1. Introduction
Advanced (001) surfaces and interfaces in the ReO
3, WO
3 complex oxides as well as BaTiO
3, SrTiO
3 and BaZrO
3 perovskites are of paramount importance due to the numerous technological applications and great potential for fundamental research caused by their phase transitions [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10]. Over the course of the last 25 years, the (001) surfaces of ReO
3 and WO
3, as well as BaTiO
3, SrTiO
3 and BaZrO
3 perovskites, have been broadly explored worldwide both from the theory and experimental sides [
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26]. All our ab initio computed BaTiO
3, SrTiO
3 and BaZrO
3 complex oxides belong to the commonly named ABO
3 perovskites. In our case, A is equal to Ba or Sr, whereas B denotes the Ti or Zr atoms [
27]. Our computed materials ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 have a huge number of applications in new and emerging technologies. For example, the novel trioxo-rhenium complex [ReO
3(phen)(H
2PO
4)]·H
2O has important antibacterial properties [
28]. Tungsten oxide nanodots (WO
3−x) exhibit remarkable antibacterial capabilities [
29]. Tungsten oxide and graphene oxide (WO
3-GO) nanocomposite is an excellent antibacterial as well as an anticancer agent [
30]. BaTiO
3 may be used as an electrical insulator and a piezoelectric substance in different kinds of microphones as well as other transducers [
31]. SrTiO
3 is an outstanding photocatalyst for the extremely important water splitting process [
32,
33]. BaZrO
3-based ceramic substances are widely used in protonic fuel cell applications [
34,
35,
36,
37,
38] as well as in hydrogen separation membranes [
39,
40].
Along with its great technological potential [
31,
41], BaTiO
3 is also a marvellous material for fundamental research since it exhibits several phase transitions [
42]. Namely, BaTiO
3 exists as one of four polymorphs [
43,
44] as a function of temperature. As the temperature lowers from high temperature to low temperature [
43,
44], the crystal symmetries of these four BaTiO
3 polymorphs are the cubic BaTiO
3 phase, tetragonal BaTiO
3 phase, orthorhombic BaTiO
3 phase and, finally, the rhombohedral BaTiO
3 phase [
43,
44]. Three of these four BaTiO
3 phases, tetragonal, orthorhombic and rhombohedral [
43,
44], display the ferroelectric effect [
43,
44]. It is worth noting that recently, the ab initio computations of the temperature effects on the structural, energetic, electronic as well as vibrational properties of four BaTiO
3 polymorphs by means of the quasi-harmonic approximations were carried out by Oliveira et al. [
45]. The ab initio study of the stability between these four BaTiO
3 phases, performed by Oliveira et al. [
45], breaks out into several contributions arising from the vibration of the lattice, electronic structure as well as volume expansion/contraction. This novel study by Oliveira et al. [
45] was helpful in order to confirm the sequence of the BaTiO
3 phase transitions as cubic → tetragonal → orthorhombic → rhombohedral [
45] and also its transition temperatures. In contrast to BaTiO
3, the SrTiO
3 and BaTiO
3 perovskites are so-called incipient ferroelectrics, and they exist only in their high symmetry cubic structure [
46,
47].
In our ab initio computations, we employed the standard cubic unit cells of BaTiO
3, SrTiO
3 and BaZrO
3 crystals containing five atoms [
46,
47]. The A-type ABO
3 perovskite atom in the cubic structure was positioned at the corner of the cube position. The ABO
3 perovskite A atom had the following fractional coordinates (0, 0, 0). The B-type ABO
3 perovskite atom in the cubic structure was positioned at the cube body center position. The B atom had the following fractional coordinates (½, ½, ½). Lastly, at the ABO
3 perovskite, cubic phase face center positions were filled with three cubic ABO
3 perovskite O atoms. The three ABO
3 perovskite O atoms had the following fractional coordinates (½, ½, 0), (½, 0, ½) and (0, ½, ½) [
48,
49,
50,
51]. All three of our ab initio computed cubic ABO
3 perovskites (BaTiO
3, SrTiO
3 and BaZrO
3) had the
space group with the space group number 221. Additionally, the ReO
3 and WO
3 crystals at their cubic symmetry structure had exactly the same space group
with the same space group number 221. The only paramount difference between the BaTiO
3, SrTiO
3 and BaZrO
3 ABO
3 perovskites as well as ReO
3 and WO
3 crystals, which had exactly the same cubic symmetry structure, the same space group
and even the same space group number 221, was missing an A-type atom in the ReO
3 and WO
3 materials.
To the best of our knowledge, only a few ab initio computations up to now exist in the world of science, dealing with the ReO
2 or WO
2-terminated polar ReO
3 and WO
3 (001) surfaces [
52,
53,
54]. It is worth noting that up to now there have been no ab initio computations performed in the world dealing with O-terminated polar ReO
3 or WO
3 (001) surfaces. For our ab initio computations, relevant experimental data [
46,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65] dealing with ReO
3, WO
3, BaTiO
3, SrTiO
3 and PbTiO
3 bulk crystals are collected in
Table 1.
The perfect cubic structure for the ReO
3 conducting oxide [
55] is stable at all temperature ranges starting from room temperature. The crystal structure of tungsten trioxide (WO
3) [
56] depends on the temperature. WO
3 has tetragonal symmetry if the temperature is above 740 °C. WO
3 is orthorhombic [
56] at the temperature range from 330 °C to 740 °C. WO
3 is monoclinic [
56] at the temperature range from 17 °C to 330 °C. Finally, WO
3 is triclinic [
56] at the temperature range from −50 °C to 17 °C. It is worth noting that the most common structure for WO
3 is monoclinic. The space group for the WO
3 monoclinic structure is P2
1/n. The experimentally measured WO
3 (Γ-Γ) bandgap is equal to 3.74 eV [
57]. In the BaTiO
3 perovskite matrix, according to the experimental measurements performed by Wemple, the room temperature (RT) bandgaps are equal to 3.38 eV and 3.27 eV [
58] for the light polarized parallel and perpendicular to the ferroelectric axis
c. The experimental SrTiO
3 Γ-Γ bandgap, according to measurements by Benthem et al. [
59], is equal to 3.75 eV (
Table 1). Finally, the experimental BaZrO
3 Γ-Γ bandgap, according to experiments performed by Robertson, is equal to 5.3 eV [
60] (
Table 1). ReO
3 is cubic at all temperatures, starting from liquid helium temperature to 673 K. The experimentally measured ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 bulk lattice constants in cubic crystal structures are listed by us in
Table 1. The objective of our contribution was to carry out the first ab initio computations for polar O-terminated ReO
3 and WO
3 (001) surfaces. We compared our ab initio computation results for polar ReO
3 and WO
3 as well as neutral BaTiO
3, SrTiO
3 and BaZrO
3 perovskite (001) surfaces and pointed out systematic tendencies in our performed computations in a way that is comfortably approachable for a broad audience of readers worldwide.
