
Racemic and Meso Crystal Structures of an Axial-Chiral Spirobi-(dinaphthoazepin)ium Salt: Emergence of an S4-Symmetric Molecule



Abstract
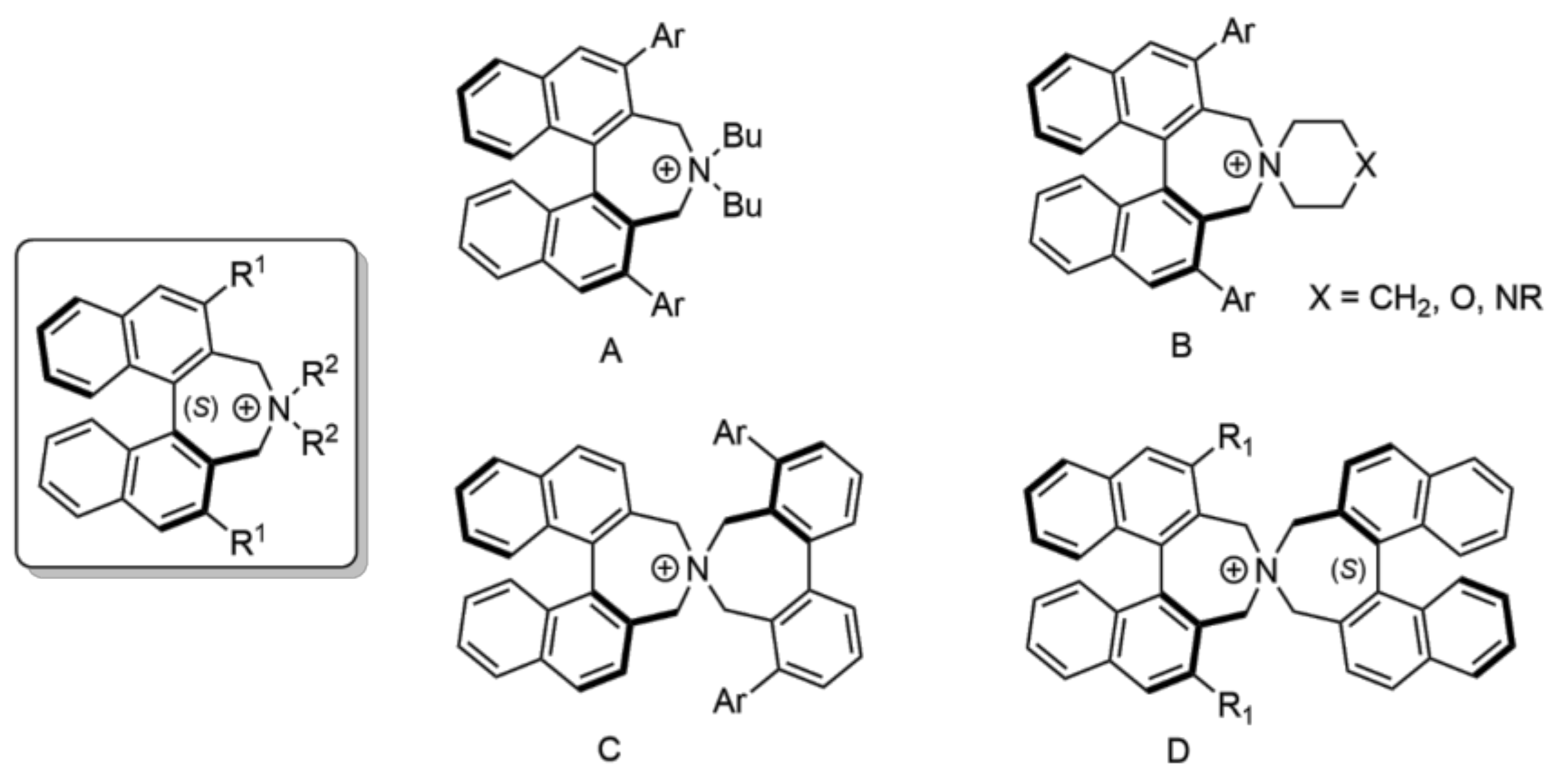
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis and Crystallisation
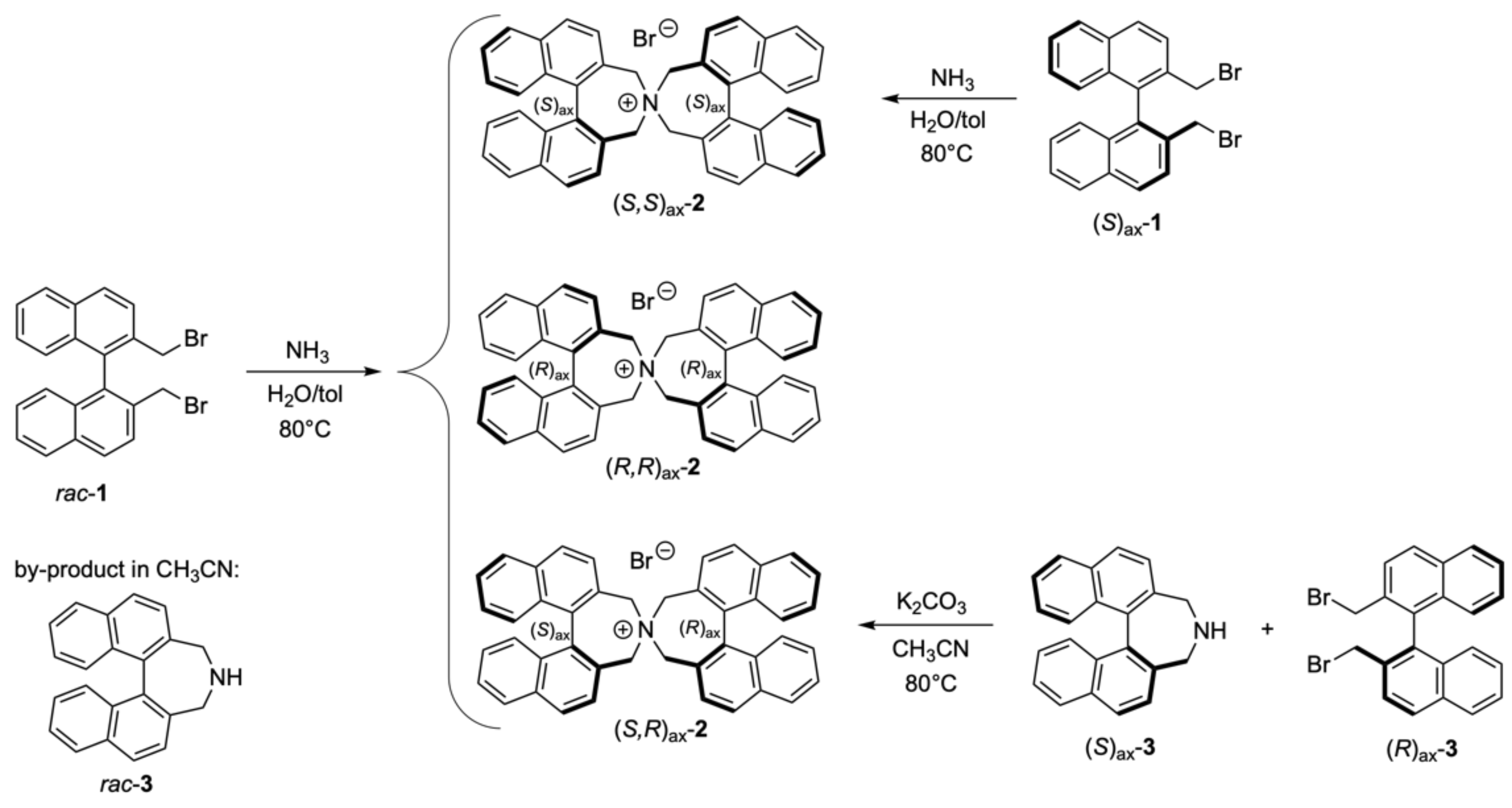
2.2.1. Synthesis of meso- and rac-3,3′,5,5′-Tetrahydro-4,4′-spirobi[dinaphtho [2,1-c:1′,2′-e]azepin]-4-ium Bromide, meso-2, and rac-2
- HRMS (ESI): calculated for C44H32N [M-Br+]: 574.2535, found 574.2548.
