
32), respectively, in accord with the rule (34). By analogy with the previous analyses, inspection of the resulting swapping one-electron densities ρ
5(
r,t) − ρ
1A1g(
r) ≡ ρ
e’2(
r,t) − ρ
1A1g(
r) and ρ
6(
r,t) − ρ
1A1g(
r) ≡ ρ
e’3(
r,t) − ρ
1A1g(
r) show that the re-optimized π/2 laser pulses labeled p=5 and 6 yield electron symmetry breaking G=D
6h → S(ρ
p=5(
r,t)) = C
2v’
2 = {E, C
2’
2, σ
h, σ
v2} and S(ρ
p=6(
r,t)) = C
2v’
3 = {E, C
2’
3, σ
h, σ
v3}, different from the previous ones.
Finally, we present two additional electron symmetry breakings in the oriented benzene, which are achieved by two different sequences of two linearly polarized laser pulses. The first example is motivated by a specific result of Ref. [
1] for laser preparation of oriented benzene in selective non-aromatic (“na”) superposition states
or
For the orientation of benzene adapted from Ref. [
1], cf.
Figure 1, the excited states |1B
2u> and |1B
1u> transform as x(x
2 − 3y
2) and y(3x
2 − y
2), respectively [
13]. Symmetry selection rules do not allow direct excitations of the ground state to these target states [
1,
13], but Ulusoy and Nest succeeded in designing sequences of three
ex and
ey linearly polarized laser pulses, or alternative re-optimized pulses which reach the goals. Our first example is stimulated by their series of three
ey polarized laser pulses which prepare the target state (50) by means of three transitions which are all symmetry allowed. The first one is a π/2 laser pulses which excites the aromatic ground state |1A
1g> to the superposition state (1/√2)*(|1A
1g> + |1E
1uy>), analogous to the present laser pulse labeled p = 2. The second one is a π laser pulse which transfers the excited state |1E
1uy> into |1E
2gx2−y2>. This state is one of two degenerate 1E
2g states {|1E
2gx2−y2>, |1E
2gxy>} with energy E
1E2g which transform as x
2 − y
2 and xy, respectively [
13]. The third one is another π laser pulse which transfers |1E
2gx2−y2> into |1B
1u>. Consequently, at the ends of the second and third laser pulses, the oriented benzene is in the superposition state (1/√2) (|1A
1g> + |1E
2gx2−y2>) and finally in the target state (50), respectively.
As set off in the Introduction, Ulusoy and Nest already noted that their three-step approach to laser control of aromaticity in oriented benzene breaks electron symmetry [
1]. Our original plan was to investigate laser-induced electron symmetry breaking already after two steps, i.e., after excitation of the ground state (|1A
1g> first to the superposition state (1/√2)*(|1A
1g> + |1E
1uy>) and then to (1/√2)*(|1A
1g> + |1E
2gx2−y2>), by means of two
ey polarized π/2 and π laser pulses, but now under the constraint of the conservation of nuclear symmetry. In the context of this manuscript, these laser pulses shall be labeled p = 7 and 8, or briefly 7&8. The constraint requires that their durations should add up to τ
7 + τ
8 ≤ 10 fs. [
7,
8].
Unfortunately, however, the plan cannot be realized, because of the following dilemma: Ulusoy and Nest’s “intermediate” excited states |1E
1uy> and |1E
2gx2−y2> are near-degenerate, cf.
Figure 3. The narrow energy gap E
1E1u − E
1E2g = ℏω
1E1u,1E2g = 0.306 eV implies the rather long period T
1E1u,1E2g = 2π/ω
1E1g,1E2g = 13.52 fs, cf. Equation (45). The condition (47) thus requires that the second π laser pulse should take too long to transfer the “intermediate” excited state |1E
1uy> into |1E
2gx2−y2>. This is far beyond the upper limit, 10 fs, for the conservation of nuclear symmetry, i.e., for our purpose, it is unacceptable.
To overcome this problem, it is essential to get rid of the near degeneracy of the levels E
1E1u and E
1E2g. For this purpose, we decided that the “first” intermediate state |1E
1uy> should be replaced by the more excited state |2E
1uy> with much higher energy E
2E1u compared to E
1E1u. The corresponding sequence of two laser-induced target transitions is illustrated schematically by two sequential arrows in
Figure 3. The previous near degeneracy is clearly lifted. Specifically, the energy gap between states |2E
1uy> and |1E
2gx2−y2> is E
2E1u − E
1E2g = 5.75 eV, with corresponding period T
2E1u,1E2g = 719 as. Likewise, the energy gap between states |2E
1uy> and |1A
1g > is E
2E1u − E
1A1g = 14.23 eV, with period T
2E1u,1A1g = 291 as. These two periods are short enough to satisfy condition (47) for two laser pulses with durations τ
7 = 2.5 fs and τ
8 = 7.5 fs, respectively. Their durations add up to t
f = τ
7 + τ
8 = 10 fs, in accord with the constraint for the conservation of nuclear symmetry [
7,
8]. The smaller value of τ
7 compared to τ
8 is suggested by condition (47), due to the smaller value of T
2E1u,1A1g compared to T
2E1u,1E2g.
The replacement of the intermediate state |1E
1uy> by the more excited state |2E
1uy> with the same IRREP raises another problem, however, namely the absolute values of the y-components of the transition matrix element |<1A
1g|d
y|2E
1uy>| = 0.161 e a
0 for the target transitions |1A
1g> → |2E
1uy> via |2E
1uy> is significantly smaller than |<1A
1g|d
y|1E
1uy>| = 2.339 e a
0 for Ulusoy and Nest’s transitions via |1E
1uy>. At the same time, the required durations τ
7 = 2.5 fs and τ
8 = 7.5 fs are an order of magnitude smaller than the values used in Ref. [
1]. Consequently, the conditions (46) and (48) for a hypothetical sequence of π/2 and π laser pulses for sequential transitions|1A
1g> → |2E
1uy> → |1E
2gx2−y2> call for exceedingly large field strengths and intensities—this is, again, unacceptable.
