
Microsatellites as Agents of Adaptive Change: An RNA-Seq-Based Comparative Study of Transcriptomes from Five Helianthus Species

 , and
, and
Abstract
:1. Introduction
- (1)
- Are microsatellite-containing genes more likely to show evidence of expression divergence among species, as compared to genes lacking microsatellites?
- (2)
- Do microsatellites-containing genes exhibit greater levels of genetic divergence compared to genes lacking microsatellites?
2. Materials and Methods
2.1. Plant Sampling and Sequencing
2.2. Post Sequencing Data Collection
2.3. Functional Annotation
2.4. Mining and Genotyping SNPs and Microsatellites
2.5. Differential Expression
2.6. Gene Ontology (GO) Enrichment Analysis
2.7. Relative Importance of Microsatellites in Species Divergence
2.8. Population Genetic Analyses
2.9. Shared Microsatellites
3. Results
3.1. SNPs and Microsatellites Mined
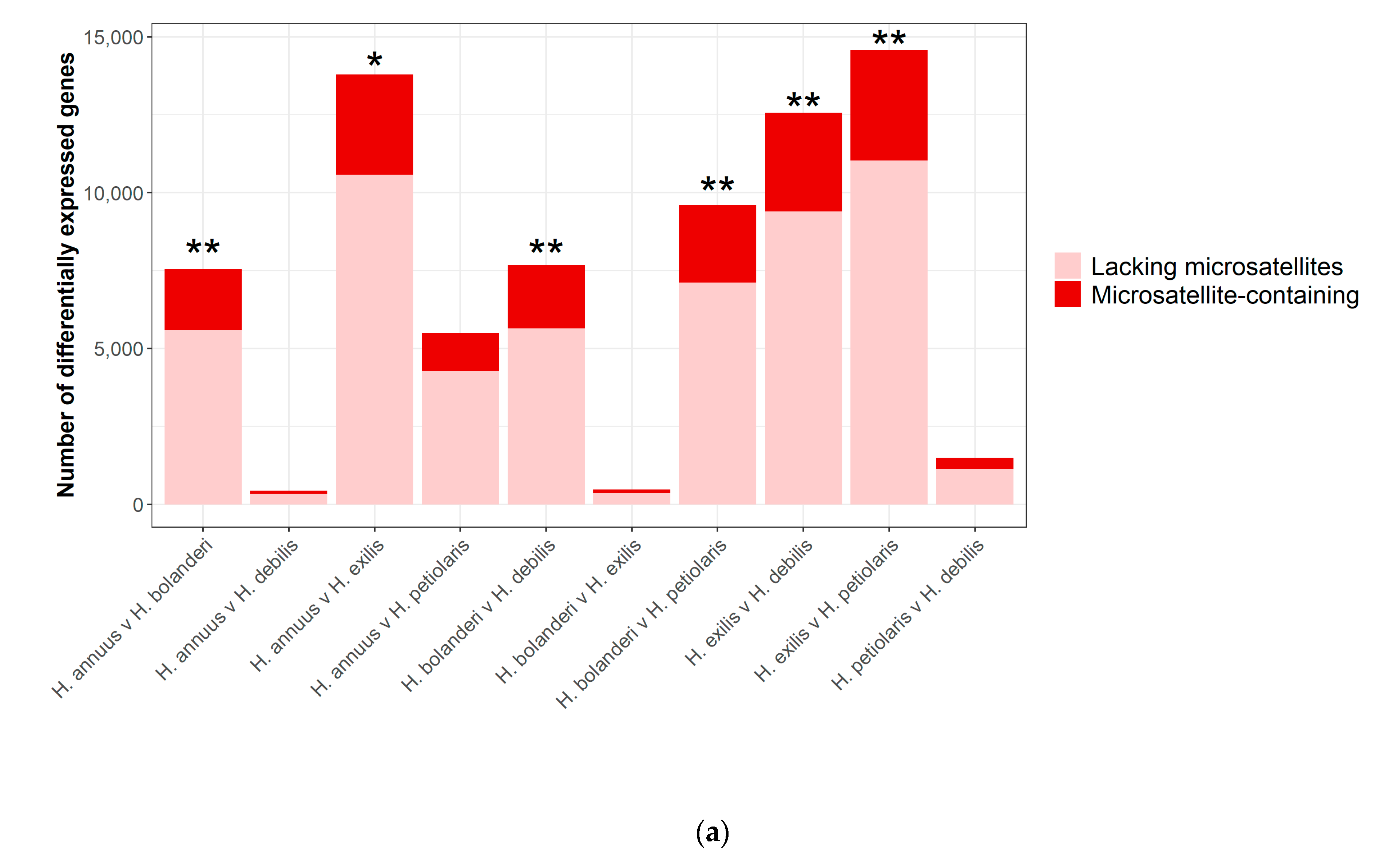
3.2. Differential Expression Analysis
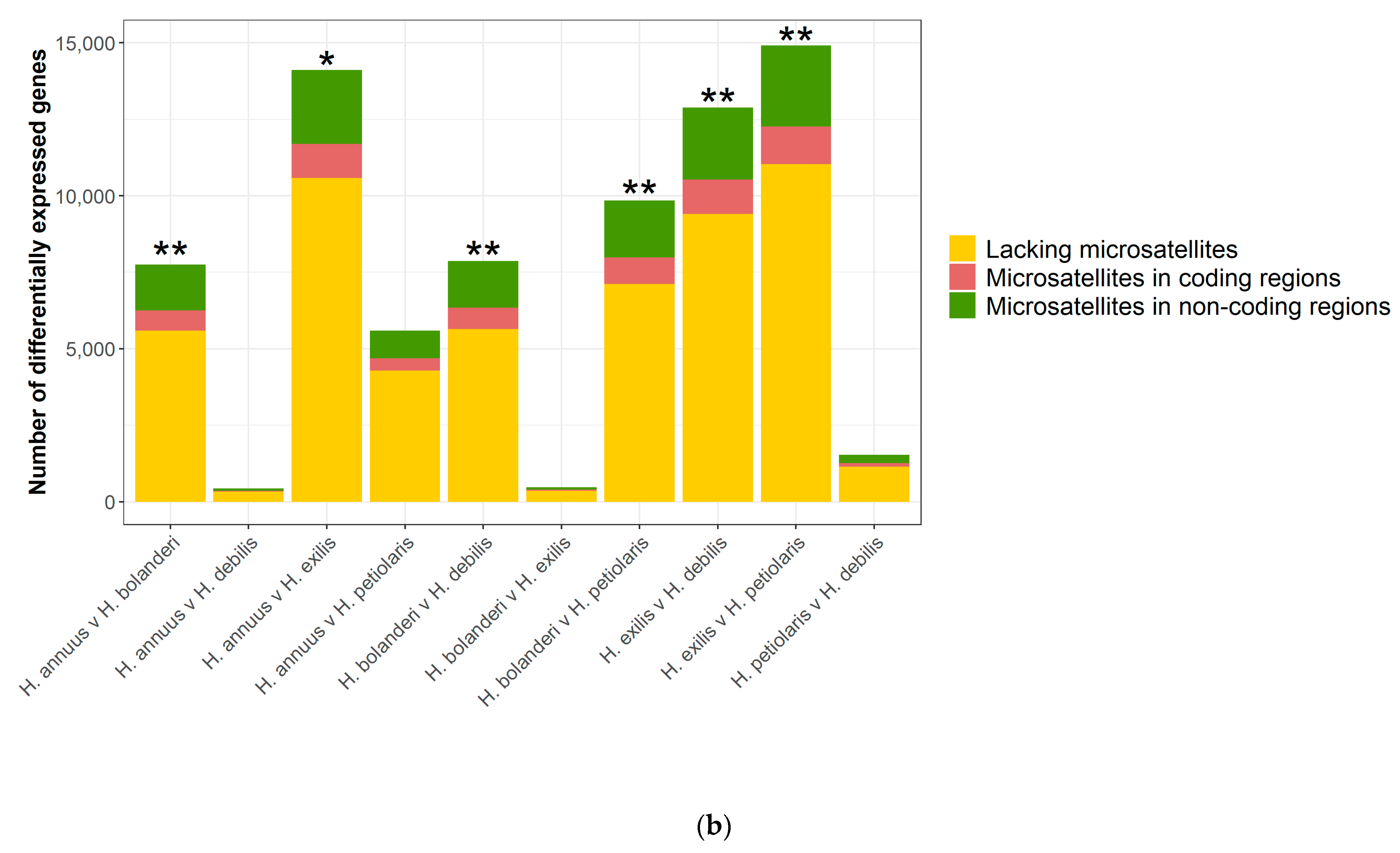
3.3. Microsatellite-Containing Differentially Expressed Genes
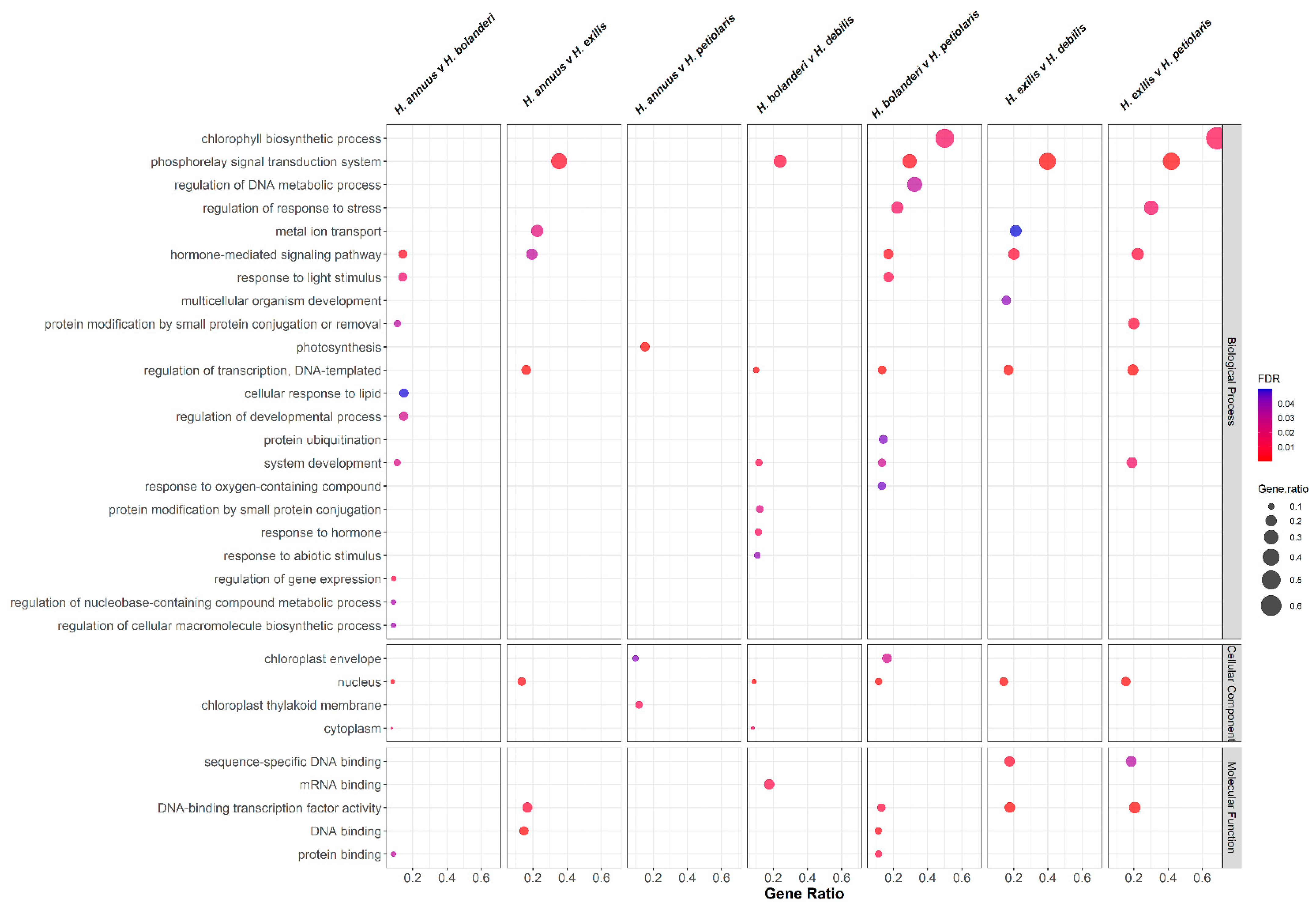
3.4. Functional Annotation and Gene Ontology Enrichment Analysis
3.5. Population Genetic Estimates
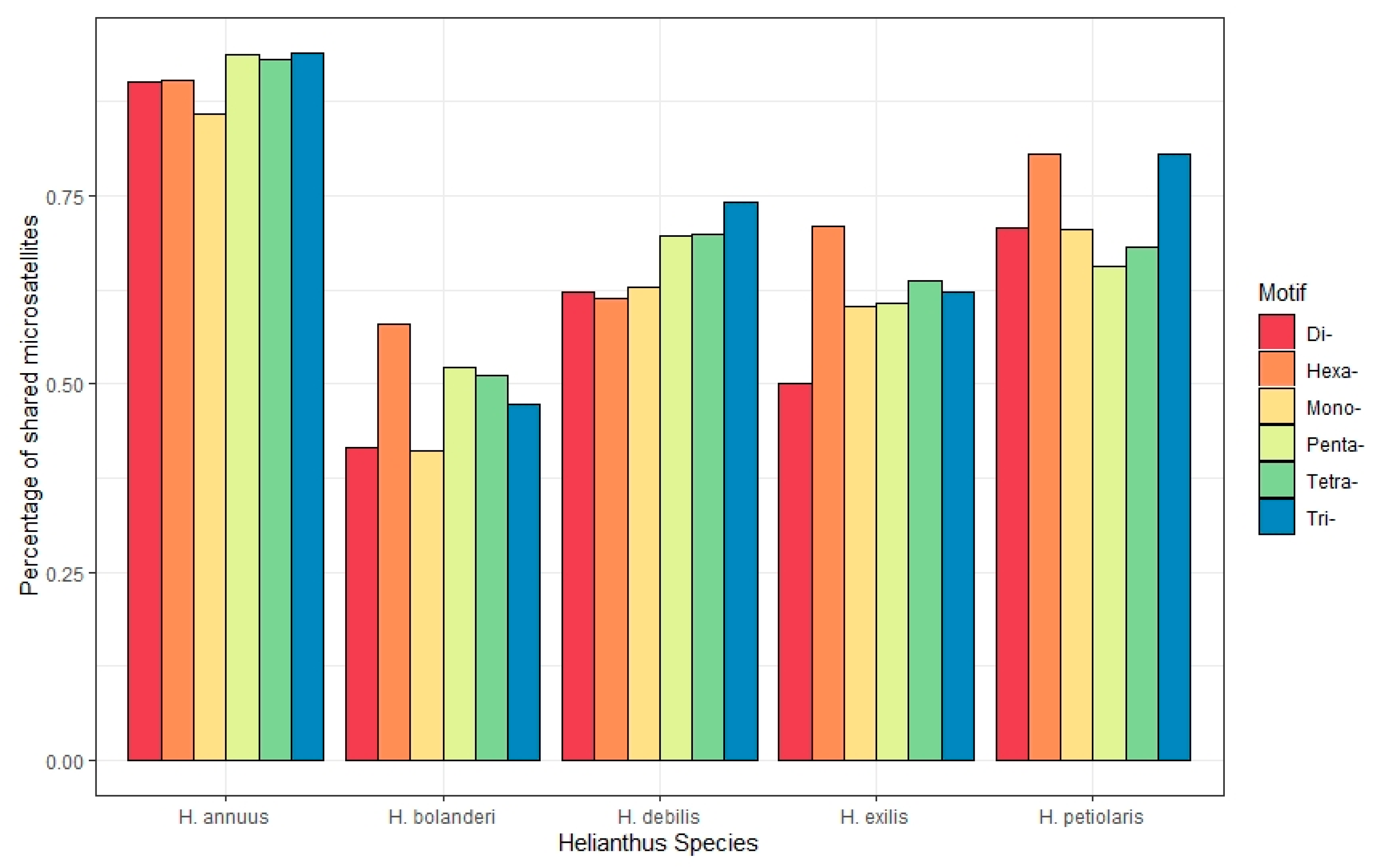
3.6. Shared Microsatellites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wray, G.A.; Hahn, M.W.; Abouheif, E.; Balhoff, J.P.; Pizer, M.; Rockman, M.V.; Romano, L.A. The Evolution of Transcriptional Regulation in Eukaryotes. Mol. Biol. Evol. 2003, 20, 1377–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, M.C.; Wilson, A.C. Evolution at Two Levels in Humans and Chimpanzees. Science 1975, 188, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittkopp, P.J.; Haerum, B.K.; Clark, A.G. Regulatory Changes Underlying Expression Differences within and between Drosophila Species. Nat. Genet. 2008, 40, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Bedford, T.; Hartl, D.L. Optimization of Gene Expression by Natural Selection. Proc. Natl. Acad. Sci. USA 2009, 106, 1133–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, D.L.; Orgogozo, V. The Loci of Evolution: How Predictable Is Genetic Evolution? Evol. Int. J. Org. Evol. 2008, 62, 2155–2177. [Google Scholar] [CrossRef] [Green Version]