- rac-2: m.p.: 330–345 °C (decomposition).
- Applying the same procedure to (R)ax-1 afforded 94% of (R,R)ax-2.
2.2.2. Synthesis of meso-3,3′,5,5′-Tetrahydro-4,4′-spirobi[dinaphtho[2,1-c:1′,2′-e]azepin]-4-ium Bromide, (R,S)ax-2
2.2.3. Synthesis of 11,13,26,26a-Tetrahydrodinaphtho[2′,1′:3,4;1′′,2′′:5,6], azepino[1,2-a]dinaphtho[2,1-d:1′,2′-f]azocine; (S)C(R,R)ax-4 (racemate)
2.2.4. Stevens Rearrangement of meso-2
2.2.5. N-Methylation of 4
2.3. X-ray Diffractometry
3. Results and Discussion
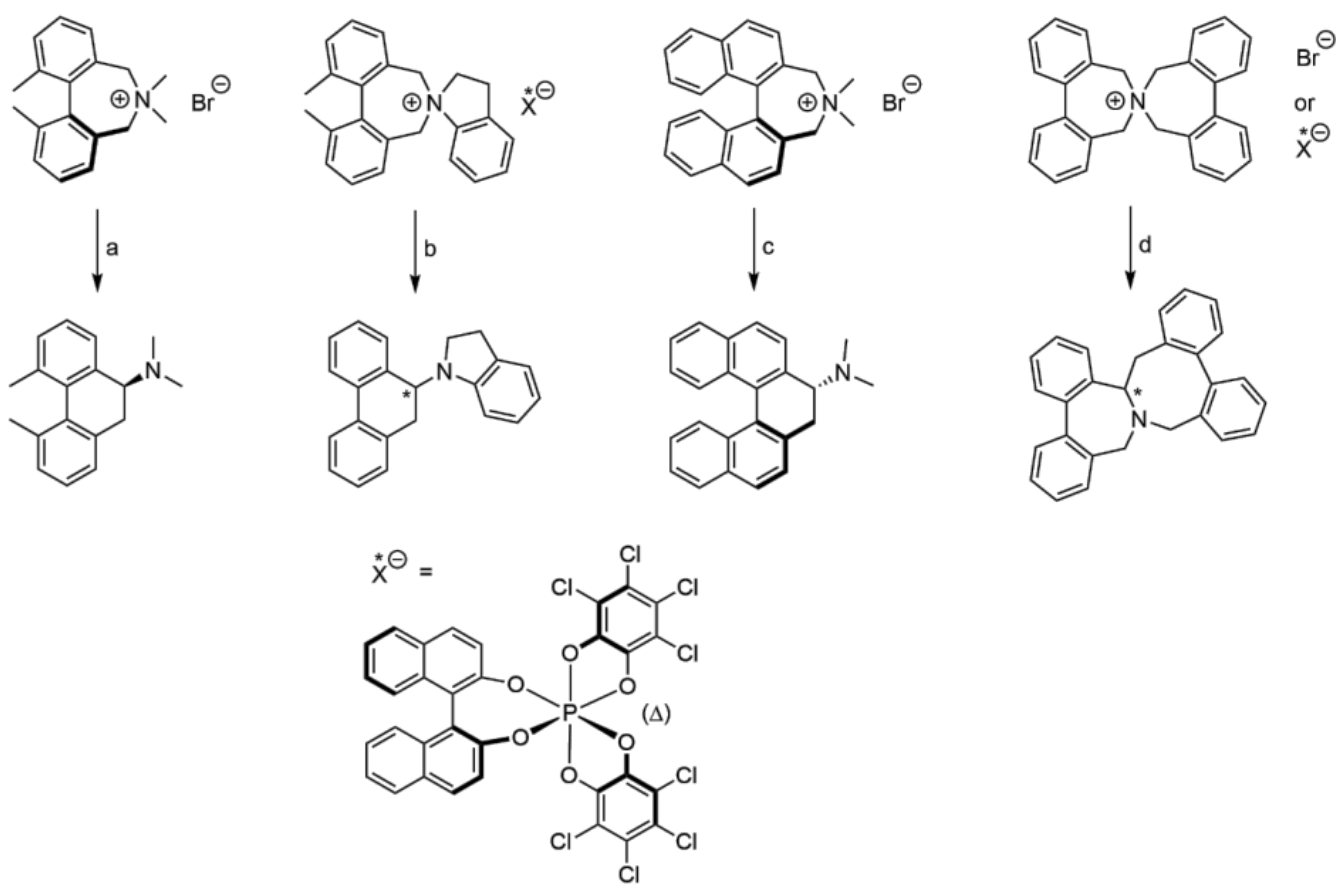