The way out of this dilemma is pointed by Ref. [
2] which shows that for the present purpose, i.e., for laser-induced electron symmetry breaking, it is not really necessary to use π/2 and π laser pulses. Instead, it suffices to employ weaker pulses p = 7 and/or 8 which prepare superposition states such as c
e’1,1A1g(τ
7) |1A
1g> + c
e’1,2E1uy(τ
7) |2E
1uy> and subsequently c
e’1&e’1,1A1g(t
f) |1A
1g> + c
e’1&e’1,1E2g(t
f) |1E
2gx2−y2>, with rather small populations of the excited states compared to the ground states, P
e’1,2E1uy(τ
7) = |c
e’1,2E1uy(τ
7)|
2 « P
e’1,1A1g(τ
7) = |c
e’1,1A1g(τ
7)|
2 and P
e’1&e’1,1E2g^x2-y2(t
f) = |c
e’1&e’1,1E2g^x2-y2(t
f)|
2 « P
e’1&e’1,1A1g(t
f) = |c
e’1&e’1,1A1g(t
f)|
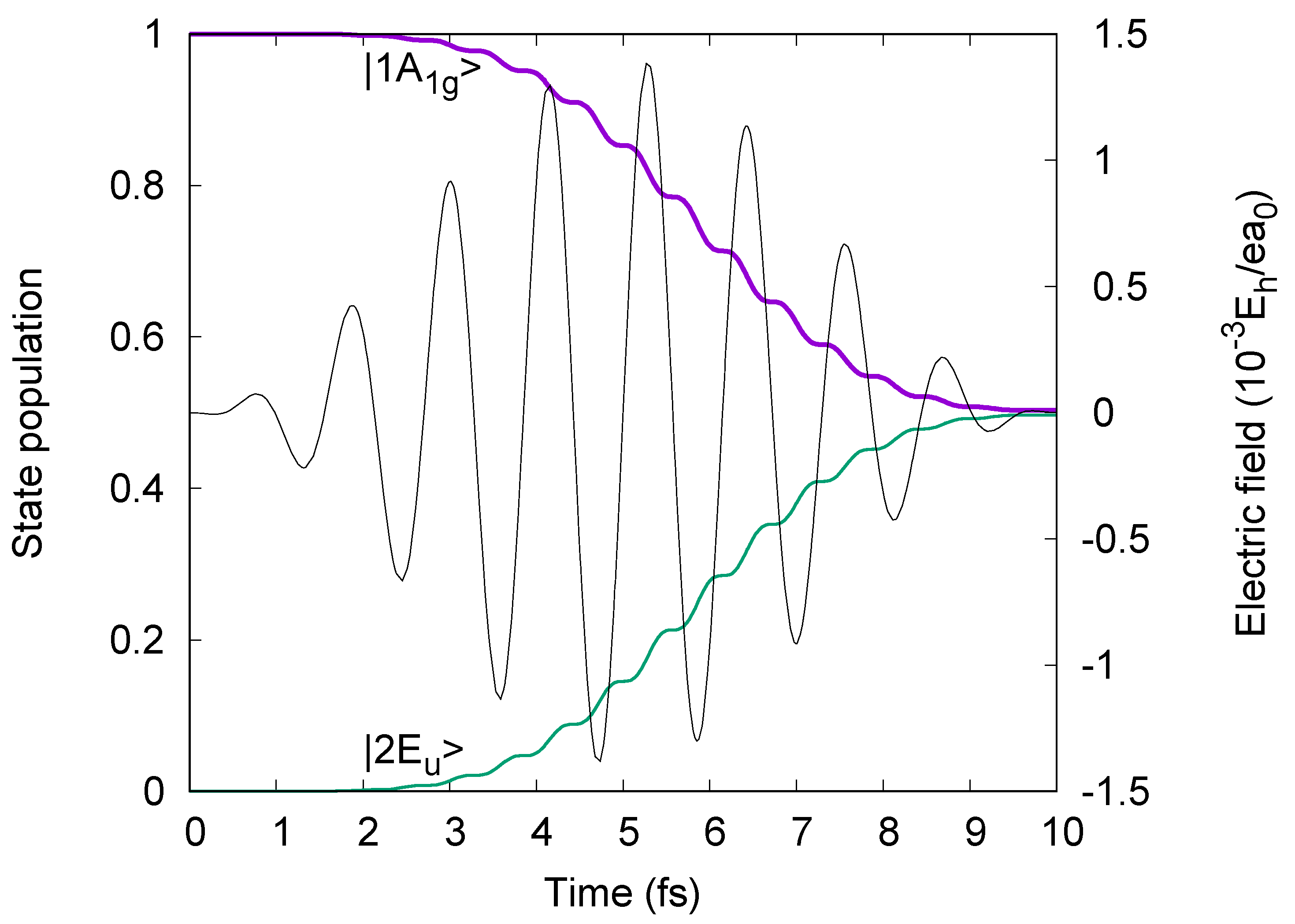
2. Consequently, we employ two sequential linearly
e7 =
e8 =
ey =
e’
1 polarized laser pulses with durations τ
7 =2.5 fs and τ
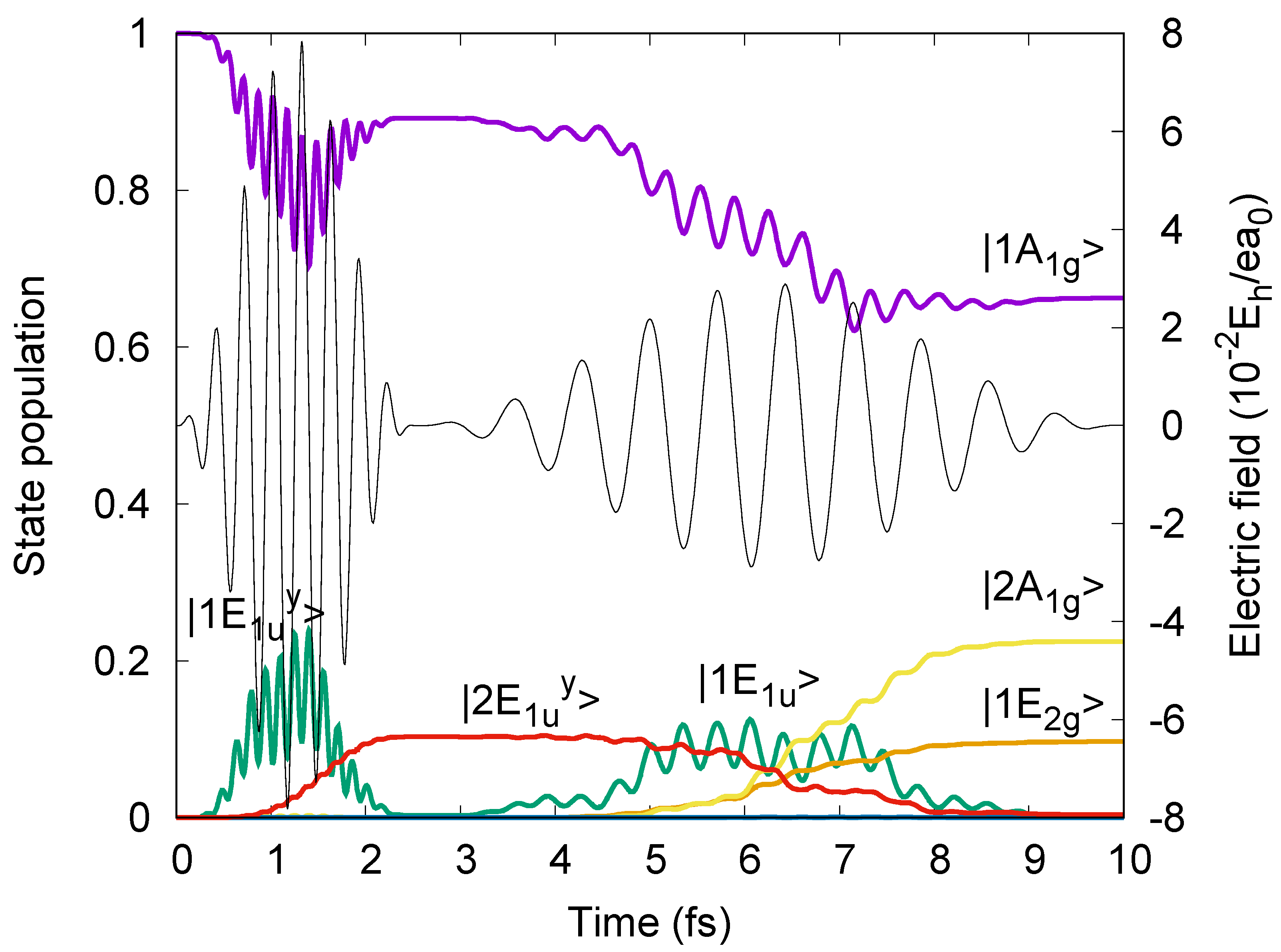
8 = 7.5 fs, with the same form of the electric fields, Equation (1), as before, with the carrier frequencies slightly detuned from the resonance frequencies, yet with electric field strengths well below the values for hypothetical π/2 and π pulses. The electric fields
p(t) of the laser pulses p = 7, 8 are illustrated in
Figure 9. The laser parameters are listed in the
Figure 9. The resulting population dynamics are also shown in
Figure 9.
Figure 9 shows that at the end (τ
7 = 2.5 fs) of the first
ep=7 =
e’
1 =
ey polarized laser pulse p = 7, the oriented benzene is excited from the initial ground state |1A
1g> to the intermediate target superposition state c
e’1,1A1g(τ
7) |1A
1g> + c
e’1,1E1uy(τ
7) |1E
1uy>, with populations P
e’1,1E1uy(τ
7) = |c
e’1,1E1uy(τ
7)|
2 = 0.105 « P
e’1,1A1g(τ
7) = |c
e’1,1A1g(τ
7)|
2 = 0.890. Subsequently, at t
f = τ
7 + τ
8 = 10 fs, the second
ep=8 =
e’
1 =
ey polarized laser pulse p = 8 yields the superposition of three eigenstates (cf. Equation (16))
with populations P
e’1&e’1,1A1g (t
f) = |c
e’1&e’1,1A1g (t
f)|
2 = 0.664, P
e’1&e’1,2A1g (t
f) = |c
e’1&e’1,2A1g (t
f)|
2 = 0.225 and P
e‘1&e‘1,1E2g^x2-y2 (t
f) = |c
e‘1&e‘1,1E2g^x2-y2 (t
f)|
2 = 0.098. The subscript “
e’
1&
e’
1” indicates that the wavefunction (51) is prepared by a sequence of two polarized laser pulses with the same polarizations,
e’
1 and again
e’
1. The population of state |2A
1g> is added unexpectedly to those of the two target states |1A
1g> and |1E
2gx2−y2>. However, this does not harm the present purpose. What matters for the new example of electron symmetry breaking is that the final state (51) is prepared as superposition of states which transform according to two different IRREPs—here this is A
1g (the same for states |1A
1g> and |2A
1g>) and E
2gx2−y2 (for state|1E
2gx2−y2>). Apparently,
Figure 9 shows that the pulses p = 7 and 8 also cause transient populations of complementary states, in particular |1E
1uy>. This is caused by non-linear effects—but this is irrelevant for the present purpose, and beyond the scope of this paper.