2. Computation Methods and Materials
We performed our forefront ab initio computations for the ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 bulk and their (001) surfaces by means of the hybrid B3PW [
66,
67] or B3LYP [
68] exchange-correlation functionals. Both these B3PW [
66,
67] as well as B3LYP [
68] hybrid exchange-correlation functionals are implemented into the very famous, world-class computational package CRYSTAL [
69], developed by Torino University, Italy. The computational package CRYSTAL [
69] utilizes 2-D isolated slab representation for the (001) surface structure first principles computations. We performed the reciprocal space integration in our first principles computations for ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 matrixes. Namely, we integrated the Brillouin zone employing the 8 × 8 × 8 times expanded Pack–Monkhorst [
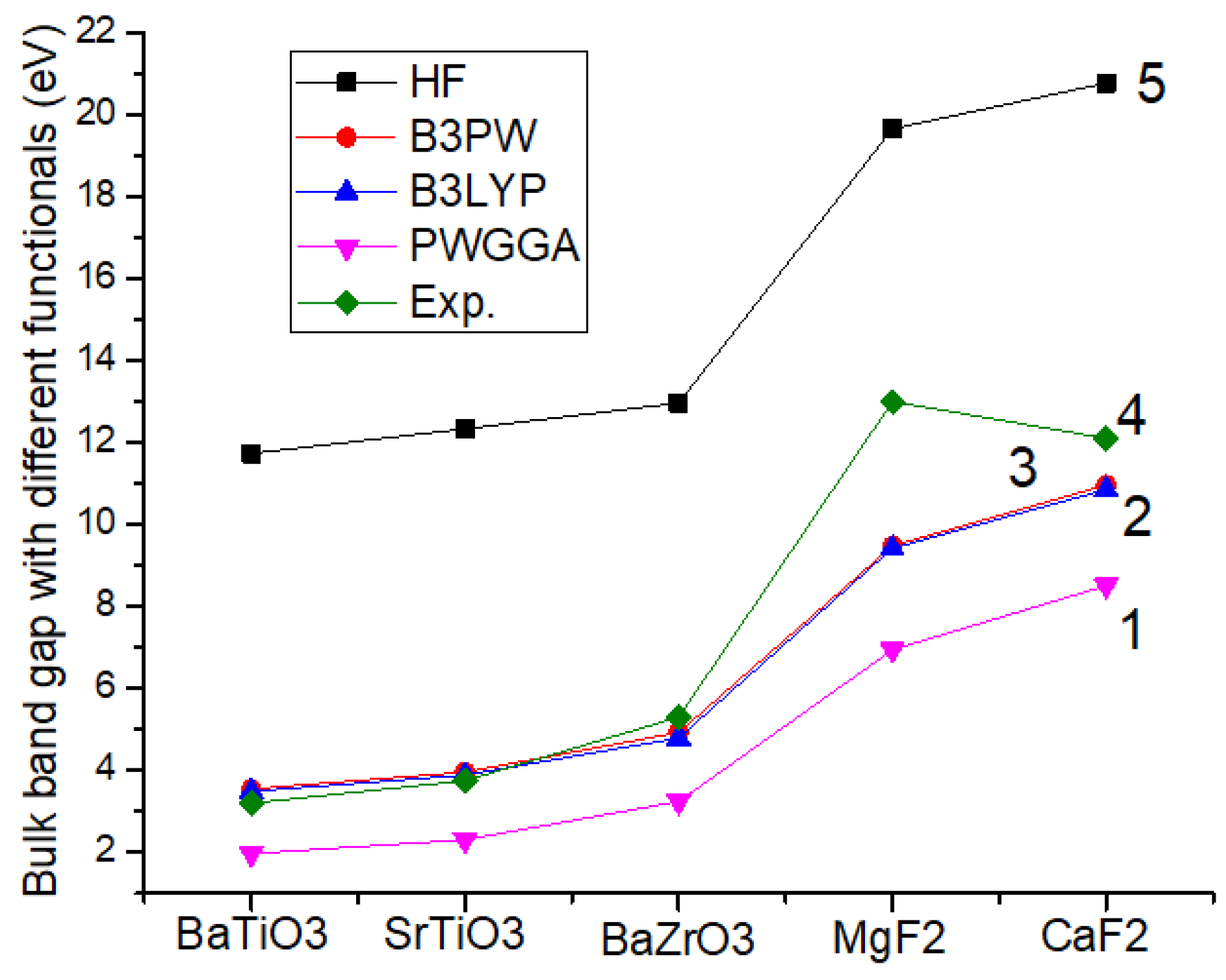
70] net for the bulk ab initio computations as well as 8 × 8 × 1 times enlargement for the (001) surface ab initio computations of these materials. In order to achieve the high accuracy of our computations, sufficiently large tolerances of 7, 8, 7, 7 and 14 were used by us for the Coulomb overlap, Coulomb penetration, exchange overlap, the first exchange pseudo-overlap as well as for the second exchange pseudo-overlap, respectively. With the goal of detecting the performance of various non-identical methods, we computed the bulk Γ-Γ bandgaps for ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 matrixes (
Table 2 and
Figure 1) and compared our ab initio computation results with the available experimental data [
14,
59,
60,
71,
72,
73,
74,
75,
76].