- Gemayel, R.; Vinces, M.D.; Legendre, M.; Verstrepen, K.J. Variable Tandem Repeats Accelerate Evolution of Coding and Regulatory Sequences. Annu. Rev. Genet. 2010, 44, 445–477. [Google Scholar] [CrossRef] [Green Version]
- Rockman, M.V.; Wray, G.A. Abundant Raw Material for Cis-Regulatory Evolution in Humans. Mol. Biol. Evol. 2002, 19, 1991–2004. [Google Scholar] [CrossRef] [Green Version]
- Bagshaw, A.T.M. Functional Mechanisms of Microsatellite DNA in Eukaryotic Genomes. Genome Biol. Evol. 2017, 9, 2428–2443. [Google Scholar] [CrossRef] [Green Version]
- Tautz, D.; Renz, M. Simple Sequences Are Ubiquitous Repetitive Components of Eukaryotic Genomes. Nucleic Acids Res. 1984, 12, 4127–4138. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic Distribution, Putative Functions and Mutational Mechanisms: A Review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef]
- Ohta, T.; Kimura, M. A Model of Mutation Appropriate to Estimate the Number of Electrophoretically Detectable Alleles in a Finite Population. Genet. Res. 1973, 22, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Kruglyak, S.; Durrett, R.T.; Schug, M.D.; Aquadro, C.F. Equilibrium Distributions of Microsatellite Repeat Length Resulting from a Balance between Slippage Events and Point Mutations. Proc. Natl. Acad. Sci. USA 1998, 95, 10774–10778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.C.; Röder, M.S.; Fahima, T.; Kirzhner, V.M.; Beiles, A.; Korol, A.B.; Nevo, E. Climatic Effects on Microsatellite Diversity in Wild Emmer Wheat (Triticum dicoccoides) at the Yehudiyya Microsite, Israel. Heredity 2002, 89, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodel, R.G.J.; Segovia-Salcedo, M.C.; Landis, J.B.; Crowl, A.A.; Sun, M.; Liu, X.; Gitzendanner, M.A.; Douglas, N.A.; Germain-Aubrey, C.C.; Chen, S.; et al. The Report of My Death Was an Exaggeration: A Review for Researchers Using Microsatellites in the 21st Century. Appl. Plant Sci. 2016, 4, 1600025. [Google Scholar] [CrossRef]
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in Different Eukaryotic Genomes: Surveys and Analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef] [Green Version]