3.1. Preparation of the Spiro Compounds
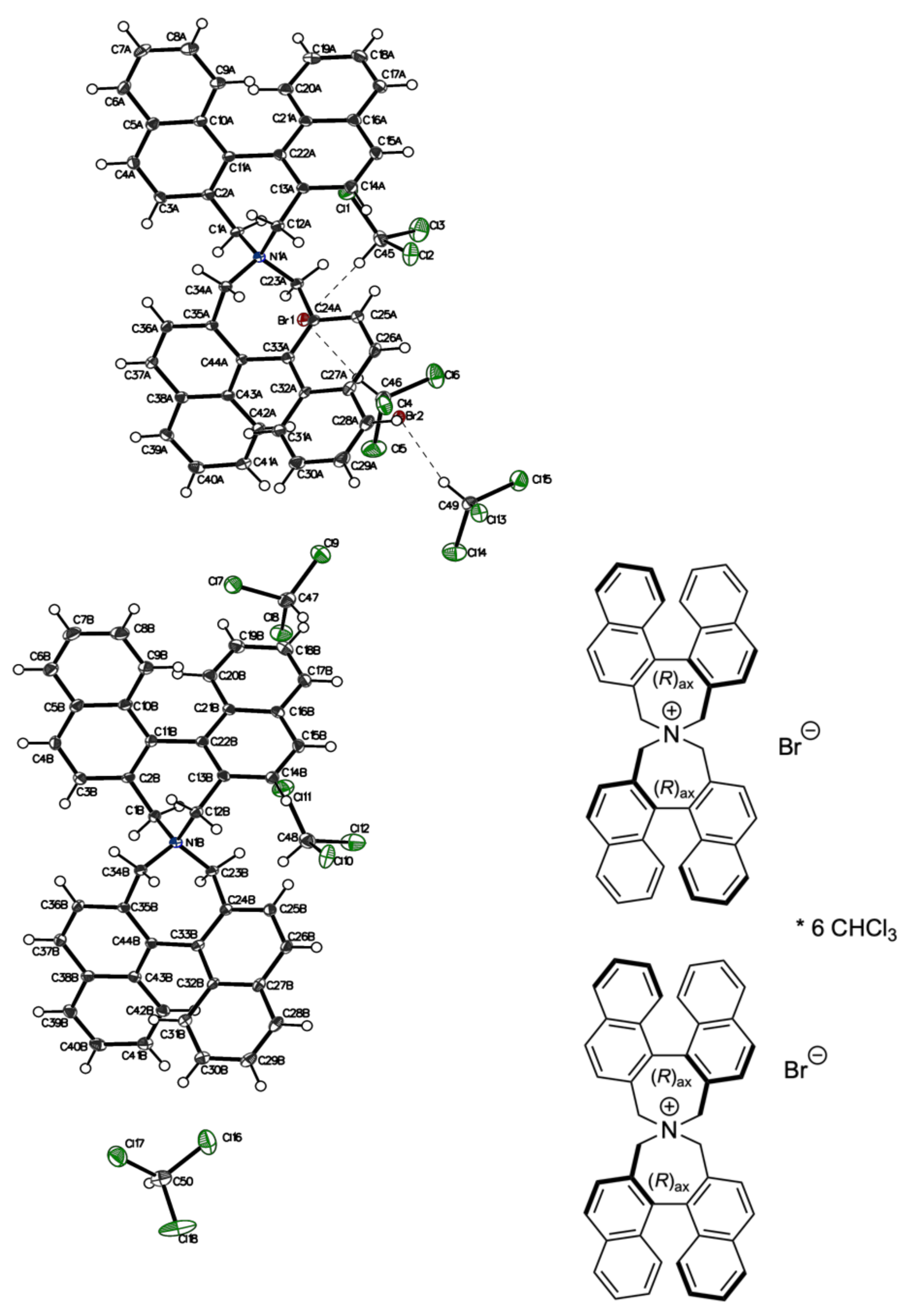
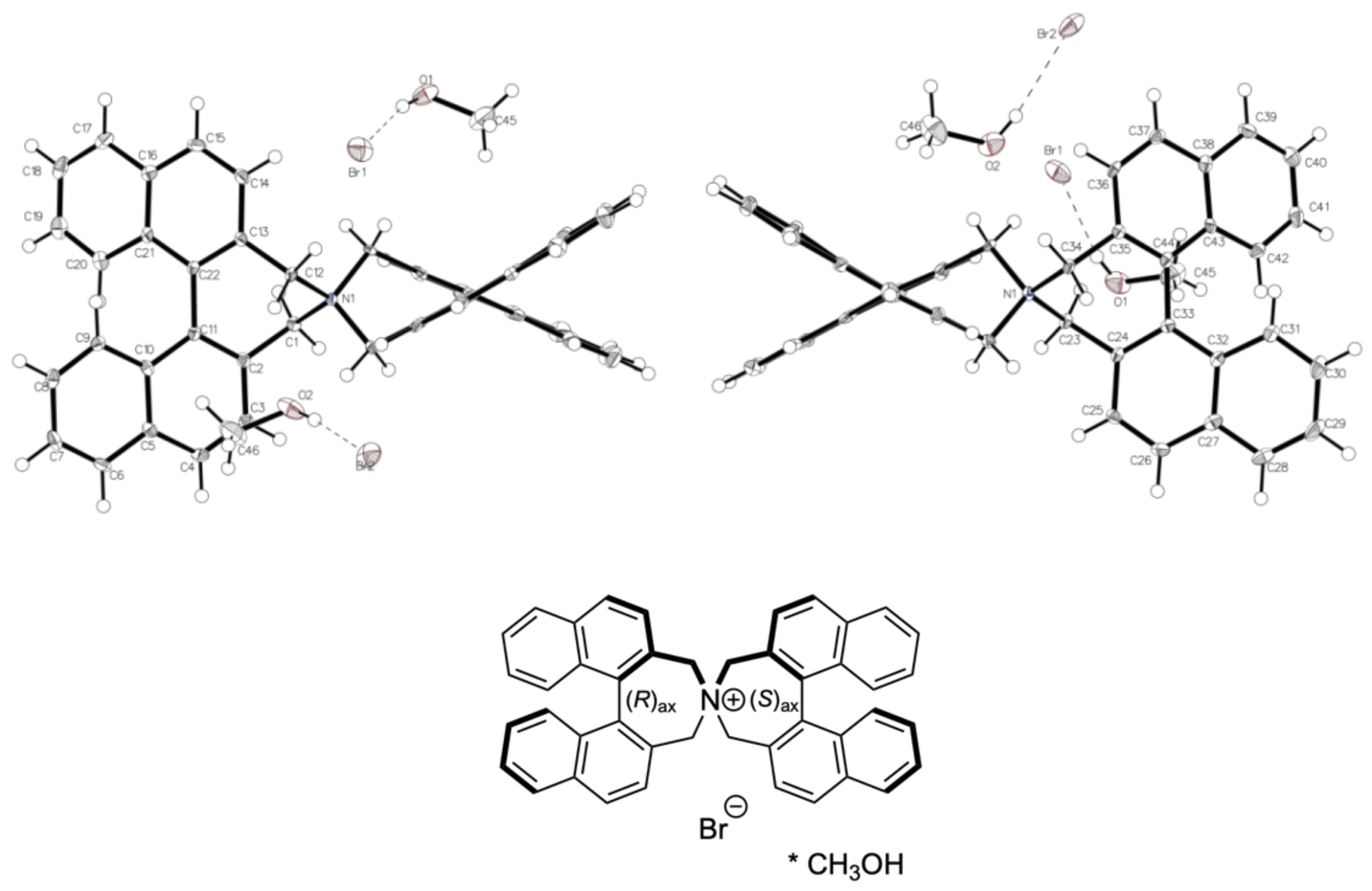
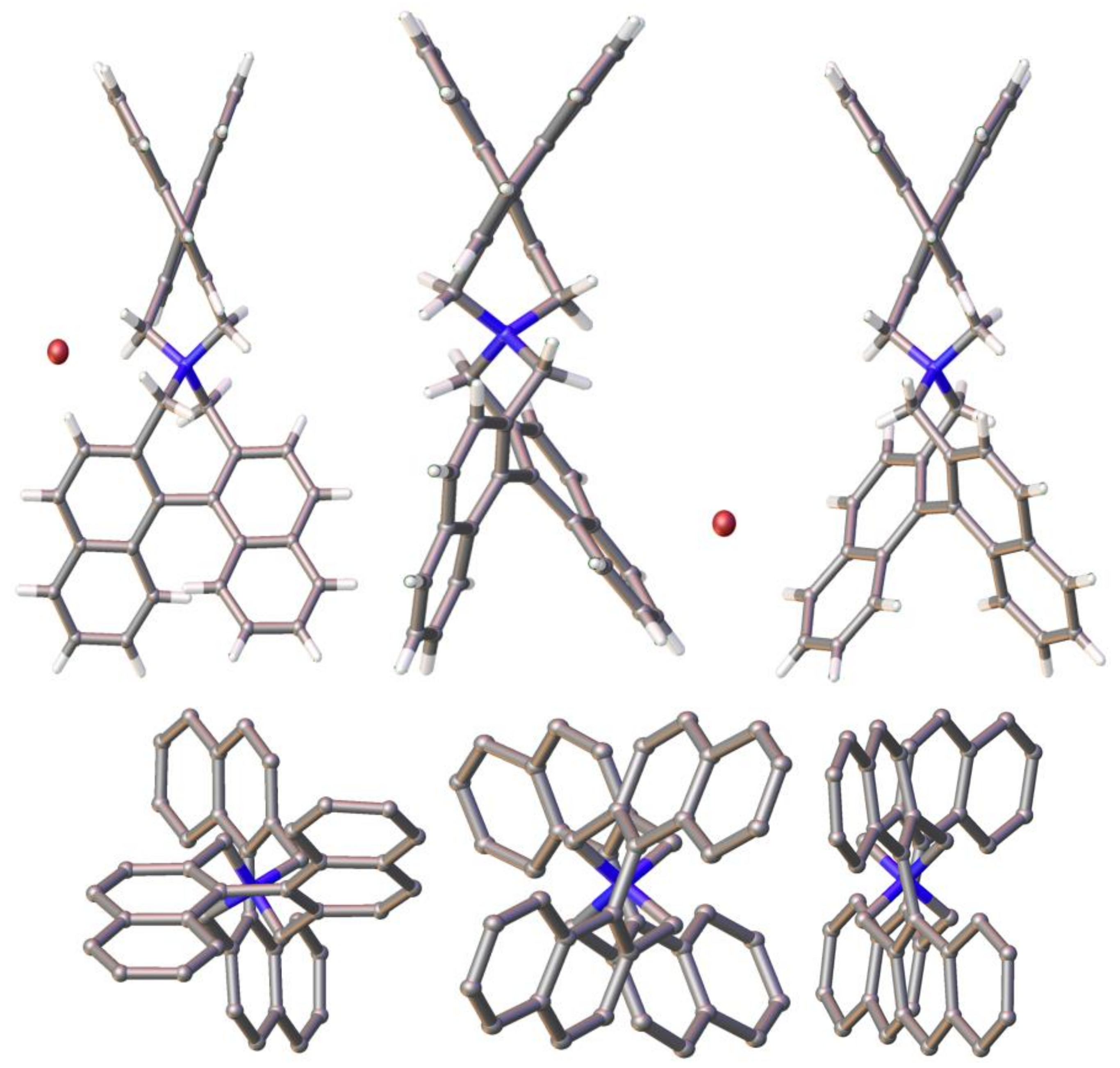
3.2. Comparison of Solid-State Structures of meso-2 and rac-2 (Racemic and Enantiomeric)
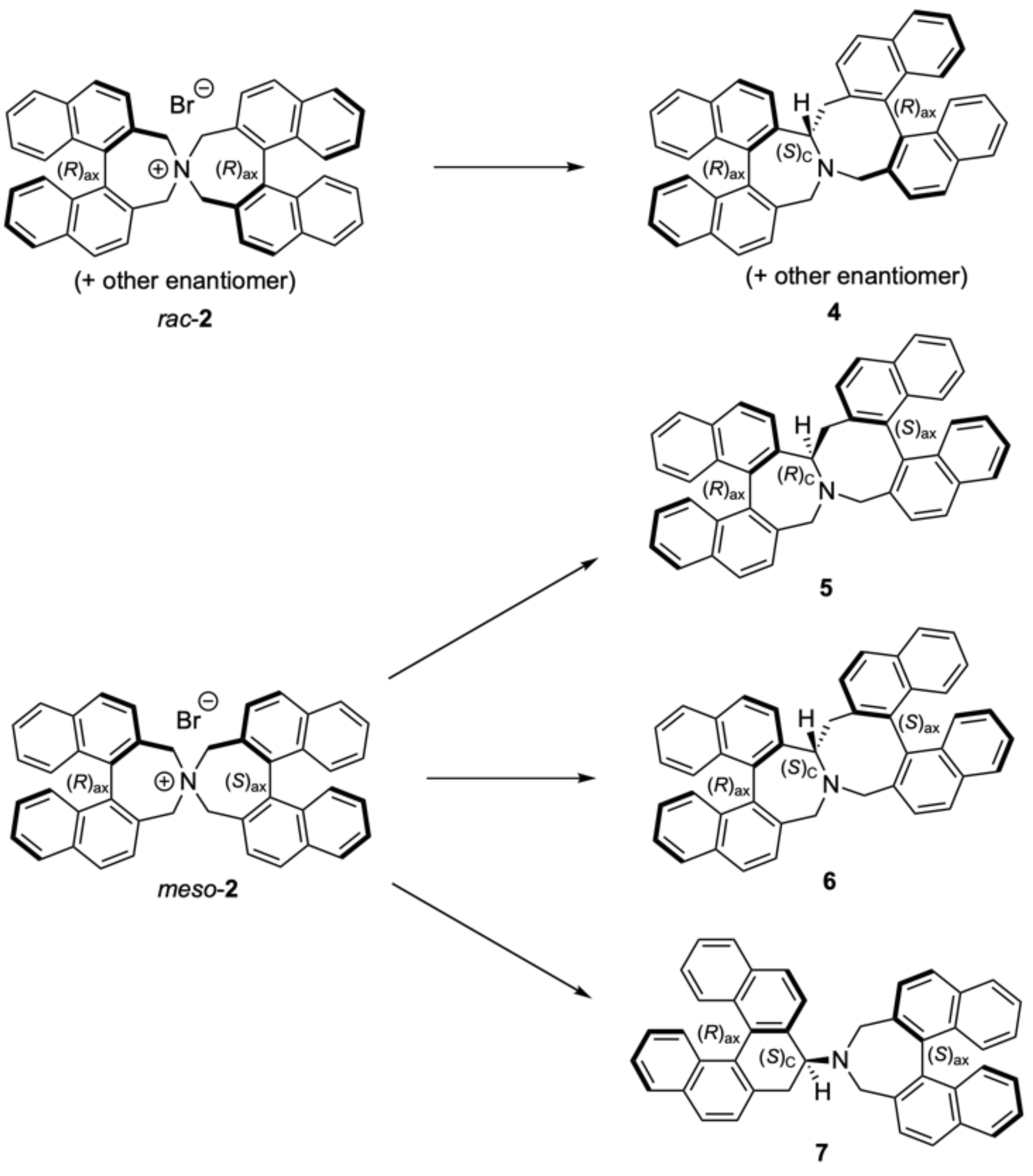
3.3. Attempted Separation of Enantiomers of 2
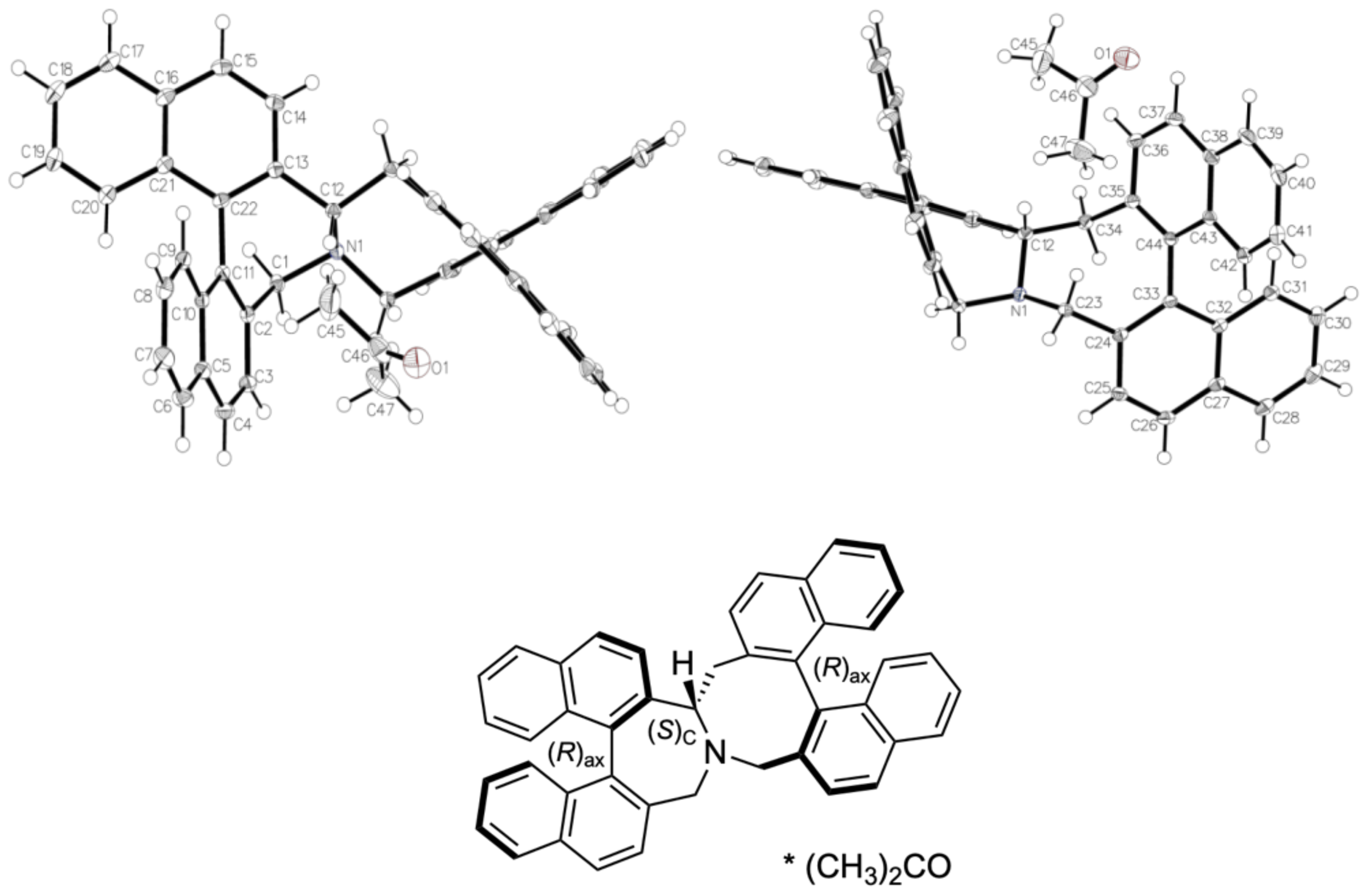
3.4. Stevens Rearrangement of 2
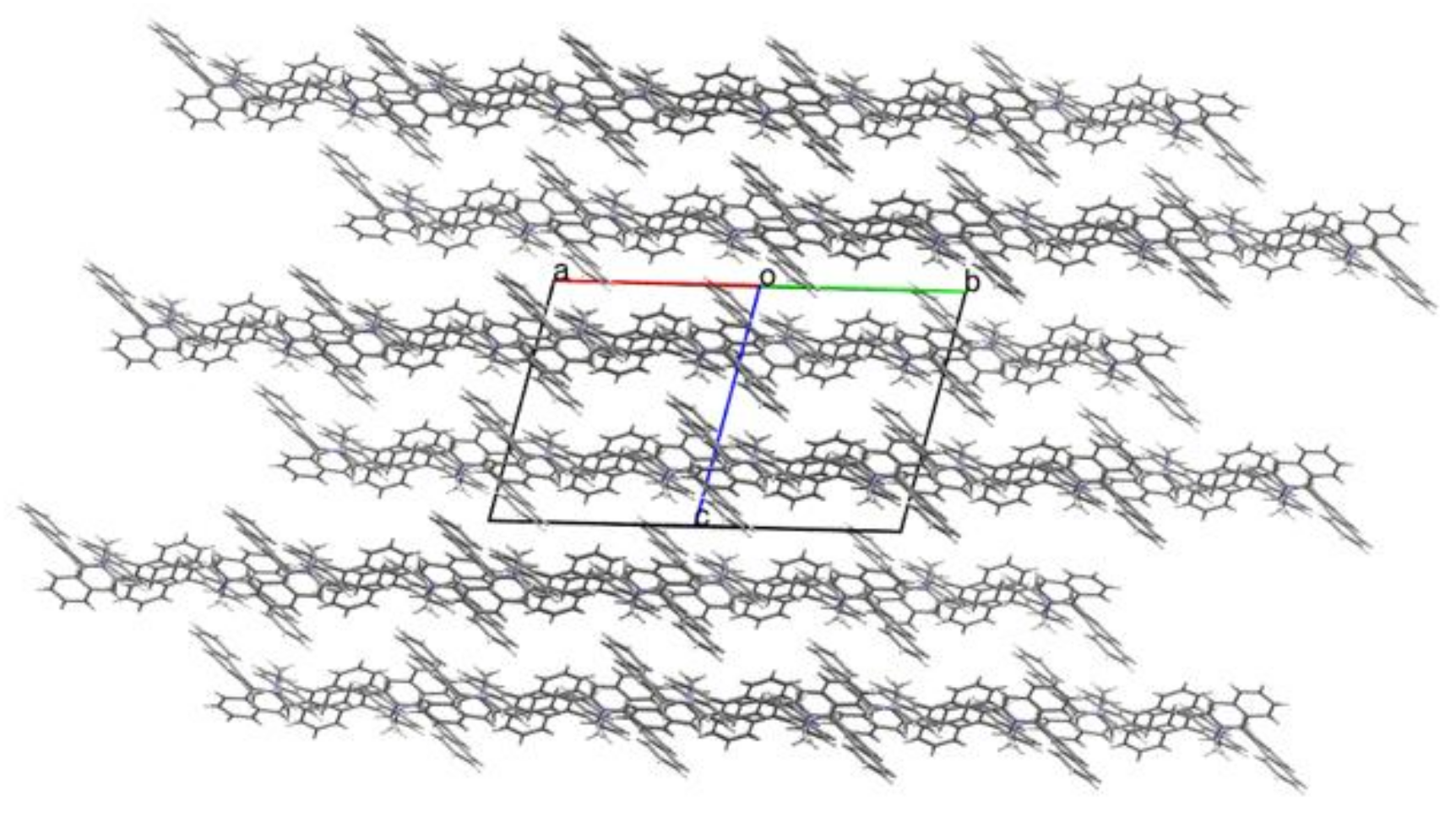
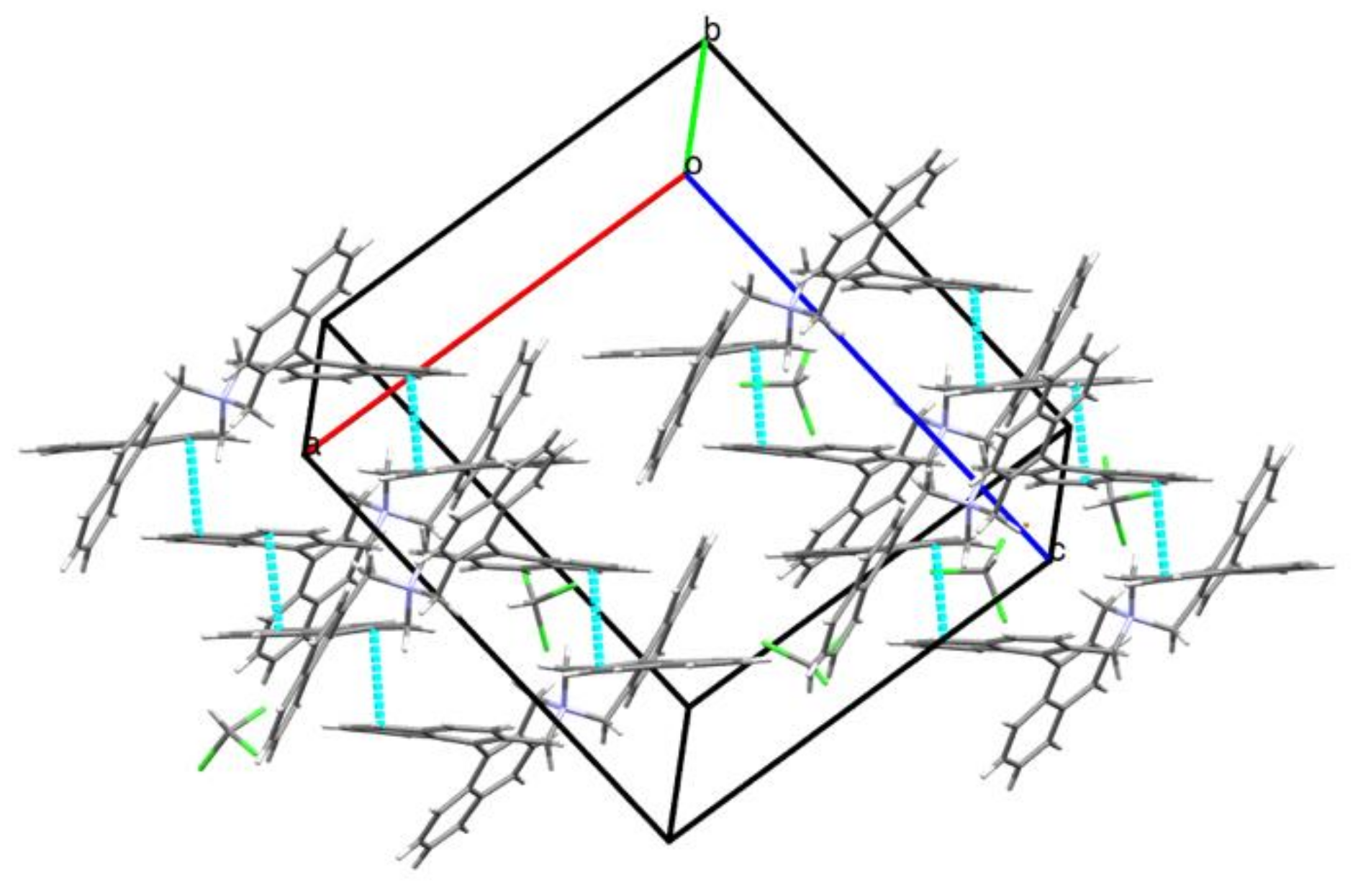
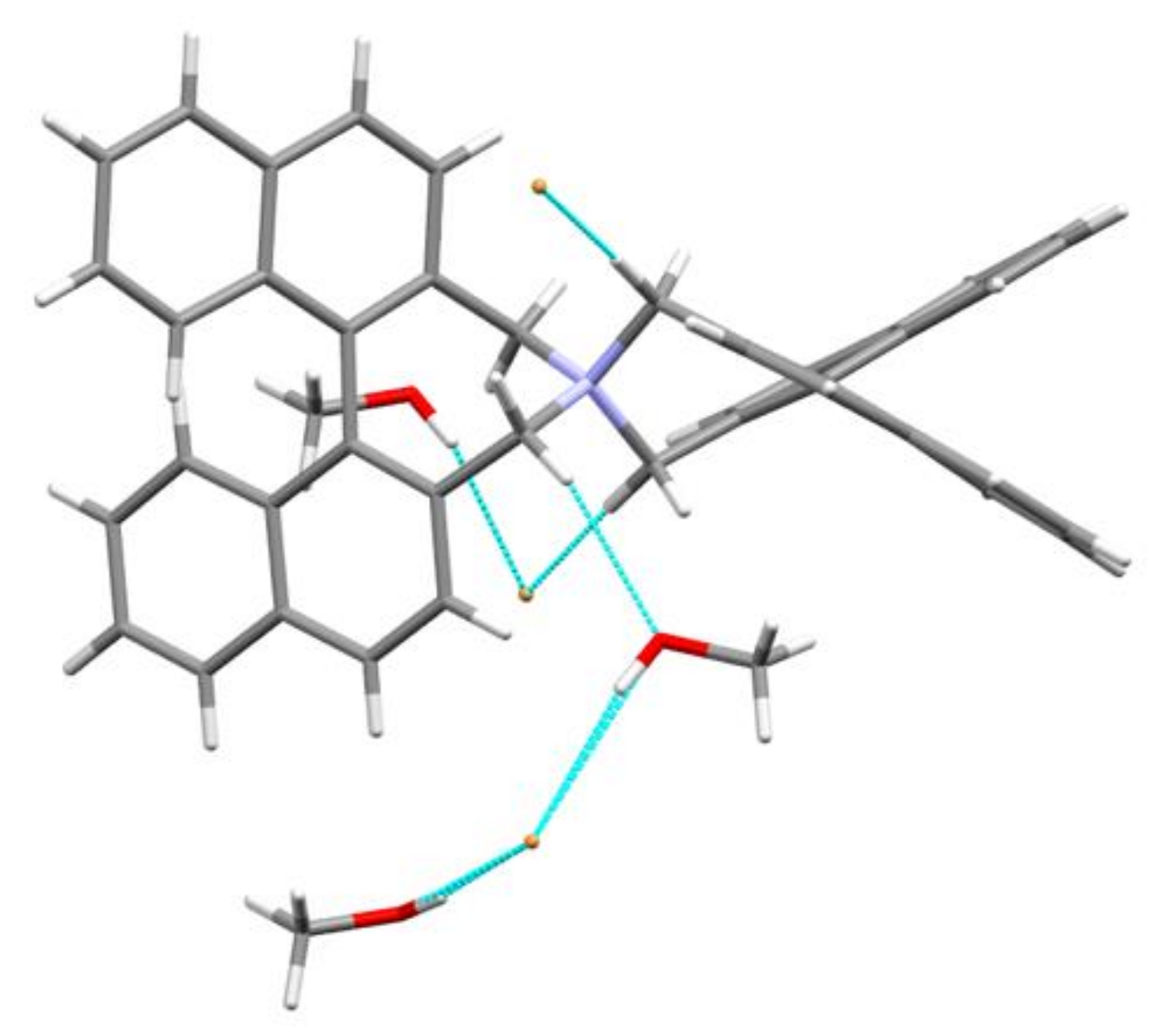
3.5. Intermolecular Structural Features