As it is possible to see from
Table 2 and
Figure 1, the Hartree–Fock (HF) [
77,
78] method always, for all computed materials, significantly overestimates the Γ-Γ bandgap. For example, our ab initio computed BaTiO
3 (Γ-Γ) bulk bandgap is overestimated by 3.67 times with respect to the experimental BaTiO
3 bulk Γ-Γ bandgap values (
Table 2). Additionally, our ab initio computed SrTiO
3 bulk Γ-Γ bandgap is overestimated by 3.29 times regarding the experimental SrTiO
3 bulk Γ-Γ bandgap value (
Table 2). In our density functional theory (DFT) computations, we used the local density approximation (LDA) with the Dirac–Slater exchange [
79] as well as the Vosko–Wilk–Nusair correlation [
80] energy functionals and a set of GGA exchange and correlation functionals as suggested by Perdew and Wang (PWGGA) [
66,
67]. On another side, the ab initio computed Γ-Γ bulk bandgaps for all five materials using the PWGGA are always considerably underestimated with respect to the experimental bulk Γ-Γ bandgap values (
Table 2 and
Figure 1). For example, our ab initio PWGGA computed MgF
2 bulk Γ-Γ bandgap value (6.94 eV) is 1.97 times underestimated with respect to the experimental MgF
2 Γ-Γ bulk bandgap value equal to (13.0 eV) [
76] (
Figure 1 and
Table 2). As we can see from
Table 2 and
Figure 1, the hybrid exchange-correlation functionals B3PW and B3LYP always allow us to achieve the best possible agreement between the ab initio computed as well as experimental bulk Γ-Γ bandgaps for all five of our first principles computed materials BaTiO
3, SrTiO
3, BaZrO
3, MgF
2 as well as CaF
2. The main reason for such a good agreement is that the hybrid B3LYP and B3PW functionals include a portion of exact exchange energy density from the HF theory (20%), while the rest of the exchange-correlation part is a mixture of various approaches (both exchange and correlation). This is the key reason why we performed all our future bulk as well as (001) surface ab initio computations by means of the B3PW or B3LYP hybrid exchange-correlation functionals (
Table 2 and
Figure 1).
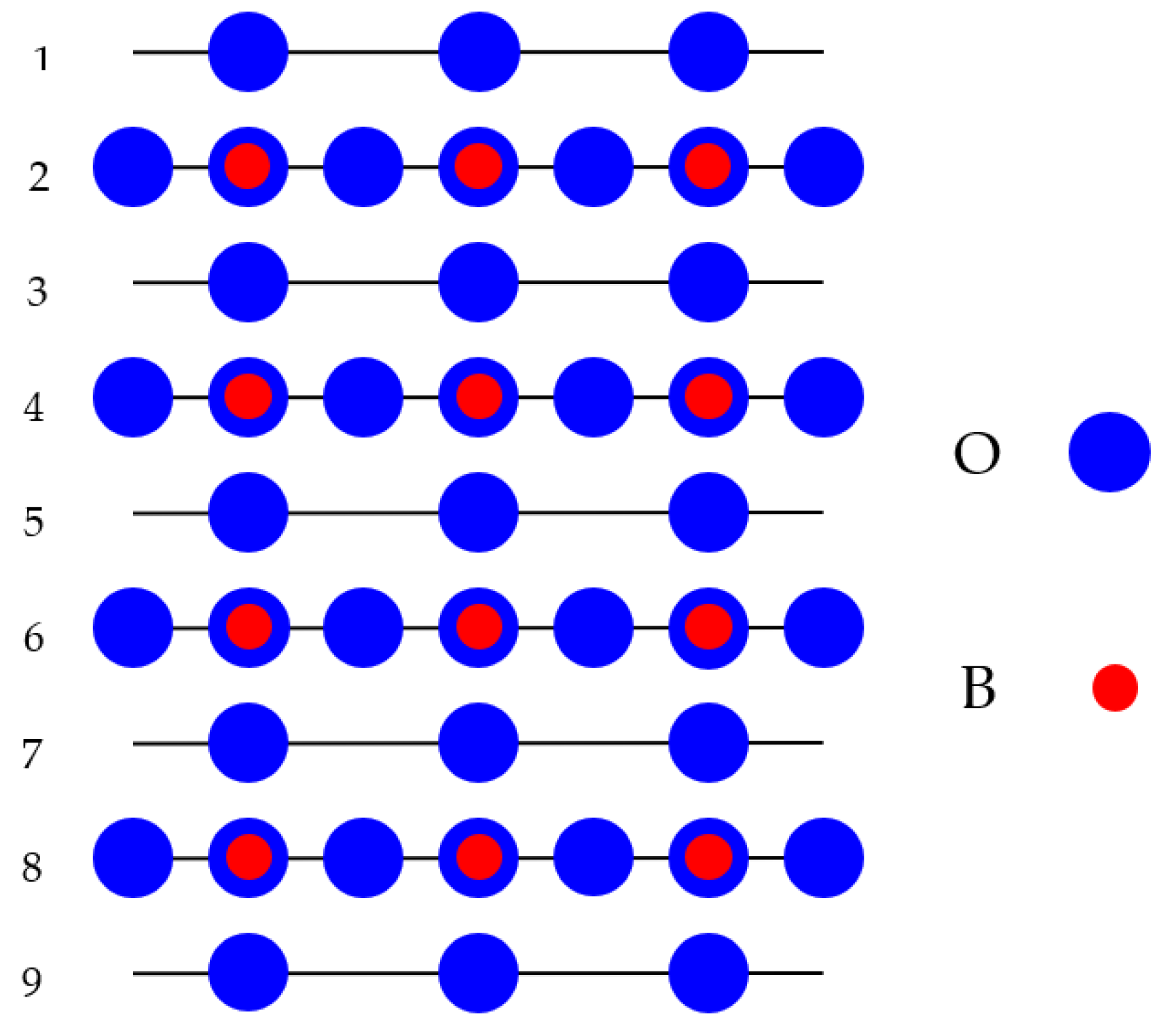
With an aim to ab initio compute the TiO
2-terminated BaTiO
3, SrTiO
3 and ZrO
2-terminated BaZrO
3 (001) surfaces, we selected nine-layer, mirror-symmetrical (001) slabs. They consisted of neutral and alternating TiO
2(ZrO
2) or AO layers (
Figure 2). These slabs were positioned perpendicular to the axis
z. Our generated nine-layer slab, used by us in ABO
3 perovskite (001) surface ab initio computations, was terminated from both sides by the TiO
2-terminated planes for BaTiO
3 and SrTiO
3 perovskites as well as by ZrO
2-terminated planes for BaZrO
3 perovskite (
Figure 2). Accordingly, our in ab initio computations employed the (001) surface model for the BO
2-terminated ABO
3 perovskites and the nine-layer slab consisted of a 23-atom supercell. Our ab initio computed ABO
3 perovskite BO
2-terminated (001) slab was non-stoichiometric (
Figure 2), and it had the following chemical equation A
4B
5O
14. With the objective to directly compare the properties of three perovskites (BaTiO
3, SrTiO
3 and BaZrO
3) as well as ReO
3 and WO
3 materials under the same conditions and, as much as possible, to reduce the computational time, we only investigated the high symmetry (
) cubic phases of these five materials. In our surface structure B3LYP and B3PW first principles computations, we allowed the atoms of the upper two or three surface layers to only relax along the
z-axis since the (001) surfaces of the perfect cubic crystals, due to symmetry restrictions, do not have any forces acting along the other
x- or
y-axes. We optimized the (001) surface atom atomic coordinated through the slab total energy minimization. For this purpose, we employed our own computer code, which implements [
81] conjugated gradients optimization technique with numerical computation of derivatives [
81].