- Mirkin, S.M. Expandable DNA Repeats and Human Disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef]
- Moxon, E.R.; Rainey, P.B.; Nowak, M.A.; Lenski, R.E. Adaptive Evolution of Highly Mutable Loci in Pathogenic Bacteria. Curr. Biol. 1994, 4, 24–33. [Google Scholar] [CrossRef]
- Kashi, Y.; King, D.G. Simple Sequence Repeats as Advantageous Mutators in Evolution. Trends Genet. 2006, 22, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Undurraga, S.F.; Press, M.O.; Legendre, M.; Bujdoso, N.; Bale, J.; Wang, H.; Davis, S.J.; Verstrepen, K.J.; Queitsch, C. Background-Dependent Effects of Polyglutamine Variation in the Arabidopsis thaliana Gene ELF3. Proc. Natl. Acad. Sci. USA 2012, 109, 19363–19367. [Google Scholar] [CrossRef] [Green Version]
- Rival, P.; Press, M.O.; Bale, J.; Grancharova, T.; Undurraga, S.F.; Queitsch, C. The Conserved PFT1 Tandem Repeat Is Crucial for Proper Flowering in Arabidopsis thaliana. Genetics 2014, 198, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Press, M.O.; Queitsch, C. Variability in a Short Tandem Repeat Mediates Complex Epistatic Interactions in Arabidopsis thaliana. Genetics 2017, 205, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Namdar-Aligoodarzi, P.; Mohammadparast, S.; Zaker-Kandjani, B.; Kakroodi, S.T.; Vesiehsari, M.J.; Ohadi, M. Exceptionally Long 5′ UTR Short Tandem Repeats Specifically Linked to Primates. Gene 2015, 569, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Ohadi, M.; Valipour, E.; Ghadimi-Haddadan, S.; Namdar-Aligoodarzi, P.; Bagheri, A.; Kowsari, A.; Rezazadeh, M.; Darvish, H.; Kazeminasab, S. Core Promoter Short Tandem Repeats as Evolutionary Switch Codes for Primate Speciation. Am. J. Primatol. 2015, 77, 34–43. [Google Scholar] [CrossRef]
- Fondon, J.W.; Garner, H.R. Molecular Origins of Rapid and Continuous Morphological Evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 18058–18063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, T.P.; Park, S.; Kim, T.S.; Booth, J.; Byer, A.; Sun, Q.; Chory, J.; Lee, K. Simple Sequence Repeats Provide a Substrate for Phenotypic Variation in the Neurospora crassa Circadian Clock. PLoS ONE 2007, 2, e795. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Qin, W.; Liu, F.; Liu, B.; Zhou, Y.; Jiang, T.; Yu, C. Sex-specific Mediation Effect of the Right Fusiform Face Area Volume on the Association between Variants in Repeat Length of AVPR 1 A RS 3 and Altruistic Behavior in Healthy Adults. Hum. Brain Mapp. 2016, 37, 2700–2709. [Google Scholar] [CrossRef]