After the sequence of two laser pulses 7 and 8 with the same polarizations
e’
1 and again
e’
1, for times t ≥ t
f = τ
7 + τ
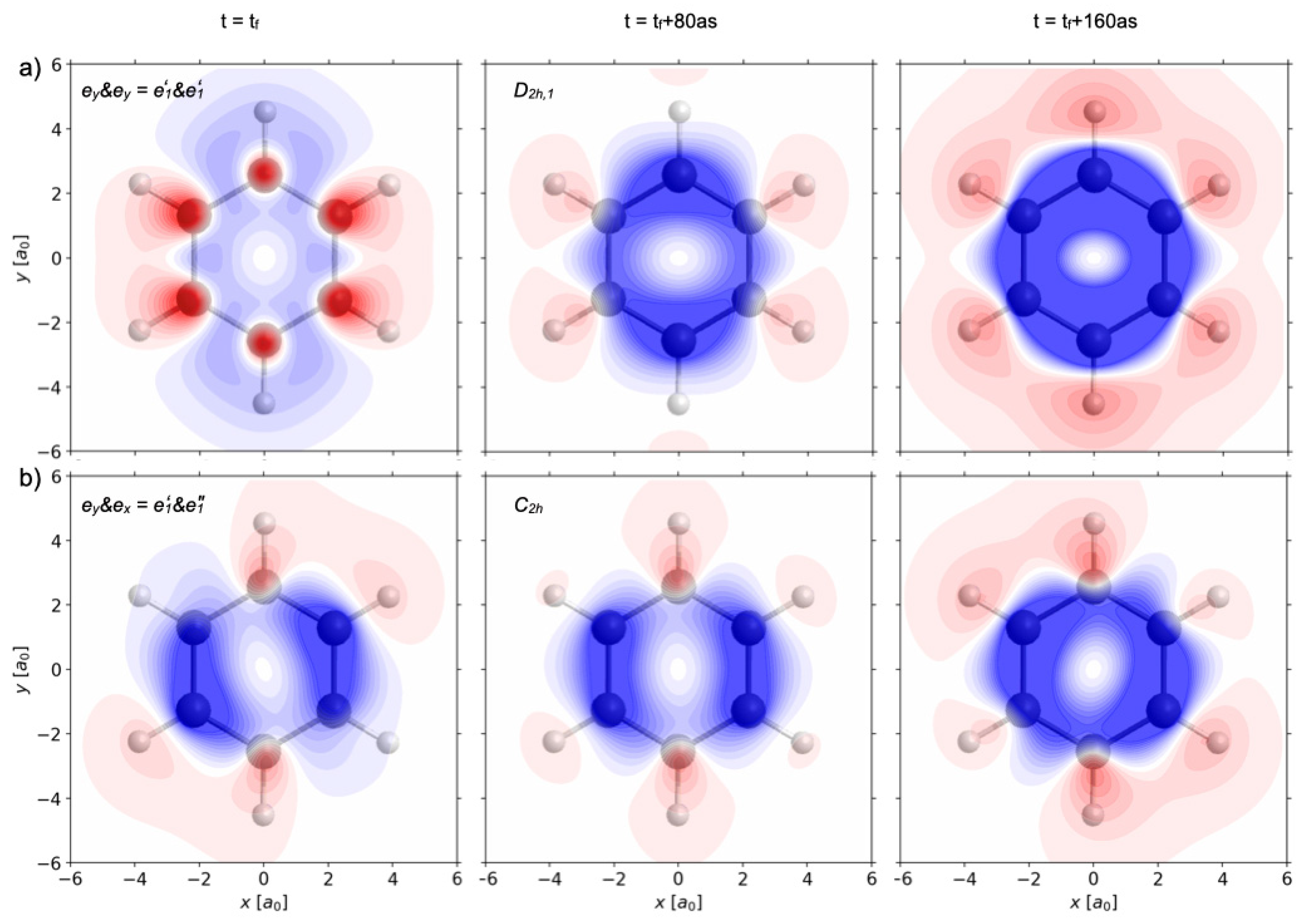
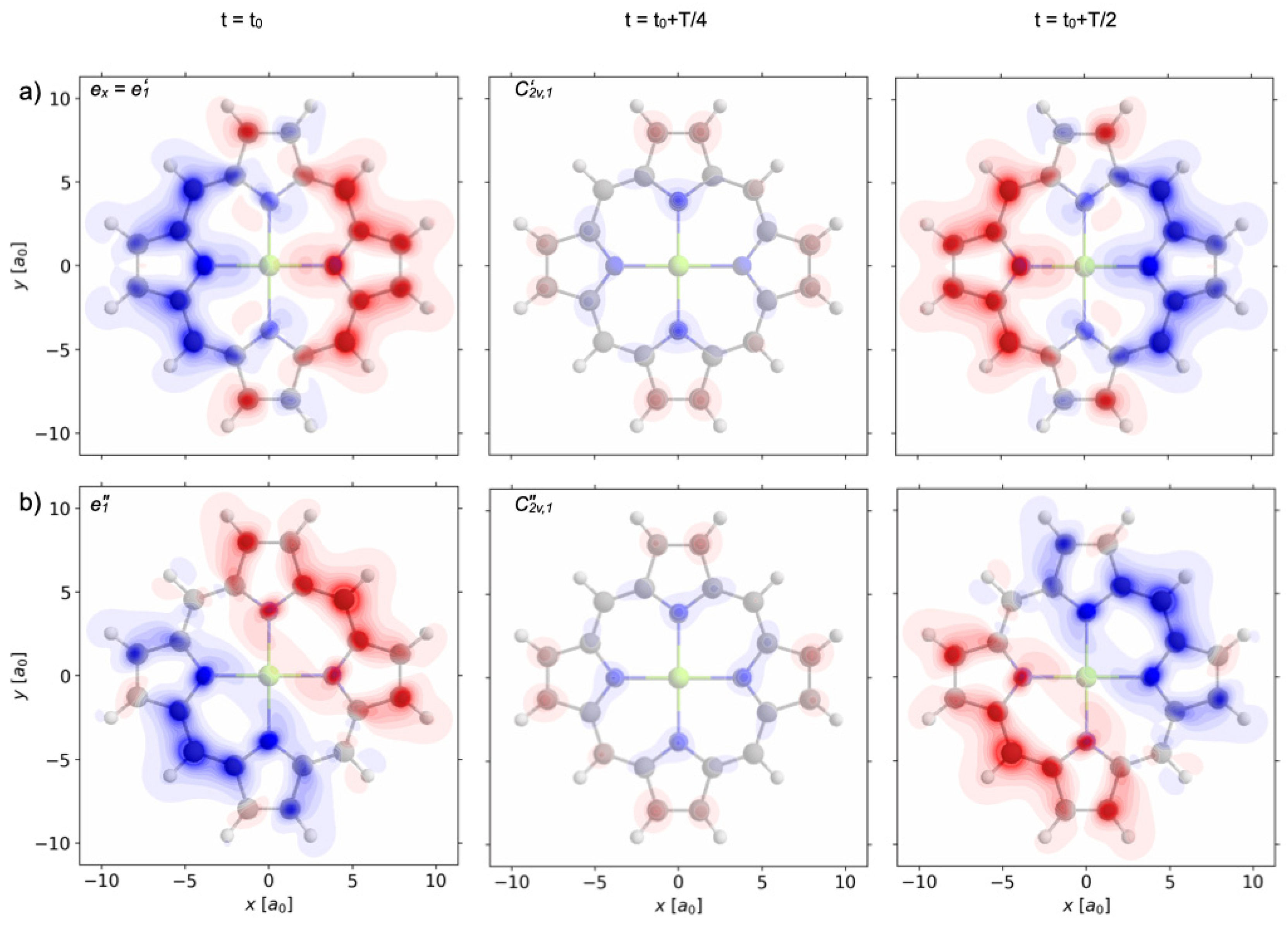
8 = 10 fs, the wavefunction (51) evolves in field-free environments, with coefficients specified in Equation (26). The corresponding swapping part of the one-electron densities ρ
e’1&e’1(
r,t) − ρ
1A1g(
r) is illustrated in
Figure 10a by snapshots analogous to those shown in
Figure 6,
Figure 7 and
Figure 8. Corresponding inspection and analyses of these snapshots reveal that ρ
e’1&e’1(
r,t) displays charge migration with conservation of the symmetry elements of the subgroup D
2h,1 = {E,C
2,C
2’
1,C
2”
1,i,σ
h, σ
v1,σ
d1} of D
6h. Hence, as resume of the combined
Figure 1 and
Figure 10a, the series of two laser pulses 7 and 8, also called
e’
1&
e’
1 (
Figure 9) yield electron symmetry breaking D
6h → S(ρ
e’1&e’1(
r,t)) =D
2h,1 in the oriented model benzene, with conservation of nuclear symmetry. This is quite different from the six previous examples which are documented in the combined
Figure 1 and
Figure 6,
Figure 7 or
Figure 8.