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shirakawa, S.; Maruoka, K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.; Maruoka, K. Recent Advances in Asymmetric Phase-Transfer Catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4266. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.; Kameda, M.; Maruoka, K. Design of N-Spiro C2-Symmetric Chiral Quaternary Ammonium Bromides as Novel Chiral Phase-Transfer Catalysts: Synthesis and Application to Practical Asymmetric Synthesis of α-Amino Acids. J. Am. Chem. Soc. 2003, 125, 5139–5151. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.; Uematsu, Y.; Maruoka, K. New, Improved Procedure for the Synthesis of Structurally Diverse N-Spiro C2-Symmetric Chiral Quaternary Ammonium Bromides. J. Org. Chem. 2003, 68, 4576–4578. [Google Scholar] [CrossRef]
- Ooi, T.; Kameda, M.; Maruoka, K. Molecular Design of a C2-Symmetric Chiral Phase-Transfer Catalyst for Practical Asymmetric Synthesis of r-Amino Acids. J. Am. Chem. Soc. 1999, 121, 6519–6520. [Google Scholar] [CrossRef]
- Ooi, T.; Uematsu, Y.; Kameda, M.; Maruoka, K. Conformationally Flexible, Chiral Quaternary Ammonium Bromides for Asymmetric Phase-Transfer Catalysis. Angew. Chem. Int. Ed. 2002, 41, 1551–1554. [Google Scholar] [CrossRef]
- Kitamura, M.; Shirakawa, S.; Maruoka, K. Powerful Chiral Phase-Transfer Catalysts for the Asymmetric Synthesis of α-Alkyl- and α,α-Dialkyl-α-amino Acids. Angew. Chem. Int. Ed. 2005, 44, 1549–1551. [Google Scholar] [CrossRef]
- Ooi, T.; Takeuchi, M.; Kameda, M.; Maruoka, K. Practical Catalytic Enantioselective Synthesis of α,α-Dialkyl-α-amino Acids by Chiral Phase-Transfer Catalysis. J. Am. Chem. Soc. 2000, 122, 5228–5229. [Google Scholar] [CrossRef]
- Hawkins, J.M.; Fu, G.C. Asymmetric Michael Reactions of 3, 5-Dihydro-4H-dinaphth [2, 1-c: 1′, 2′-e] azepine with Methyl Crotonate. J. Org. Chem. 1986, 51, 2820–2822. [Google Scholar] [CrossRef]
- Maigrot, N.; Mazeleyrat, J.P. New and Improved Synthesis of Optically Pure (R)-and (S)-2,2′-Dimethyl-1, 1′-binaphthyl and Related Compounds. Synthesis 1985, 3, 317–320. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Bruker SAINT, version 8.38B; Bruker AXS: Billerica, MA, USA, 2019.