- Xu, G.; Goodridge, A.G. Act Repeat in the Promoter of the Chicken Malic Enzyme Gene is Essential for Function at an Alternative Transcription Start Site. Arch. Biochem. Biophys. 1998, 358, 83–91. [Google Scholar] [CrossRef]
- Ng, T.K.; Lam, C.Y.; Lam, D.S.; Chiang, S.W.; Tam, P.O.; Wang, D.Y.; Fan, B.J.; Yam, G.H.; Fan, D.S.; Pang, C.P. AC and AG Dinucleotide Repeats in the PAX6 P1 Promoter are Associated with High Myopia. Mol. Vis. 2009, 15, 2239–2248. [Google Scholar]
- Quilez, J.; Guilmatre, A.; Garg, P.; Highnam, G.; Gymrek, M.; Erlich, Y.; Joshi, R.S.; Mittelman, D.; Sharp, A.J. Polymorphic Tandem Repeats within Gene Promoters Act as Modifiers of Gene Expression and DNA Methylation in Humans. Nucleic Acids Res. 2016, 44, 3750–3762. [Google Scholar] [CrossRef] [Green Version]
- Gymrek, M.; Willems, T.; Guilmatre, A.; Zeng, H.; Markus, B.; Georgiev, S.; Daly, M.J.; Price, A.L.; Pritchard, J.K.; Sharp, A.J.; et al. Abundant Contribution of Short Tandem Repeats to Gene Expression Variation in Humans. Nat. Genet. 2015, 48, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Fotsing, S.F.; Margoliash, J.; Wang, C.; Saini, S.; Yanicky, R.; Shleizer-Burko, S.; Goren, A.; Gymrek, M. The Impact of Short Tandem Repeat Variation on Gene Expression. Nat. Genet. 2019, 51, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Ranathunge, C.; Wheeler, G.L.; Chimahusky, M.E.; Perkins, A.D.; Pramod, S.; Welch, M.E. Transcribed Microsatellite Allele Lengths Are Often Correlated with Gene Expression in Natural Sunflower Populations. Mol. Ecol. 2020, 29, 1704–1716. [Google Scholar] [CrossRef] [PubMed]