Our last example of electron symmetry breaking in oriented benzene is achieved by another sequence of two laser pulses—this time they are labeled 7 and 9. The first laser pulse (p = 7) is the same as the first laser pulse in the previous series of laser pulses 7 and 8, cf.
Figure 9. The second pulse (p = 9) has the same electric field as the previous pulse labeled p=8, i.e.,
p=9(t) =
p=8(t), cf.
Figure 9, but the laser polarization is
e9 =
e”
1 =
ex instead of
e8 =
e’
1 =
ey. Hence the series of laser pulses 7 and 9 will also be denoted
e’
1&
e”
1. The resulting population probabilities P
e’1&e“1,m (t) of eigenstates |m> are shown in
Figure 9. Since the two series of laser pulses 7 and 8 and 7 and 9 employ the same first pulse (p = 7), they yield the same populations during the time 0 ≤ t < τ
7 = 2.5 fs of the first laser pulse. During the time τ
7 = 2.5 fs ≤ t ≤ t
f = τ
7 + τ
8 = τ
7 + τ
9 = 10 fs of the second pulses, p = 8 or 9, the equality of their electric fields
p=8(t) =
p=9(t) but orthogonality of the laser polarizations
e8 =
e’
1 =
ey ⊥
e9 =
e”
1 =
ex yields the same populations P
e’1&e“1,m”(t) = P
e’1&e’1,m’(t) for the non-degenerate states |m”> = |m’> = |1A
1g> and |2A
1g>, but require proper modifications of the quantum numbers of the degenerate states, i.e., the series of pulses 7 and 8 excite the transient and final states |1E
1uy> and |1E
2gx2−y2> whereas pulses 7 and 9 excite |1E
1ux> and |1E
2gxy>, respectively. Accordingly, at the end (t = t
f = τ
7 + τ
9 = 10 fs) of the sequence of laser pulses 7 and 9, also called
e’
1&
e”
1, the oriented benzene is prepared in the superposition state
with the same populations of the states |1A
1g>, |2A
1g>, |1E
2gxy> as those (|1A
1g>, |2A
1g>, |1E
2gx2−y2>) for pulses 7 and 8.
The field-free time evolution of the wave function (52) after the sequence laser pulses 7 and 9, also called
e’
1&
e”
1, i.e., for t ≥ t
f = 10 fs, yields the swapping one-electron density ρ
e’1&e’1(
r,t) − ρ
1A1g(
r) which is illustrated in
Figure 10b by snapshots, analogous to those shown in
Figure 10a for the laser pulses 7 and 8. They reveal a new type of charge migration, with the conservation of the symmetry elements of the subgroup C
2h = {E,C
2,i,σ
h} of D
6h. Hence, as a result of the combined
Figure 1 and
Figure 10b, the series of laser pulses 7 and 9 with polarizations
e’
1 and
e”
1 (
Figure 9) yield electron symmetry breaking D
6h → S(ρ
e’1&e”1(
r,t)) = C
2h in the oriented model benzene, with the conservation of nuclear symmetry.
To summarize, all the different electron symmetry breakings D6h → S(ρp(r,t)) from the point group G = D6h to different subgroups S(ρp(r,t)) of G, p = 1, …, 9 are achieved by means of different linearly polarized laser pulses, or two sequential laser pulses, which excite the initial electronic ground state |1A1g> to different superpositions (16) of the electronic ground state plus one (p = 1, …, 6) or two different excited states (p = 7&8 and 7&9). An important common property of the different sets of the electronic ground and excited states which constitute the different superpositions is that (at least) two of them have different IRREPs, e.g., A1g and E1u or A1g and E2g. In the case of doubly degenerate IRREPs, preparations of superposition states with different sets of basis functions yield different point subgroups S(ρp(r,t)) of G.
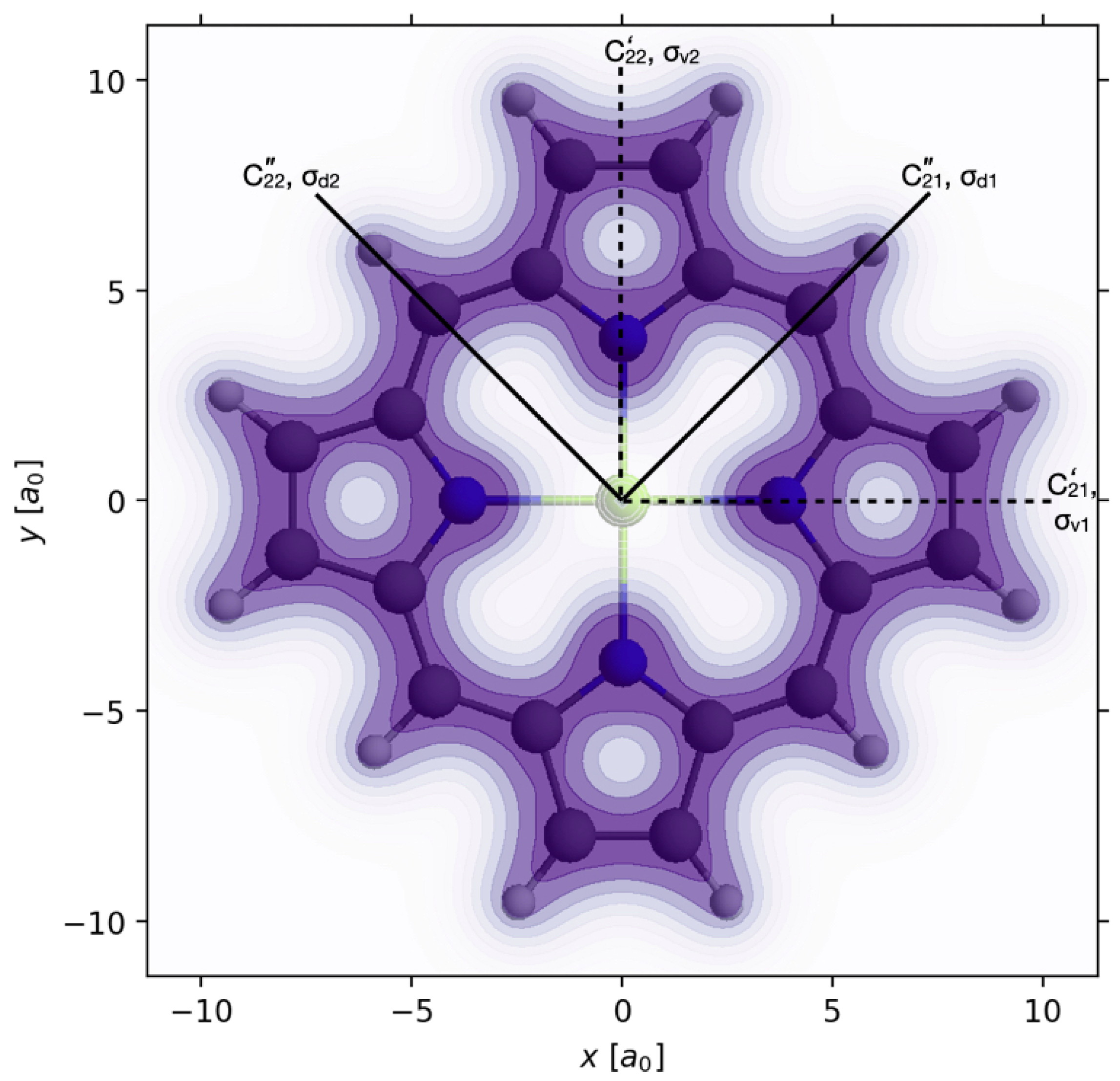
3.2. Electron Symmetry Breaking and Charge Migration Induced by Linearly Polarized Laser Pulses in Oriented Mg-Porphyrin with D4h Nuclear Scaffold
This Section heightens the impression which was gained in the previous
Section 3.1, namely different well designed short linearly polarized laser pulses labeled p = 1, 2, 3, … can achieve different types of charge migrations with different electron symmetry breakings, from the point group of the electronic ground state G to various subgroups S(ρ
p(
r,t)) of G. For this purpose, we present additional examples, with applications to another oriented molecule with nuclear point group symmetry G different from D
6h. Specifically, we choose oriented Mg-porphyrin with G = D
4h, cf.