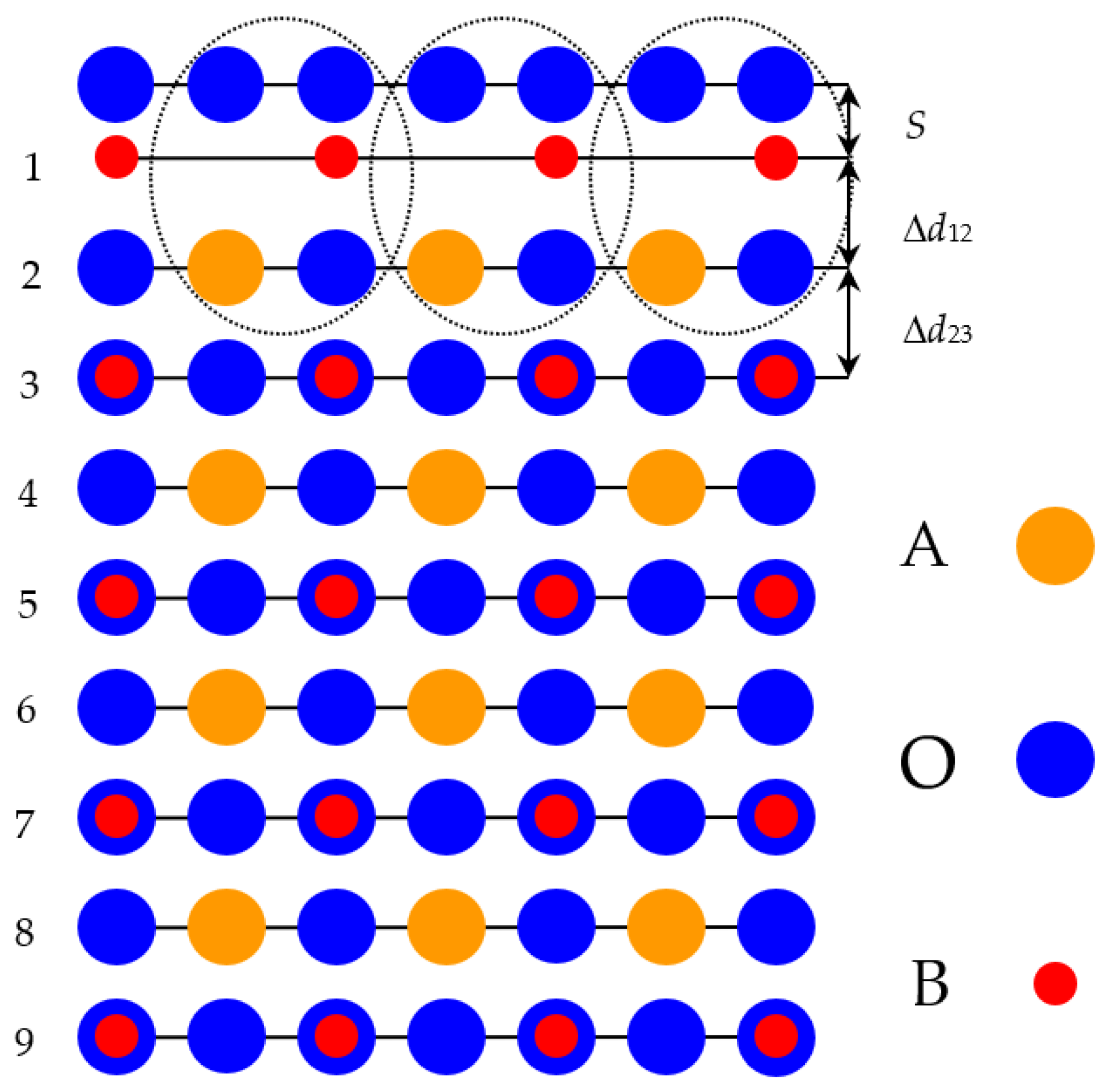
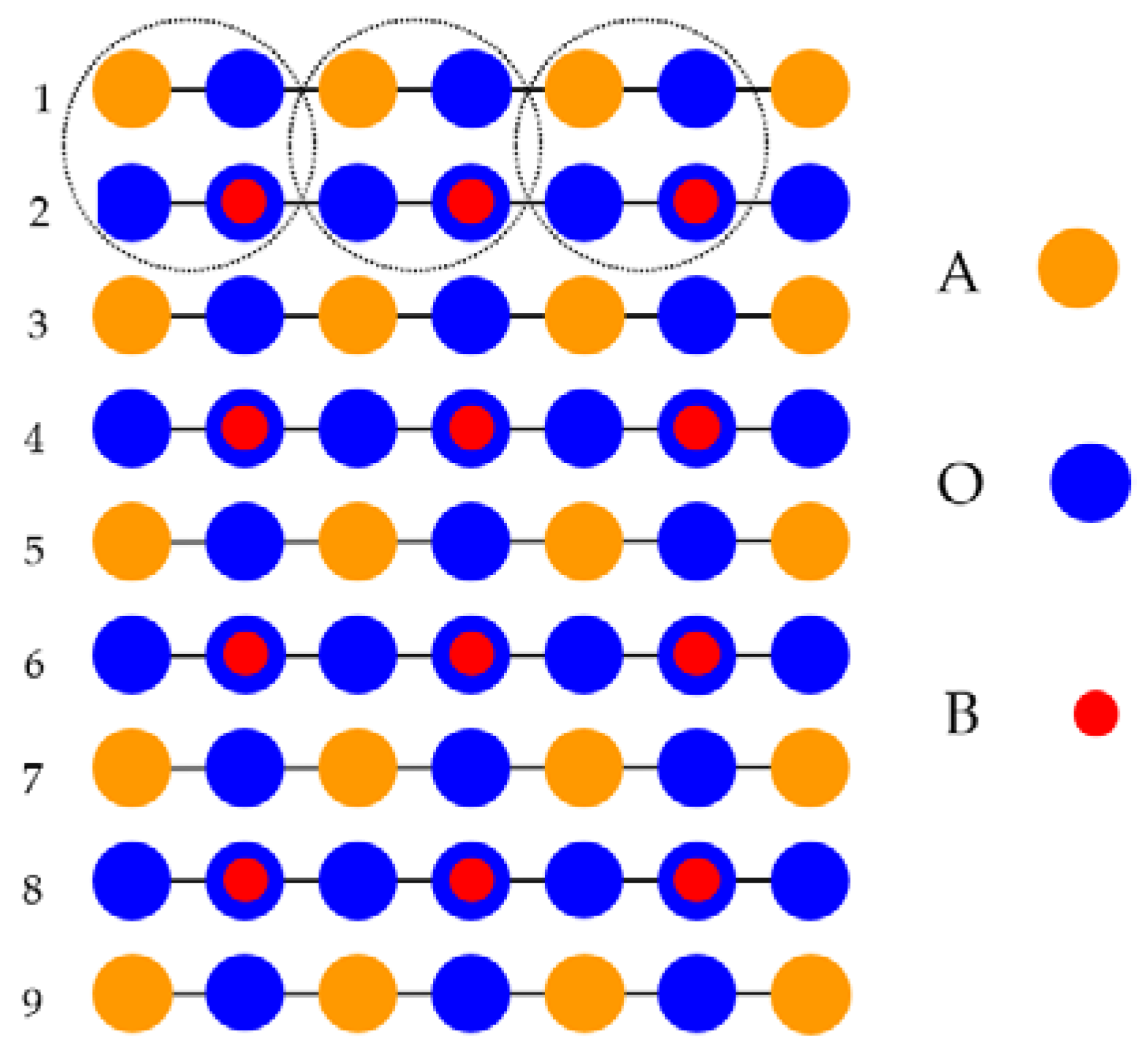
Just opposite to our ab initio computed neutral BaTiO
3, SrTiO
3 and BaZrO
3 (001) surfaces, which are built up from the neutral BO
2 or AO layers (
Figure 2), the WO
2 or ReO
2-terminated polar WO
3 or ReO
3 (001) surfaces were formed from charged (
Figure 3) WO
2 (ReO
2) or O layers. This is much more demanding to compute at the ab initio level for the polar WO
2 or ReO
2-terminated WO
3 or ReO
3 (001) surfaces (
Figure 3) than the neutral (
Figure 2) ABO
3 perovskite BO
2-terminated (001) surfaces [
8,
52,
82,
83,
84]. For example, in our ab initio computations, the ReO
2-terminated polar ReO
3 (001) surface consisted of nine alternating ReO
2 or O layers (
Figure 3). This means that this ReO
2-terminated ReO
3 polar (001) surface contained 19 atoms and had the chemical equation Re
5O
14.
In our ab initio computations, we employed the neutral atomic basis sets for all three atoms entering the WO
3 and ReO
3 crystals. Namely, we used the neutral W atom basis set from the reference [
85] for the W atom. Additionally, for the Re atom, we used the neutral atom basis set from the reference [
69]. Finally, for the O atom, again, we used the neutral O atom basis set, developed by Piskunov et al., from reference [
71]. Using the neutral atomic basis sets for all three Re, W and O atoms, we determined in our ab initio computations that both ReO
2 and WO
2-terminated ReO
3 and WO
3 (001) surfaces have a total charge of our employed nine-layer slab equal to zero. In our ab initio computations, we utilized the well-known classical Mulliken population analysis for the description of the ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 effective atomic charges