- Stephens, J.D.; Rogers, W.L.; Mason, C.M.; Donovan, L.A.; Malmberg, R.L. Species Tree Estimation of Diploid Helianthus (Asteraceae) Using Target Enrichment. Am. J. Bot. 2015, 102, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, R.L.; Rieseberg, L.H. Divergence Is Focused on Few Genomic Regions Early in Speciation: Incipient Speciation of Sunflower Ecotypes. Evolution 2013, 67, 2468–2482. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Andrew, R.L.; Owens, G.L.; Ostevik, K.L.; Rieseberg, L.H. Multiple Chromosomal Inversions Contribute to Adaptive Divergence of a Dune Sunflower Ecotype. Mol. Ecol. 2020, 29, 2535–2549. [Google Scholar] [CrossRef]
- Todesco, M.; Owens, G.L.; Bercovich, N.; Légaré, J.-S.; Soudi, S.; Burge, D.O.; Huang, K.; Ostevik, K.L.; Drummond, E.B.M.; Imerovski, I. Massive Haplotypes Underlie Ecotypic Differentiation in Sunflowers. Nature 2020, 584, 602–607. [Google Scholar] [CrossRef]
- Ranathunge, C.; Wheeler, G.L.; Chimahusky, M.E.; Kennedy, M.M.; Morrison, J.I.; Baldwin, B.S.; Perkins, A.D.; Welch, M.E. Transcriptome Profiles of Sunflower Reveal the Potential Role of Microsatellites in Gene Expression Divergence. Mol. Ecol. 2018, 27, 1188–1199. [Google Scholar] [CrossRef]
- Renaut, S.; Owens, G.L.; Rieseberg, L.H. Shared Selective Pressure and Local Genomic Landscape Lead to Repeatable Patterns of Genomic Divergence in Sunflowers. Mol. Ecol. 2014, 23, 311–324. [Google Scholar] [CrossRef]
- Renaut, S.; Grassa, C.J.; Moyers, B.T.; Kane, N.C.; Rieseberg, L.H. The Population Genomics of Sunflowers and Genomic Determinants of Protein Evolution Revealed by RNAseq. Biology 2012, 1, 575–596. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Renaut, S.; Grassa, C.J.; Yeaman, S.; Moyers, B.T.; Lai, Z.; Kane, N.C.; Bowers, J.E.; Burke, J.M.; Rieseberg, L.H. Genomic Islands of Divergence Are Not Affected by Geography of Speciation in Sunflowers. Nat. Commun. 2013, 4, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofler, R.; Schlötterer, C.; Lelley, T. SciRoKo: A New Tool for Whole Genome Microsatellite Search and Investigation. Bioinformatics 2007, 23, 1683–1685. [Google Scholar] [CrossRef] [Green Version]
- Highnam, G.; Franck, C.; Martin, A.; Stephens, C.; Puthige, A.; Mittelman, D. Accurate Human Microsatellite Genotypes from High-Throughput Resequencing Data Using Informed Error Profiles. Nucleic Acids Res. 2013, 41, e32. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B. The Sunflower Genome Provides Insights into Oil Metabolism, Flowering and Asterid Evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef]