Figure 2 and
Figure 4, and demonstrate different electron symmetry breakings D
4h→ S(ρ
p(
r,t)) by means of different linearly polarized laser pulses. The oriented model Mg-porphyrin is suggested by Ref. [
15] which discovers the stimulation of a specific type of charge migration, namely electron circulation, by means of a circularly polarized laser pulse. As a byproduct, Ref. [
15] has a series of snapshots of the time-dependent one-electron density after the laser-pulse. In retrospect, those snapshots clearly document electron symmetry breaking D
4h → C
s = {E, σ
h} by a short laser pulse, with the conservation of the nuclear symmetry D
4h. However, this effect was not noted in Ref. [
15]—the word “symmetry” is not even mentioned therein. In contrast with Ref. [
15], here we are heading for the explicit discovery of electron symmetry breaking D
4h → S(ρ
p(
r,t)) in oriented Mg-porphyrin, by means of selective linearly polarized laser pulses. The effects are analogous to those for D
6h → S(ρ
p(
r,t)) electron symmetry breaking in oriented benzene, as documented in
Section 3.1. This analogy allows a rather brief presentation of the results for Mg-porphyrin, without any repetition of the detailed derivations of
Section 3.1. Moreover, we restrict the presentation to just two cases of electron symmetry breaking D
4h → S(ρ
p(
r,t)) by means of two different laser pulses labeled p = 1, 2. The rich examples D
6h → S(ρ
p(
r,t)), p = 1, …, 9, for benzene should then suffice to stimulate the reader’s imagination to invent additional cases, also for oriented Mg-porphyrin.
As in
Section 3.1, we start with the demonstration of electron symmetry breaking in oriented Mg-porphyrin, by means of a
ep=1 =
ex =
e’
1 polarized re-optimized π/2 laser pulse. The laser pulse labeled p = 1 has the familiar form of the electric field
p=1(t), Equation (1). It is illustrated in
Figure 11, with the laser parameters in
Figure 11. The resulting population dynamics are also shown in
Figure 11. Apparently, the first laser pulse p = 1 with duration τ
1 = 10 fs excites the oriented Mg-porphyrin from the initial (t = 0) electronic ground state |1A
1g> to the superposition state
*(|1A
1g> + |2E
ux>). The notation |2E
ux> refers to the doublet {|2E
ux>, |2E
uy>} ≡ {|2E
u’
1>, |2E
u’
2>} of two orthogonal degenerate states with IRREP E
u with the second-lowest energy E
2Eu which transform as x and y, respectively [
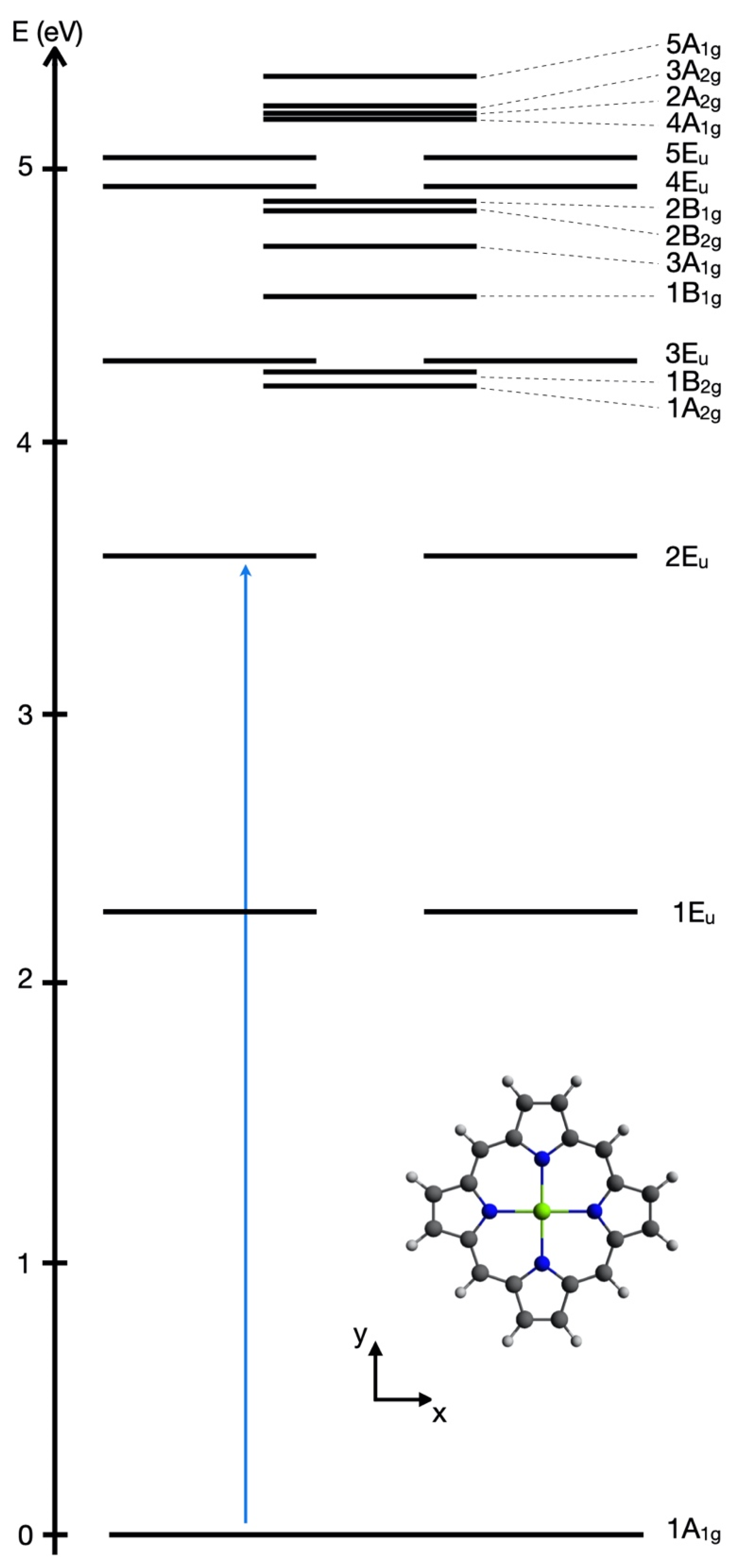
13]. The target transitions is illustrated schematically by the vertical arrow from the energy level E
1A1g to E
2Eu in
Figure 4. The energy gap between their eigenenergies is ΔE
1A1g,2Eu = E
2Eu − E
1A1g = 3.60 eV with corresponding transition frequency ω
1A1g,2Eu = ΔE
1A1g,2Eu /ℏ and period T = T
1A1g,2Eu = 2π/ω
1A1g,2Eu = h/ΔE
1A1g,2Eu = 1.147 fs, cf. Equations (42) and (43).
The subsequent (t ≥ τ
1 = 10 fs) time evolution of the swapping one-electron density ρ
p=1(
r,t) − ρ
1A1g(
r) is illustrated in
Figure 12a by three snapshots, analogous to the previous
Figure 6 for oriented benzene. Apparently, the oriented Mg-porphyrin exhibits periodic charge migration from the left to the right, and back, with period T. Analogous inspection and analyses show that the migrating one-electron density conserves three symmetry elements, namely C
2’
1, σ
h, σ
v1, cf.
Figure 2. Accordingly, the combined
Figure 2 and
Figure 12a document electron symmetry breaking induced by the
e’
1 polarized laser pulse p = 1, namely D
4h → S(ρ
p=1(
r,t)) = C
2v’
1 = {E, C
2’
1, σ
h, σ
v1}.
The second example for electron symmetry breaking in oriented Mg-porphyrin employs another re-optimized π/2 laser pulse labeled p = 2. It has the same electric field as the first pulse p = 1, i.e.,
2(t) =
1(t), cf.
Figure 11, but different polarization
e2 =
e”
1 = (1/√2)* (
ex +
ey) = C
8 e”
1. It prepares the superposition state (1/√2)*(|1A
1g> + |2E
u”
1>], analogous to the superposition state (1/√2)*(|1A
1g> + |2E
u’
1>] which is prepared by the e’
1 polarized pulse p = 1. The
e’
1 (p = 1) and
e”
1 (p = 2) polarized pulses yield the same population dynamics, cf.