q and also their chemical bond populations
P [
86,
87,
88,
89].
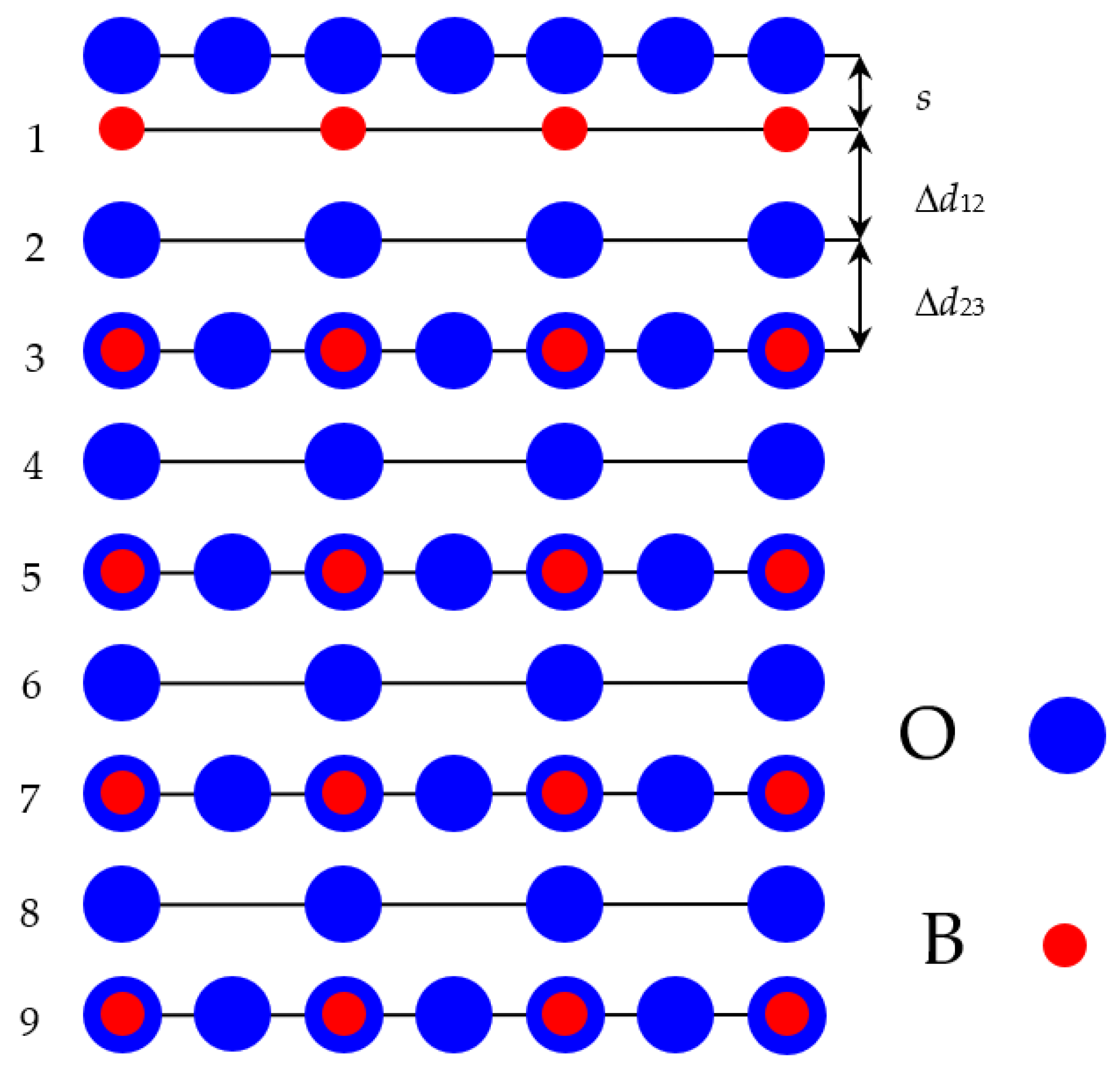
For our ab initio computations of AO-terminated BaTiO
3, SrTiO
3 and BaZrO
3 (001) neutral surfaces, we used, from both sides, AO-terminated mirror-symmetrical slabs containing nine alternating AO and BO
2 layers (
Figure 4). These AO-terminated ABO
3 perovskite (001) nine-layer slabs consisted of a supercell containing 22 atoms (
Figure 4). They were non-stoichiometric, and they had the following unit cell equation A
5B
4O
13 (
Figure 4). The only striking difference between the AO-terminated BaTiO
3, SrTiO
3 and BaZrO
3 (001) slabs (
Figure 4) and O-terminated ReO
3 and WO
3 slabs was the missing O atom in the ReO
3 and WO
3 (001) slabs (
Figure 5). Thereby, the O-terminated ReO
3 and WO
3 nine-layer (001) slabs consisted of alternating O-BO
2-O-BO
2-O-BO
2-O-BO
2-O layers (
Figure 5). They contained 17 atoms and had the following unit cell equation B
4O
13 (
Figure 5).
3. Ab Initio Computation Results for ReO3, WO3, BaTiO3, SrTiO3 and BaZrO3 Bulk Properties
As an opening of our first principles computations, by means of the B3LYP or B3PW hybrid exchange-correlation functionals, we computed the ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 bulk lattice constants [
52,
53,
54,
90,
91,
92,
93]. It is worth noting that we performed all our ab initio computations by B3LYP hybrid exchange-correlation functional B3LYP for ReO
3 and WO
3 matrixes as well as by B3PW hybrid exchange-correlation functional for BaTiO
3, SrTiO
3 and BaZrO
3 perovskites. Our ab initio computed bulk lattice constants for ReO
3 (3.758 Å), WO