- Wren, J.D.; Forgacs, E.; Fondon, J.W.; Pertsemlidis, A.; Cheng, S.Y.; Gallardo, T.; Williams, R.S.; Shohet, R.V.; Minna, J.D.; Garner, H.R. Repeat Polymorphisms within Gene Regions: Phenotypic and Evolutionary Implications. Am. J. Hum. Genet. 2000, 67, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Morgante, M.; Hanafey, M.; Powell, W. Microsatellites Are Preferentially Associated with Nonrepetitive DNA in Plant Genomes. Nat. Genet. 2002, 30, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Liu, J. A Genome-Wide Analysis of Simple Sequence Repeats in Maize and the Development of Polymorphism Markers from next-Generation Sequence Data. BMC Res. Notes 2013, 6, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassone, F.; Beilina, A.; Carosi, C.; Albertosi, S.; Bagni, C.; Li, L.; Glover, K.; Bentley, D.; Hagerman, P.J. Elevated FMR1 MRNA in Premutation Carriers Is Due to Increased Transcription. RNA 2007, 13, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Joshi-Saha, A.; Reddy, K.S. Repeat Length Variation in the 5ʹUTR of Myo-Inositol Monophosphatase Gene Is Related to Phytic Acid Content and Contributes to Drought Tolerance in Chickpea (Cicer arietinum L.). J. Exp. Bot. 2015, 66, 5683–5690. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Kuo, P.; Hsu, C.; Tsai, S.; Chen, M.; Lin, C.; Sun, H.S. Microsatellite in the 3′ Untranslated Region of Human Fibroblast Growth Factor 9 (FGF9) Gene Exhibits Pleiotropic Effect on Modulating FGF9 Protein Expression. Hum. Mutat. 2007, 28, 98. [Google Scholar] [CrossRef] [PubMed]
- Lischer, H.E.L.; Excoffier, L. PGDSpider: An Automated Data Conversion Tool for Connecting Population Genetics and Genomics Programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- R Core Team. A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2012. [Google Scholar]
- Goudet, J. Hierfstat, a Package for R to Compute and Test Hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Jarne, P.; Lagoda, P.J.L. Microsatellites, from Molecules to Populations and Back. Trends Ecol. Evol. 1996, 11, 424–429. [Google Scholar] [CrossRef]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A Program for Detecting, Evaluating, and Reconstructing Potential Coding Regions in EST Sequences. Proc. ISMB 1999, 99, 138–148. [Google Scholar]