Figure 11. The periodic charge migration which is initiated by the
e”
1 (p = 2) polarized pulse is illustrated in
Figure 12b. It has the same period T as for the charge migration initiated by pulse p = 1, but it is from the lower left to the upper right of the oriented scaffold, and back. Apparently, it conserves three symmetry elements, namely C
2”
1, σ
h, σ
d1, cf.
Figure 2. Accordingly, the combined
Figure 2 and
Figure 12b document electron symmetry breaking induced by the
e”
1 polarized laser pulse p = 2, namely D
4h→ S(ρ
p=2(
r,t)) = C
2v”
1 = {E, C
2”
1, σ
h, σ
d1}.
The summary of this
Section 3.2 with application to oriented Mg-porphyrin is analogous to the summary for
Section 3.1 for oriented benzene. Different electron symmetry breakings D
4h → S(ρ
p(
r,t)) from the point group G = D
4h to different subgroups S(ρ
p(
r,t)) of G, p = 1, 2 are achieved by means of different linearly polarized laser pulses which excite the initial electronic ground state |1A
1g> to different superpositions (16) of the electronic ground state plus an excited state with different IRREP E
u. Preparations of superposition states with different basis functions yield different point subgroups S(ρ
p(
r,t)) of G.
3.3. Symmetry of One-Electron Densities of Superposition States in Oriented Benzene
Section 3.1 shows that different short laser pulses, or two sequential laser pulses with linear polarization(s)
e and with total durations t
f = 10 fs, initiate different charge migrations in the oriented benzene which are represented by different one-electron densities ρ
e(t > t
f) with different electron symmetries S(ρ
e(t)) depending on
e. All those electron symmetry breakings G = D
6h → S(ρ
e(t)) conserve nuclear symmetry D
6h. Until now, the symmetries S(ρ
e(t)) were determined by inspection of the symmetry elements of G which are conserved in ρ
e(t > t
f).
The purpose of this Section is to derive a recipe for the rigorous determination of the subgroup S(ρ
e(t > t
f)), starting from the superposition state (16) which was prepared by the laser pulse, or two sequential laser pulses with polarization(s)
e, without any empirical inspection of the symmetry elements of ρ
e(t). For this purpose, to simplify the notation, we rewrite the superposition state (16) as
i.e., we keep in mind that the wavefunctions |Ψ> and |Ψ
k> as well as the expansion coefficients c
m and c
n,k depend on the laser polarization(s)
e and on the time after the laser pulse, t > t
f, cf. Equation (26), but this dependence is no longer written explicitly. Equation. (53) also replaces the energy quantum number m by two quantum numbers n,k. Here, k denotes the IRREP Γ
m = Γ
k of the basis function |m> = |n,k> ≡ |n, Γ
k >, and n specifies the energetic order of the basis functions with IRREP Γ
k. The wave functions
in Equation (53) are in general linear combinations of basis functions |n, Γ
k > which have the same IRREP Γ
k and non-zero coefficients c
n,k. The wave function |Ψ> and the basis functions |m> = |n, Γ
k > in Equations (53) and (54) are normalized, whereas the |Ψ
k> are not normalized. In fact, normalization of the |Ψ
k> is not necessary, for the present purpose.
The derivation of the subgroup S(ρ
e(t > t
f)) of the one electron density for t > t
f after the laser pulse(s) with polarization(s)
e proceeds in three steps, cf.
Appendix A: First we determine the subgroup S(|Ψ
e(t > t
f)>) of the superposition state (53). Next we derive the subgroup S(Ψ
e(t > t
f)
* Ψ
e(t > t
f)) of the electron density Ψ
e(t > t
f)
* Ψ
e(t > t
f), and finally the target subgroup S(ρ
e(t > t
f)) of the one-electron density ρ
e(t > t
f). The derivations are general, in principle, but the presentation focuses on the present applications (cf.
Section 3.1) where the laser pulses prepare superposition states (16), (53) with non-vanishing contributions of the ground state |1A
1g> plus possibly excited states |nA
1g> with the same IRREP A
1g plus one excited basis function |n,k> ≡ |n, Γ
k ≠A
1g > with IRREP Γ
k ≠A
1g, thus
The
Appendix A proves that for such cases (and also for various other cases), the subgroups are all identical,
and the IRREP of ρ
e(t > t
f) in the subgroup S(ρ
e(t > t
f)) is always the totally symmetric one. For this reason, the subsequent presentation focuses on the derivation of the subgroup S(|Ψ
e(t > t
f)>). Once S(|Ψ
e(t > t
f)>) is determined, the theorem (56) tells us that the target subgroup S(ρ
e(t > t
f)) is the same as S(|Ψ
e(t > t
f)>).
The general theory is presented in
Appendix A. It ends up with a simple recipe. It is demonstrated here for the cases which were presented in
Section 3.1. The results are summarized in
Table 3. This
Table 3 may be recognized as a collection of special parts of the character tables of the subgroups S(|Ψ
e(t > t
f)>) of the superpositions |Ψ
e(t > t
f)>, Equation (55), namely for the characters of the totally symmetric IRREPs. These characters are all equal to 1. Thus
Table 3 lists the number “1” for all symmetry operations g ϵ S(|Ψ
e(t > t
f)>). Turning the table, the task is to determine the symmetry elements g with “marks 1” in
Table 3. These sets of symmetry operations identify the subgroups S(|Ψ
e(t > t
f)>). At the same time, the theorem (56) for superpositions (55) makes those subgroups S(|Ψ
e(t > t
f)>) equal to the target subgroup S(ρ
e(t > t
f)) of the one-electron density ρ
e(t > t
f).
Here we present step-by-step applications of the general theory (cf.
Appendix A) to two (out of eight) examples which were presented in
Section 3.1.
Table 3 also lists analogous results for the other six cases of
Section 3.1, and also for the non-aromatic states (49), (50) of the oriented benzene which are prepared by three linearly polarized laser pulses or by re-optimized laser pulses designed by Ulusoy and Nest [
1].
Our first example is for the electronic superposition state |Ψ
e”1(t > t
f)> = c
e”1,A1g(t) |1A
1g> + c
e”1,1E1ux(t) |1E
1ux> which is prepared by the
e”
1 =
ex polarized π/2 laser pulse shown in
Figure 5, cf.
Section 3.1. Using the simplified notation of Equations (53) and (55), this superposition state is rewritten
It consists of two partial waves with IRREPs A
1g and E
1ux of the group G=D
6h. According to the general theory (cf.
Appendix A), the subgroup S(|Ψ>) of |Ψ> is the subset of the symmetry operations g of G which satisfy the same symmetry properties for all components,
with the same characters χ(g) for all wave functions |Ψ
k> in the expansion (53). In the present case (57), S(|Ψ>) consists of all g which satisfy g|A
1g> = χ(g) |A
1g> as well as g|E
1ux> = χ(g) |E
1ux >. The characters χ(g) of the totally symmetric IRREP A
1g are all equal to 1. Hence the operations g of G must satisfy the symmetry property g|E
1ux> =|E
1ux >,
Since the electronic wave function |E
1ux > transforms as x, the operations g of G must satisfy g x = x, or g
ex =
ex. There are only four operations g of G which map x on x, or the unit vector
ex on
ex, namely g = E, C
21”, σ
h and σ
d1, cf.
Figure 1. The set of operations {E, C
21”, σ
h, σ
d1} is the subgroup C
2v”
1, cf.
Table 1. Hence S(|Ψ>) = C
2v”
1. Accordingly, the entry |Ψ> = |A
1g> + |E
1ux> in
Table 3 marks the characters “1” for the operations g = E, C
21”, σ
h and σ
d1, and these yield the subgroup S(|Ψ>) = C
2v”
1 with totally symmetric IRREP A
1g. The theorem (56) implies that the corresponding one-electron density ρ
e(t > t
f) has the same symmetry C
2v”
1.
Our second example is for the superposition state |Ψ
e’1&e”1(t >t
f)> = c
e’1&e”1,1A1g (t) |1A
1g> + c
e’1&e”1,2A1g (t) |2A
1g> + c
e‘1&e“1,1E2g^xy (t) |1E
2gxy>, cf. Equation (52), which is prepared by the series of two laser pulses with different polarizations
e’
1 and
e”
1, cf.