3 (3.775 Å), BaTiO
3 (4.008 Å), SrTiO
3 (3.904 Å) and BaZrO
3 (4.234 Å) perovskites are in a fine agreement with the obtained experimental measurements (
Table 1). For example, our ab initio B3LYP computed ReO
3 bulk lattice constant (3.758 Å) is only overestimated by approximately 0.29% with respect to the experimental ReO
3 bulk lattice constant equal to 3.747 Å (
Table 1). Additionally, our ab initio B3PW computed BaTiO
3 bulk lattice constant (4.008 Å) is almost in a perfect agreement with the experimentally measured BaTiO
3 bulk lattice constant (4.004 Å) (
Table 1).
As we can see from
Table 3, our ab initio computed atomic charges for all atoms are considerably smaller than the generally accepted classical ionic charges in ABO
3 perovskites for Ba or Sr atoms (+2
e), for Ti or Zr atoms (+4
e), or for O atoms (−2
e). Additionally, our ab initio computed Re and W atom effective charges in ReO
3 or WO
3 materials (+2.382
e or +3.095
e, respectively) are considerably smaller than the Re or W classical ionic charges equal to (+6
e). It is worth noting that the absolute values of O atom charges in the ABO
3 perovskites BaTiO
3, SrTiO
3 and BaZrO
3 (−1.388
e, −1.407
e, and −1.316
e, respectively) are always larger than the absolute values of O atom charges in the ReO
3 or WO
3 crystals (−0.794
e or −1.032
e, respectively). Just opposite, our ab initio computed chemical bond populations between the Re and O as well as W and O atoms in the ReO
3 and WO
3 materials (
Table 3) (+0.212
e and +0.142
e, respectively) are always considerably larger than the respective B-O atom chemical bond populations in the BaTiO
3, SrTiO
3 and BaZrO
3 perovskites (+0.098
e, +0.088
e and +0.108
e, respectively).