- Lottaz, C.; Iseli, C.; Jongeneel, C.V.; Bucher, P. Modeling Sequencing Errors by Combining Hidden Markov Models. Bioinformatics 2003, 19, ii103–ii112. [Google Scholar] [CrossRef] [Green Version]
- Sonay, T.B.; Carvalho, T.; Robinson, M.D.; Greminger, M.P.; Krützen, M.; Comas, D.; Highnam, G.; Mittelman, D.; Sharp, A.; Marques-Bonet, T.; et al. Tandem Repeat Variation in Human and Great Ape Populations and Its Impact on Gene Expression Divergence. Genome Res. 2015, 25, 1591–1599. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Bhatia, S. A Polymorphic (GA/CT) n-SSR Influences Promoter Activity of Tryptophan Decarboxylase Gene in Catharanthus roseus L. Don. Sci. Rep. 2016, 6, 33280. [Google Scholar] [CrossRef] [Green Version]
- Kramer, M.; Sponholz, C.; Slaba, M.; Wissuwa, B.; Claus, R.A.; Menzel, U.; Huse, K.; Platzer, M.; Bauer, M. Alternative 5′untranslated Regions Are Involved in Expression Regulation of Human Heme Oxygenase-1. PLoS ONE 2013, 8, e77224. [Google Scholar] [CrossRef] [PubMed]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated Regions of MRNAs. Genome Biol. 2002, 3, 0004.1–0004.10. [Google Scholar] [CrossRef]
- Searle, S.; Blackwell, J.M. Evidence for a Functional Repeat Polymorphism in the Promoter of the Human NRAMP1 Gene that Correlates with Autoimmune versus Infectious Disease Susceptibility. J. Med. Genet. 1999, 36, 295–299. [Google Scholar] [PubMed]
- Bayele, H.K.; Peyssonnaux, C.; Giatromanolaki, A.; Arrais-Silva, W.W.; Mohamed, H.S.; Collins, H.; Giorgio, S.; Koukourakis, M.; Johnson, R.S.; Blackwell, J.M. HIF-1 Regulates Heritable Variation and Allele Expression Phenotypes of the Macrophage Immune Response Gene SLC11A1 from a Z-DNA–Forming Microsatellite. Blood 2007, 110, 3039–3048. [Google Scholar] [CrossRef] [Green Version]
- Taka, S.; Gazouli, M.; Politis, P.K.; Pappa, K.I.; Anagnou, N.P. Transcription Factor ATF-3 Regulates Allele Variation Phenotypes of the Human SLC11A1 Gene. Mol. Biol. Rep. 2013, 40, 2263–2271. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
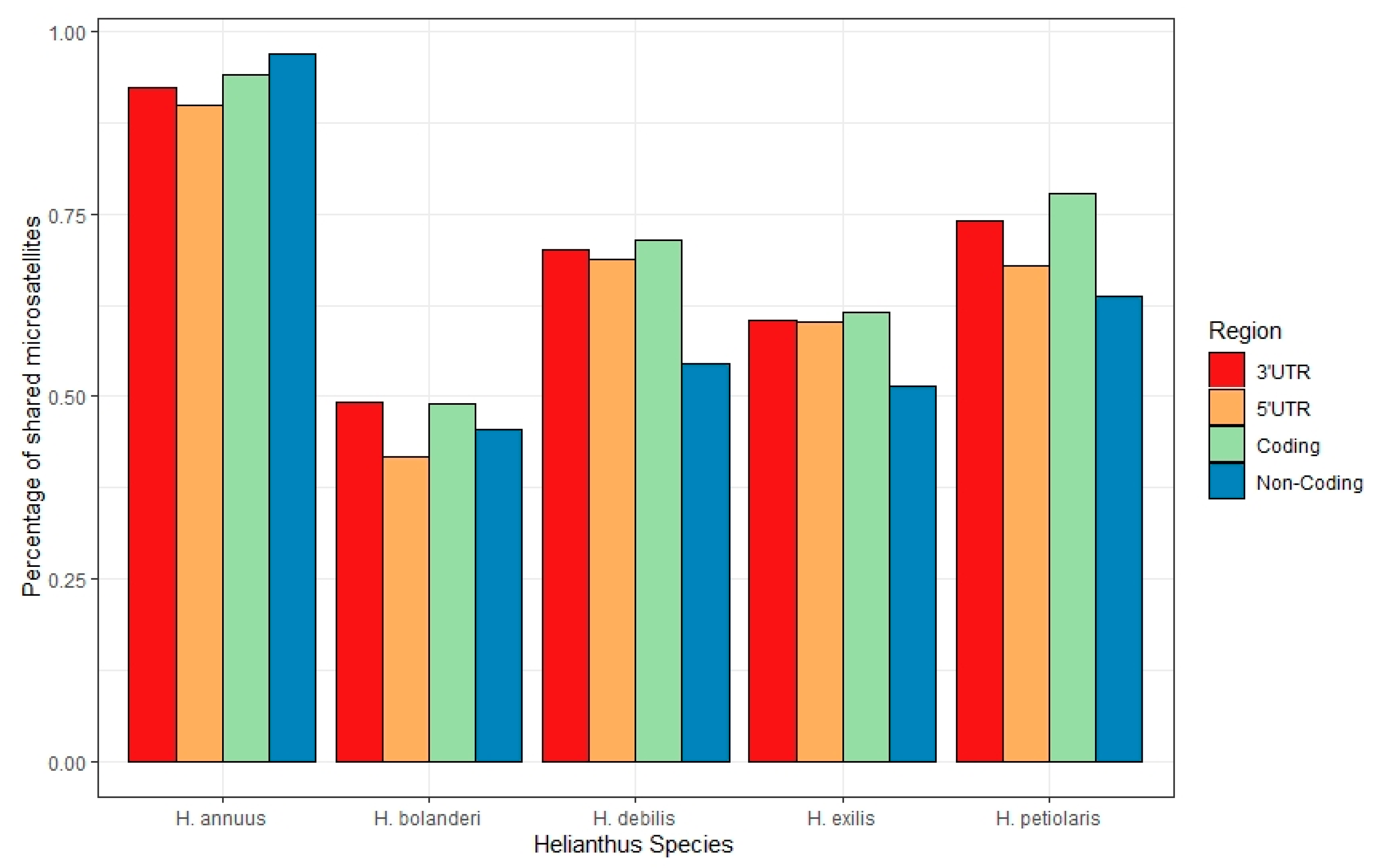{kind=link}