Figure 9. Using again the simplified notation of Equations (53) and (55), this superposition state is rewritten
where
It consists of two partial waves with IRREPs A
1g and E
2gxy of the group G=D
6h. According to the general theory (cf.
Appendix A), and by analogy with the first example, the subgroup S(|Ψ>) of |Ψ> is the subset of the symmetry operations g of G which satisfy the symmetry property g|E
2gxy> =|E
2gxy>,
Since the electronic wave function |E
2gxy > transforms as xy, the operations g of G must satisfy g xy = xy. There are only four operations g of G which map xy on xy, namely g = E, C
2z, i and σ
h, cf.
Figure 1. The set of operations {E, C
2z, i, σ
h} is the subgroup C
2h, cf.
Table 1. Hence S(|Ψ>) = C
2h. Accordingly, the entry |Ψ> = |A
1g> + |E
2gxy> in
Table 3 marks the characters “1” for the operations g = E, C
2z, i, σ
h, and these yield the subgroup S(|Ψ>) = C
2h with totally symmetric IRREP A
g. The theorem (56) implies that the corresponding one-electron density ρ
e(t > t
f) has the same symmetry S(ρ
e(t > t
f)) = C
2h.
To summarize, the applications of the general theory (cf.
Appendix A) to the cases (57) and (60) demonstrate a simple recipe for the approach from the laser-driven electronic superposition state (53) via its symmetry subgroup (58) to the same symmetry subgroup of the one-electron density. The same approach can be applied to all other cases which were presented in
Section 3.1. The resulting electron symmetry breakings due to laser pulses p are also listed in
Table 3. They are in perfect agreement with the empirical results which were obtained in
Section 3.1, by inspection of the symmetry elements of the one-electron densities.
Table 3 also presents analogous results for the symmetry subgroups of the one-electron densities of two non-aromatic superposition states (49) and (50) which are prepared by laser pulses designed by Ulusoy and Nest [
1]. Accordingly, our result S(|A
1g> + |B
1u>) = D
3h’ for the superposition state |A
1g> + |B
1u> and for its one-electron density confirms their assignment. However, for the other case, our approach yields S(|A
1g> + |B
2u>) = D
3h”, different from their result D
3d [
1]. In retrospect, close inspection of their cartoons of the one-electron density and of the corresponding Lewis structures verifies the present result.
3.4. Symmetry of One-Electron Densities of Superposition States in Oriented Mg-Porphyrin
This Section for the derivation of the symmetries of one-electron densities of laser-driven superposition states in oriented Mg-porphyrin (D
4h) is entirely analogous to the corresponding
Section 3.3 for oriented benzene (D
6h). The previous
Section 3 has detailed presentations of two applications of the general theory (cf.
Appendix A) to two different superposition states which are prepared by a linearly polarized laser pulse, or by two sequential linearly polarized pulses, as illustrated in
Figure 5 and
Figure 9, respectively, cf.
Section 3.1. The results are summarized in
Table 3, together with the results for six additional cases. The present
Section 3.4 summarizes the corresponding results for four applications to laser-driven superposition states in Mg-porphyrin in
Table 4, complemented by the analogous step-by-step presentation of one example.
Specifically, let us derive the symmetry subgroup of the one-electron density of the superposition state |Ψ
e’1(t > t
f)> = c
e’1,A1g(t) |1A
1g> + c
e’1,1Eux(t) |1E
ux> which is prepared by the
e’
1 =
ex polarized π/2 laser pulse shown in
Figure 11, cf.
Section 3.2. Using the simplified notation of Equations (53) and (55), this superposition state is rewritten
It consists of two partial waves with IRREPs A
1g and E
ux ≡ E
u1’ of the group G = D
4h; the two equivalent notations remind of the two notations of the laser polarizations,
ex =
e’
1. According to the general theory (cf.
Appendix A) and its application in
Section 3.3, the symmetry subgroup S(|Ψ>) of the wavefunction (63) consists of the operations g of G which satisfy the symmetry property g|E
ux> =|E
ux>,
Since the electronic wave function |E
1ux > ≡ |E
u1’> transforms as x, the operations g of G must satisfy g x = x, or g
e’
1 =
e’
1. There are four operations g of G which map x on x, or the unit vector
e’
1 on
e’
1, namely g = E, C
21’, σ
h and σ
v1, cf.
Figure 2. Accordingly, the entry |Ψ> = |A
1g> + |E
u1’> in
Table 4 marks the characters “1” for the operations g = E, C
21’, σ
h and σ
v1, and these yield the subgroup S(|Ψ>) = C
2v’
1 with totally symmetric IRREP A
1g. The theorem (56) implies that the corresponding one-electron density ρ
e’1(t > t
f) has the same symmetry C
2v’
1.
Table 4 summarizes this result, together with three additional applications. The empirical results which were derived by inspection of the symmetry elements of the one-electron densities of two laser-driven superposition states in
Section 3.2 (cf.
Figure 12) are confirmed by the group-theoretical results of
Table 4.
Table 4 also comprises the results for two cases of electron symmetry breaking in the oriented Mg-porphyrin which have not been demonstrated in
Section 3.2. We use these cases to explain two applications of the general approach (cf.
Appendix A) which was exemplified in
Section 3.3 and
Section 3.4. First, it provides an efficient and firm method to determine the subgroup S(ρ
e(t > t
f)) of electron symmetry breaking G → S(ρ
e(t > t
f)) by a laser pulse. As example, consider the (hypothetical) preparation of the superposition state |Ψ
e”2(t > t
f)> = c
e”2,A1g(t) |1A
1g > + c
e”2,1Eu2”(t) |1E
u2” > by means of a
e”
2 polarized π/2 laser pulse, labeled p = 3. It would represent a new type of charge migration. The traditional way of determining the related electron symmetry breaking G = D
4h → S(ρ
e”2(t > t
f)) would cost quantum dynamics simulations of the laser excitation |1A
1g> → |Ψ
e”2(t > t
f)>, calculation of the one-electron density ρ
e”2(t > t
f), an inspection of ρ
e”2(t > t
f) to determine the symmetry elements of G which are conserved in ρ
e”2(t > t
f), and finally, the determination of the subgroup S(ρ
e”2(t > t
f)) as the set of the corresponding symmetry operations, cf.
Section 3.1 and
Section 3.2. In contrast, the recipe which was exemplified in
Section 3.3 and
Section 3.4 just calls for rewriting the target wavefunction as
and then to determine the symmetry group of this wavefunction as the set of symmetry operations g which satisfy g |E
u2”> =|E
u2”>. The recipe of
Section 3.3 and
Section 3.4 yields the result
without any inspection of the one-electron density ρ
e”2(t > t
f), cf.
Table 4.
The second application is for laser control of electron symmetry. This task is motivated, e.g., by the Woodward–Hoffmann rules for the control of chemical reactions by preparations of the reactants with different electron symmetries in the electronic ground and excited states [
35]. For example, let us assume that the goal is to design the laser pulse, or series of laser pulses with total duration t
f ≤ 10 fs and polarization(s)
e which achieve(s) electron symmetry breaking G = D
4h → S(ρ
e(t > t
f)) = C
2v’
2 in the oriented Mg-porphyrin. The recipe of
Section 3.3 and
Section 3.4 suggests that for this purpose, the laser pulse(s) should prepare the superposition state
which consists of basis functions with IRREP A
1g and another IRREP Γ
k ǂ A
1g such that
cf.
Table 1 and
Table 4 and
Figure 2.
The first challenge then is to determine the IRREP Γ
k of the wavefunction | Γ
k > which satisfies Equation (68). For this purpose, one should check the D
4h character Table for the characters for the one-dimensional (1d) IRREPs and—in the case of 2d IRREPs—also the diagonal elements of the 2 × 2 representation matrices for all symmetry operations of D
4h, see, e.g., Ref. [
13]. The target IRREP Γ
k must have characters or diagonal elements of the representation matrices equal to “1” precisely for the symmetry elements {g=E,C
2‘
2, σ
h, σ
v2}.
Table 4 shows that this condition is satisfied exclusively by the basis function |nE
u2’> for IRREP Γ
k = E
u. Hence the wavefunction | Γ
k > in the target superposition state (67) should be a linear combination of basis functions |nE
u2’>. Likewise, the other wavefunction | A
1g > in the superposition state (67) should be a linear combination of basis functions |nA
1g>. These two conditions for Equation (67) allow many choices. The simplest linear combination is
with time-dependent coefficients, cf. Equation (26).