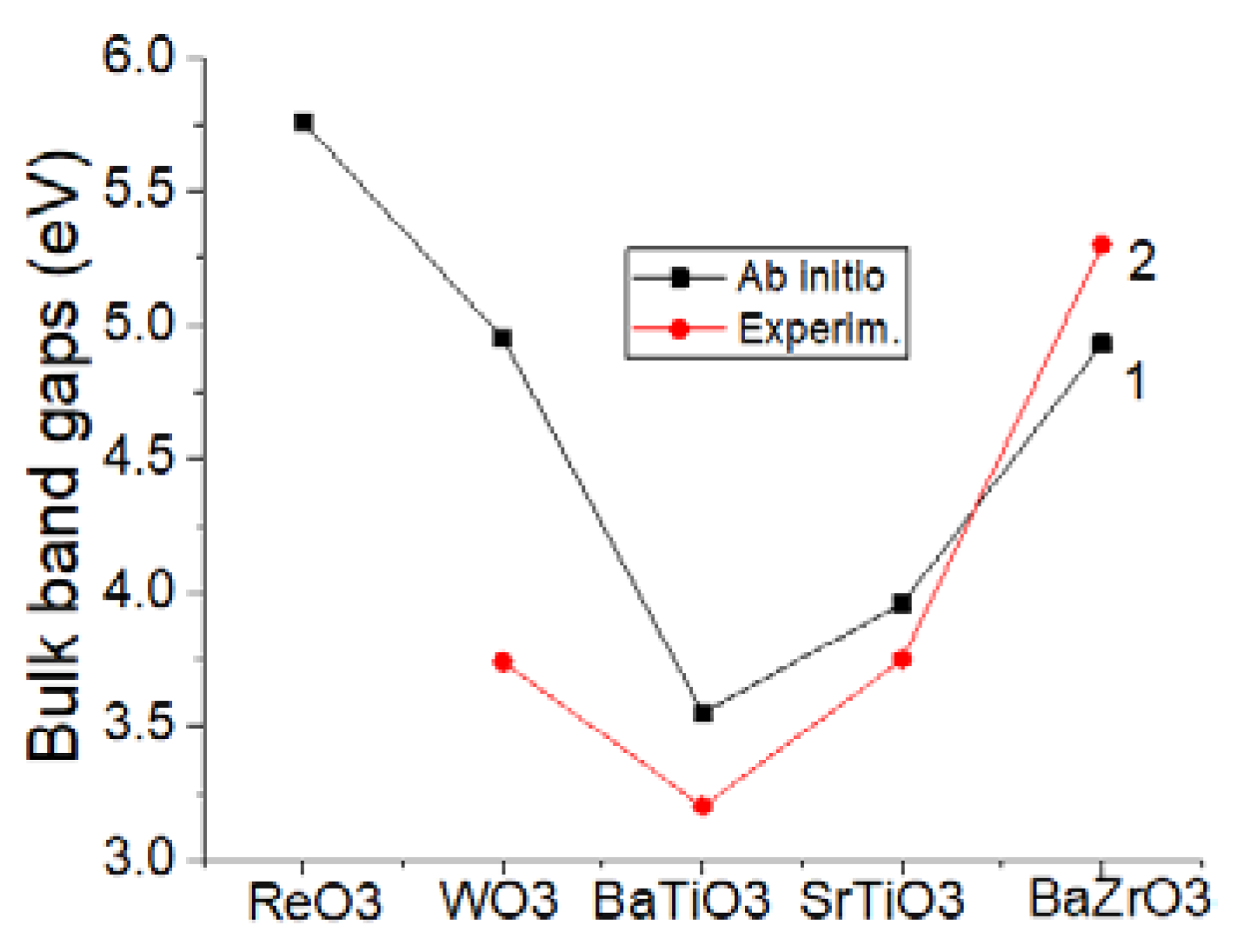
As a next step, by means of B3LYP or B3PW (
Table 4 and
Figure 6) hybrid exchange-correlation functionals, at the ab initio level, we computed the ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 bulk Γ-Γ bandgaps. As we can see from
Table 4 and
Figure 6, our B3LYP computed ReO
3 bulk Γ-Γ bandgap is equal to 5.76 eV. This is a theoretical prediction since, to the best of our knowledge, there is no experimental bandgap yet detected for the ReO
3 matrix at Γ-point. Our computed WO
3 bulk Γ-Γ bandgap (4.95 eV) by 1.21 eV exceeds the WO
3 experimentally detected [
56] bulk Γ-Γ bandgap (
Table 4). Determined by means of the B3PW hybrid exchange-correlation functional ab initio, our computed BaTiO
3 (3.55 eV), SrTiO
3 (3.96 eV) and BaZrO
3 (4.93 eV) bulk Γ-Γ bandgaps are in a fair agreement with the experimentally measured bulk Γ-Γ bandgaps for BaTiO
3, SrTiO
3 and BaZrO
3 perovskites (3.2 eV, 3.75 eV and 5.3 eV, respectively) (
Table 4 and
Figure 6).
4. Ab Initio Computation Results for the BO2 and O-Terminated ReO3, WO3, BaTiO3, SrTiO3 and BaZrO3 (001) Surfaces
As we can see from our ab initio computation results for ReO
2 and WO
2-terminated ReO
3 and WO
3, as well as BO
2-terminated BaTiO
3, SrTiO
3 and BaZrO
3 (001) surfaces, collected in
Table 5, for all five our computed materials ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3, the upper layer atoms relax inwards, in the direction towards the bulk (
Table 5). The only exception from this systematic trend is the upward shift of the WO
2-terminated WO
3 (001) surface upper layer O atom by 0.42% of
a0 (
Table 5). Just opposite, all second layer atoms relax upwards, with the single exception of the ReO
2-terminated ReO
3 (001) surface second layer O atom, which relaxes inwards by 0.32% of the ReO
3 cubic lattice constant
a0 (
Table 5). Again, all third layer ReO
2 and WO
2-terminated ReO
3 and WO
3, as well as ZrO
2-terminated BaZrO
3 (001) surface atoms, relax inwards (
Table 5). It is worth noting that for all our calculated ReO
2 and WO
2-terminated ReO
3 and WO
3 as well as BO
2-terminated BaTiO
3, SrTiO
3 and BaZrO
3 perovskite (001) surfaces, in all three layers the metal atom displacement magnitudes are always larger than the O atom displacements (
Table 5).
It is worth noting that we are the first in the world to perform ab initio computations for O-terminated ReO
3 and WO
3 (001) surfaces (
Table 6). As we can see from our ab initio computation results for O-terminated ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 (001) surfaces, all upper-layer atoms relax inwards (
Table 6). The only single exception from this systematic trend is the upwards relaxation of the SrO-terminated SrTiO
3 (001) surface upper layer O atom by +0.84% (
Table 6) of the SrTiO
3 bulk lattice constant
a0. In contrast, almost all second layer atoms relax upwards. The only two exceptions are the inward relaxation of O-terminated ReO
3 and WO
3 (001) second layer O atoms by (−0.53 and −0.11% of
a0, respectively) (
Table 6). Finally, all our ab initio calculated third layer atoms relax inwards, towards the bulk. It is worth noting that for both upper O-terminated ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 (001) surface layers, the metal atom displacement magnitudes are always larger than the O atom relaxation shifts (
Table 6).