| Pairwise Species Comparison | Number of Differentially Expressed (DE) Genes | Microsatellites in DE Genes | |
|---|---|---|---|
| In Non-Coding Regions | In Coding Regions | ||
| H. annuus v. H. bolanderi | 7539 | 1490 | 669 |
| H. annuus v. H. debilis | 433 | 79 | 27 |
| H. annuus v. H. exilis | 13,789 | 2416 | 1120 |
| H. annuus v. H. petiolaris | 5490 | 910 | 401 |
| H. bolanderi v. H. debilis | 7671 | 1526 | 701 |
| H. bolanderi v. H. exilis | 470 | 76 | 37 |
| H. bolanderi v. H. petiolaris | 9593 | 1865 | 865 |
| H. exilis v. H. debilis | 12,567 | 2350 | 1132 |
| H. exilis v. H. petiolaris | 14,576 | 2653 | 1233 |
| H. petiolaris v. H. debilis | 1492 | 266 | 122 |
| Pairwise Comparison | Mean FST for Genes Lacking Microsatellites | Mean FST for Microsatellite-Containing Genes | Wilcoxon Rank Sum Test p-Value |
|---|---|---|---|
| H. annuus v. H. argophyllus | 0.413 | 0.449 | 2.95 × 10−10 |
| H. annuus v. H. debilis | 0.385 | 0.426 | 8.04 × 10−13 |
| H. annuus v. H. petiolaris | 0.326 | 0.357 | 2.17 × 10−10 |
| H. debilis v. H. argophyllus | 0.545 | 0.586 | 4.16 × 10−10 |
| H. petiolaris v. H. argophyllus | 0.506 | 0.538 | 4.50 × 10−9 |
| H. petiolaris v. H. debilis | 0.281 | 0.309 | 2.82 × 10−12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranathunge, C.; Pramod, S.; Renaut, S.; Wheeler, G.L.; Perkins, A.D.; Rieseberg, L.H.; Welch, M.E. Microsatellites as Agents of Adaptive Change: An RNA-Seq-Based Comparative Study of Transcriptomes from Five Helianthus Species. Symmetry 2021, 13, 933. https://0-doi-org.brum.beds.ac.uk/10.3390/sym13060933
Ranathunge C, Pramod S, Renaut S, Wheeler GL, Perkins AD, Rieseberg LH, Welch ME. Microsatellites as Agents of Adaptive Change: An RNA-Seq-Based Comparative Study of Transcriptomes from Five Helianthus Species. Symmetry. 2021; 13(6):933. https://0-doi-org.brum.beds.ac.uk/10.3390/sym13060933
Chicago/Turabian StyleRanathunge, Chathurani, Sreepriya Pramod, Sébastien Renaut, Gregory L. Wheeler, Andy D. Perkins, Loren H. Rieseberg, and Mark E. Welch. 2021. "Microsatellites as Agents of Adaptive Change: An RNA-Seq-Based Comparative Study of Transcriptomes from Five Helianthus Species" Symmetry 13, no. 6: 933. https://0-doi-org.brum.beds.ac.uk/10.3390/sym13060933