The second challenge is to design the laser pulse, or series of laser pulses, with polarization(s)
e which prepares the target state (69), within less than t
f = 10 fs. Again, there is no unique solution for this task. In the present case, experience with |Ψ
e’1(t > t
f) > = c
e1’,1A1g|1A
1g > + c
e1’,Eu1’|1E
u1’ > (cf.
Section 3.2,
Figure 11) suggests that one should employ the corresponding π/2 laser pulse, labeled p = 4, with polarization
e4 =
e’
2 =
ey and with the same electric field
(t) as the one shown in
Figure 11.
3.5. Symmetry of One-Electron Densities of Oriented Molecules Driven by One or Several Linearly Polarized Laser Pulses
The previous
Section 3.1,
Section 3.2,
Section 3.3 amd
Section 3.4 contain ten plus four examples of electron symmetry breaking G → S(ρ
e(t > t
f)) by short (t
f = 10 fs) laser pulses, or two sequential laser pulses with polarizations
e in the oriented benzene (G = D
6h) and Mg-porphyrin (G = D
4h). The results are summarized in
Table 3 and
Table 4. Comparison with
Table 2 reveals the rule
The corresponding orders |...| of the groups and subgroups satisfy
The equality in the middle of relations (70), (71) is adapted from Equation (56). It holds for superposition states (53) which are prepared by the laser pulse(s) such that they contain the ground state and/or an excited basis function with the IRREP A
1g of the initial state, plus one (or more) excited states with different IRREPs, see, e.g., Equation (55) and all the examples in
Table 3 and
Table 4.
The right hand side of the relations (70), (71) simply means that S(ρ
e(t > t
f))=S(|Ψ
e(t > t
f)>) is a subgroup of G. It may even be the same as G: In this case, the laser pulse does not break the symmetry G at all. This may be called the “largest case”, i.e., |S(ρ
e(t > t
f))| = |S(|Ψ
e(t > t
f)>)| = |G|. This case holds for well-designed sequences of laser pulses with polarization vectors
e which break and restore electron symmetry [
2,
3,
4], or which induce attosecond charge migration represented by superpositions of electronic eigenstates with the same IRREP as the ground state, see, e.g., Refs. [
36,
37,
38]. The left hand side of the relations (70) and (71) is an important restriction. It means that the subgroup S(ρ
e(t > t
f)) = S(|Ψ
e(t > t
f)) of laser-broken electron symmetry must contain the subgroup S(H
e(t)) of the time-dependent electronic Hamiltonian of the molecule interacting with the laser pulse(s), cf.
Section 2.5 and
Table 2. The subgroup S(ρ
e(t > t
f)) = S(|Ψ
e(t > t
f)) may even be the same as S(H
e(t)). This may be called the “smallest case”, i.e., |S(ρ
e(t > t
f))| = |S(|Ψ
e(t > t
f))| = |S(H
e(t))|. In general, however, the order of the subgroup S(ρ
e(t > t
f)) = S(|Ψ
e(t > t
f)) can be larger than S(H
e(t)), and smaller than G. Comparison of all results in
Table 3 and
Table 4 with
Table 2 confirm the rule (70) and (71) for all ten plus four examples of
Section 3.1,
Section 3.2,
Section 3.3 and
Section 3.4The purpose of this
Section 3.5 is to prove the relation (70), and to discuss some consequences. The proof of the equality in the middle of relation (70) is in
Appendix A, cf. Equation (56). The right-hand side of the relations (70) and (71) is proven by empirical evidence: The previous investigations [
2,
3,
4] show that series of two laser pulses with the same polarizations can break and restore the original symmetry of the electronic ground state. From the viewpoint of this paper, Refs. [
2,
3,
4] present the “largest cases” where S(ρ
e(t > t
f)) = S(|Ψ
e(t > t
f)) = G.
For the proof of the left-hand side of the relations (70) and (71), we use the fact that the Hamilton operator H
e(t) commutes with the group elements g ϵ S(H
e(t))
cf.
Section 2.5, Equations (11) and (13). The initial (t = 0) wavefunction is the electronic ground state, Equation (15). Since it is totally symmetric with respect to all symmetry elements of the group G,
After the laser pulse (t > t
f), the laser-driven wave function has evolved to
with time evolution operator
cf. Equation (25). Since the symmetry operations g ϵ S(H
e(t)) commute with H
e(t), cf. Equation (72), they also commute with U
e(t). Hence
This means that |Ψe(t)> has at least the symmetry S(He(t)) of the time-dependent Hamilton operator He(t). However, at times t ≥ tf after the laser pulse, the relation (76) may also hold for additional group elements g~ ϵ G which do not belong to S(He(t)). Let S(|Ψe(t > tf)>) = {g, g~} be the subgroup of G which contains the elements g ϵ S(He(t)) and g~ ∉ S(He(t)) which satisfy the relation (76). Now there may be two cases: either there are such “additional elements” g~, or not. Accordingly, either |S(He(t))| = |S(|Ψe(t > tf))| and S(He(t)) = S(|Ψe(t > tf)), or |S(He(t))| < |S(|Ψe(t > tf))| and S(He(t)) ⊆ S(|Ψe(t > tf)), respectively. This completes the proof of the left hand side of the relations (70) and (71). In any case, the IRREP of |Ψe(t)> is always the totally symmetric IRREP of the subgroup S(|Ψe(t)>), because all elements g ϵ S(He(t)) and the other ones (if any) g~ satisfy the equation (76), i.e., all the characters of |Ψe(t)> are equal to 1.
The rule (70) and (71) has (at least) two consequences, not only for the present examples, electron symmetry breaking by short (typically t
f ≤ 10 fs) laser pulses, or two sequential laser pulses with polarization(s)
e in the oriented benzene of Mg-porphyrin, but by analogy also in other oriented molecules with symmetry group G of the nuclear scaffold. On the one hand, if one starts from the electronic ground state with totally symmetric IRREP and applies laser pulse(s) with polarization vector(s)
e, then the knowledge of the symmetry S(H
e(t)) of the time-dependent electronic Hamilton operator H
e(t) of the molecule interacting with the laser pulse(s) (in semiclassical dipole approximation), does
not suffice to determine the electron symmetry breaking G → S(ρ
e(t > t
f)) = S(|Ψ
e(t > t
f)). On the other hand, one has some important information about S(ρ
e(t > t
f)) = S(|Ψ
e(t > t
f)), namely it has to be a subgroup of G,
and it must contain the symmetry group S(H
e(t)) as a subgroup. For example, if one applies an
e =
ey =
e1’ polarized laser pulse (or series of pulses) on the oriented benzene, then S(H
e(t)) = C
2v’
1, cf.
Table 2. Hence it is impossible to design any electric fields
(t) of
ey polarized laser pulses which would achieve electron symmetry breakings D
6h → C
s, C
2, or C
i because these hypothetical target subgroups have order 2, below the order 4 of S(H
e(t)), which is the minimum according to the rule (71). Instead, there is a large variety of possible symmetry breakings D
6h → S(ρ
e(t > t
f)) = S(|Ψ
e(t > t
f)) which satisfy the rule (70), (71), from the “smallest” case S(ρ
e(t > t
f)) = S(|Ψ
e(t > t
f)) = C
2v’
1 (cf.
Table 3, entry for |Ψ
e(t > t
f)> = |A
1g> + |E
1uy>) via larger subgroups D
2h,1 (cf.
Table 3, |Ψ
e(t > t
f)> = |A
1g> + |E
1ux2−y2>) or D
3h’ (cf.
Table 3, |Ψ
e(t > t
f)> = |A
1g> + |B
1u>) to G = D
6h, in the extreme case of symmetry restoration—these examples are all in accord with the rule (70), (71). Likewise, if one applies an
e =
ex =
e1” polarized laser pulse (or series of pulses) on the oriented benzene, then S(H
e(t)) = C
2v”
1, and the rule (70), (71) again opens many possibilities, from the smallest possible subgroup C
2v”
1 (cf.
Table 3, entry for |Ψ
e(t > t
f)> = |A
1g> + |E
1ux>) via the larger subgroup D
3h” (cf.
Table 3, |Ψ
e(t > t
f)> = |A
1g> + |B
2u>) to G = D
6h. However, the rule (70), (71) also excludes many hypothetical cases, not only the subgroups of order 2 which must be discarded because this is below the order 4 of the minimum group C
2v”
1, but also larger ones such as D
3d’ or D
3d”—such symmetry groups (suggested in Ref. [
1]) are excluded because they do not contain the subgroup S(H
e(t)) = C
2v”
1, i.e., they violate the theorem (70).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