- Sheldrick, G.M. SADABS; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Huebschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXS; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXL; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Kano, T.; Lan, Q.; Wang, X.; Maruoka, K. Effects of Aromatic Substituents on Binaphthyl-Based Chiral Spiro-Type Ammonium Salts in Asymmetric Phase-Transfer Reactions. Adv. Synth. Catal. 2007, 349, 556–560. [Google Scholar] [CrossRef]
- Hashimoto, T.; Tanaka, Y.; Maruoka, K. Symmetrical 4,4′,6,6′-tetraarylbinaphthyl-substituted ammonium bromide as a new, chiral phase-transfer catalyst. Tetrahedron Asym. 2003, 14, 1599–1602. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Suzuki, T. Tetrabenzo[8]circulene: Aromatic Saddles from Negatively Curved Graphene. J. Am. Chem. Soc. 2013, 135, 14074–14077. [Google Scholar] [CrossRef]
- Lutz, M.R.; Zeller, M.; Sarsah, S.R.S.; Filipowicz, A.; Wouters, H.; Becker, D.P. Synthesis, crystal structure and rearrangements of orthocyclophane cyclotetraveratrylene (CTTV) tetraketone. Supramol. Chem. 2012, 24, 803–809. [Google Scholar] [CrossRef]
- Ahn, H.C.; Yun, S.; Choi, K. A proline-based macrocyclic amide with S4 symmetry. Chem. Lett. 2008, 37, 10–11. [Google Scholar] [CrossRef]
- Coles, S.J.; Davies, D.B.; Eaton, R.J.; Hursthouse, M.B.; Kilic, A.; Shaw, R.A.; Uslu, A. Stereogenic properties of spiranes combined with four equivalent conventional centers of chirality. Dalton Trans. 2007, 20, 2040–2047. [Google Scholar] [CrossRef]
- Kozhushkov, S.I.; Kostikov, R.R.; Molchanov, A.P.; Boese, R.; Benet-Buchholz, J.; Schreiner, P.R.; Rinderspacher, C.; Ghiviriga, I.; De Meijere, A. Tetracyclopropylmethane: A unique hydrocarbon with S4 symmetry. Angew Chem. Int. Ed. 2001, 40, 180–183. [Google Scholar] [CrossRef]
- Mislow, K. Nonactin and the coupe du roi. Croatica Chem. Acta 1985, 58, 353–358. [Google Scholar]
- Murray-Rust, P.; Riddel, F.G. The Stability and Conformation of the 1,3,6,8-Tetraazatricyclo[4.4.1.13,8]dodecane System: The Structure of the Condensation Product of 1,2-Diaminocyclohexane and Formaldehyde. Can. J. Chem. 1975, 53, 1933–1935. [Google Scholar] [CrossRef] [Green Version]
- Stará, I.G.; Starý, I.; Závada, J. Nucleophilic Cleavage of 4,5-Dihydro-3H-dinaphth[2,1-c:1′,2′-e]azepinium Quaternary Salts. A Convenient Approach to New Axially Dissymmetric and Axially Asymmetric Ligands. J. Org. Chem. 1992, 57, 6966–6969. [Google Scholar] [CrossRef]
- Stará, I.G.; Starý, I.; Tichý, M.; Závada, J.; Hanus, V. Stereochemical Dichotomy in the Stevens Rearrangement of Axially Twisted Dihydroazepinium and Dihydrothiepinium Salts. A Novel Enantioselective Synthesis of Pentahelicene. J. Am. Chem. Soc. 1994, 116, 5084–5088. [Google Scholar] [CrossRef]
- Wittig, G.; König, G.; Clauss, K. 1,2,3,4,7,8,9,10-Tetrabenzo-cyclododecahexaen. Liebigs Ann. Chem. 1955, 593, 127. [Google Scholar] [CrossRef]
- Vial, L.; Lacour, J. Conformational Preference and Configurational Control of Highly Symmetric Spiro[dibenzazepinium] Cation. Org. Lett. 2002, 4, 3939–3942. [Google Scholar] [CrossRef]
- Vial, L.; Gonçalves, M.-H.; Morgantini, P.-Y.; Weber, J.; Bernardinelli, G.; Lacour, J. Unusual Regio- and Enantioselective [1,2]-Stevens Rearrangement of a Spirobi[dibenzazepinium] Cation. Synlett 2004, 9, 1565–1568. [Google Scholar] [CrossRef]
- Gonçalves-Farbos, M.-H.; Vial, L.; Lacour, J. Enantioselective [1,2]-Stevens rearrangement of quaternary ammonium salts. A mechanistic evaluation. Chem. Commun. 2008, 7, 829–831. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rac-2 | Meso-2 | 4 | |
|---|---|---|---|
| M (g/mol) | 1012.72 | 718.70 | 631.77 |
| Space group | P-1 | C2/c | P-1 |
| a (Å) | 15.9737 (13) | 30.408 (2) | 11.5087 (4) |
| b (Å) | 17.9202 (13) | 14.8246 (12) | 11.5335 (4) |
| c (Å) | 18.7489 (19) | 16.0429 (11) | 13.5262 (5) |
| α (°) | 117.544 (2) | 90 | 75.6052 (14) |
| β (°) | 93.715 (4) | 103.653 (2) | 76.2673 (14) |
| γ (°) | 108.646 (2) | 90 | 77.4586 (15) |
| V (Å3) | 4362.0 (7) | 7027.6 (9) | 1665.30 (10) |
| Z | 4 | 8 | 2 |
| Rint | 0.073 | 0.064 | 0.036 |
| R1 (I > 2σ(I)) | 0.057 | 0.055 | 0.044 |
| wR2 (all data) | 0.191 | 0.151 | 0.119 |
 | (R,S)ax-2 (meso) | (R,R)ax-2 [3] | (R,R)ax/(S,S)ax-2 (rac) |
|---|---|---|---|
| Torsion angle C2-C11-C22-C13 [°] | 51.7(4) | 50.7 | 50.6(5) |
| Torsion angle C24-C33-C44-C35 [°] | 48.9(4) | 55.5 | 49.0(5) |
| Bond angle C1-N1-C12 [°] | 109.8(2) | 111.3 | 110.9(3) |
| Bond angle C23-N1-C34 [°] | 109.4(2) | 109.9 | 110.6(3) |
| Bond angle C1-N1-C34 [°] | 109.5(2) | 108.9 | 112.1(3) |
| Bond angle C12-N1-C23 [°] | 109.0(2) | 108.9 | 112.2(3) |
| Bond angle C2-C1-N1 [°] | 112.0(3) | 110.4 | 113.8(3) |
| Bond angle C13-C12-N1 [°] | 112.1(3) | 110.4 | 112.6(3) |
| Bond angle C24-C23-N1 [°] | 111.7(3) | 110.9 | 113.5(3) |
| Bond angle C35-C34-N1 [°] | 111.6(3) | 110.9 | 112.9(3) |
| Distance C1-N1 [Å] | 1.536(4) | 1.522 | 1.525(3) |
| Distance C12-N1 [Å] | 1.534(5) | 1.522 | 1.531(4) |
| Distance C23-N1 [Å] | 1.531(4) | 1.540 | 1.526(5) |
| Distance C34-N1 [Å] | 1.527(4) | 1.540 | 1.521(5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honegger, P.; Gajic, N.; Prado-Roller, A.; Widhalm, M. Racemic and Meso Crystal Structures of an Axial-Chiral Spirobi-(dinaphthoazepin)ium Salt: Emergence of an S4-Symmetric Molecule. Symmetry 2021, 13, 1365. https://0-doi-org.brum.beds.ac.uk/10.3390/sym13081365
Honegger P, Gajic N, Prado-Roller A, Widhalm M. Racemic and Meso Crystal Structures of an Axial-Chiral Spirobi-(dinaphthoazepin)ium Salt: Emergence of an S4-Symmetric Molecule. Symmetry. 2021; 13(8):1365. https://0-doi-org.brum.beds.ac.uk/10.3390/sym13081365
Chicago/Turabian StyleHonegger, Philipp, Natalie Gajic, Alexander Prado-Roller, and Michael Widhalm. 2021. "Racemic and Meso Crystal Structures of an Axial-Chiral Spirobi-(dinaphthoazepin)ium Salt: Emergence of an S4-Symmetric Molecule" Symmetry 13, no. 8: 1365. https://0-doi-org.brum.beds.ac.uk/10.3390/sym13081365