Comparison of our ab initio calculation results with other calculations as well as available experimental data for SrO-terminated SrTiO
3 (001) surface [
94,
95,
96,
97,
98] are listed in
Table 7. As we can see from
Table 7, our ab initio B3PW computed surface rumpling amplitudes
s for SrO-terminated SrTiO
3 (001) surfaces (+5.66% of
a0) are in a qualitative agreement with other computation results ranging from (+5.8% of
a0) to (+8.2% of
a0) as well as in a qualitative agreement with available experimental data (
Table 7). Additionally, our ab initio investigation calculated that changes in the interlayer distances Δ
d12 and Δ
d23 are in qualitative agreement with other calculation results and most experiments (
Table 7). Unfortunately, our computed changes in interlayer distances Δ
d12 disagree with the RHEED experiment (
Table 7), but our computed changes in interlayer distance Δ
d23 disagree with the SXRD experiment (
Table 7). Nevertheless, since the LEED, RHEED and SXRD experiments do not always agree with each other, even with respect to signs, we can not take these LEED, RHEED and SXRD experiments (
Table 7) too seriously.
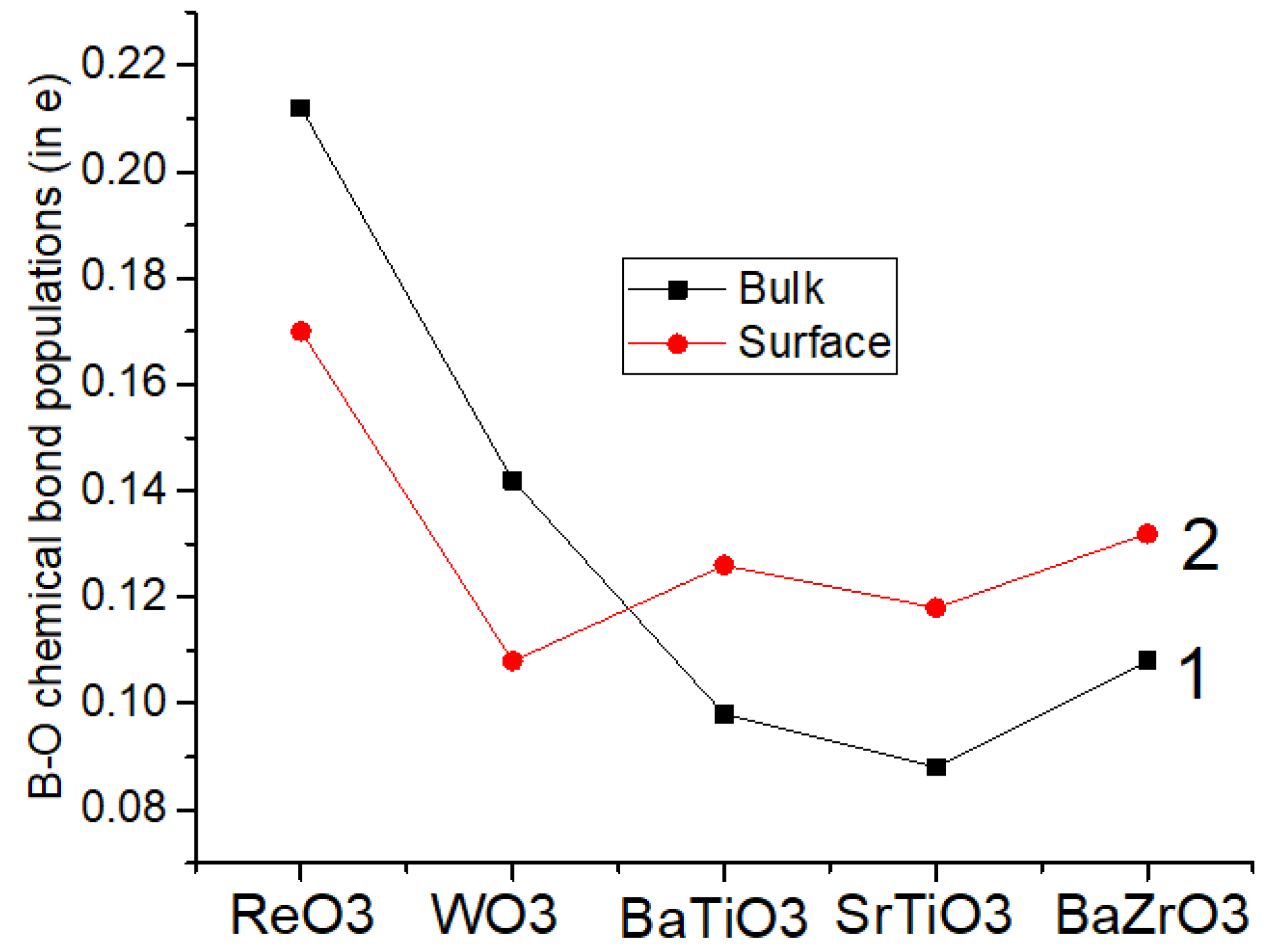
Our ab initio computed that B-O atom chemical bond populations in the BaTiO
3, SrTiO
3 and BaZrO
3 perovskite bulk (
Table 8 and
Figure 7) are always smaller than near their BO
2-terminated (001) surfaces. Just opposite, the Re-O and W-O chemical bond populations in the ReO
3 (0.212
e) and WO
3 (0.142
e) bulk (
Table 8 and
Figure 7) are slightly larger than near the ReO
2 and WO
2-terminated ReO
3 as well as WO
3 (001) surfaces (0.170
e and 0.108
e, respectively) (
Table 8 and
Figure 7). Nevertheless, the largest chemical bond populations in the ReO
3 and WO
3 matrixes are among the upper layer Re atom and the second layer O atom (0.262
e) as well as among the upper layer W atom and the second layer O atom (0.278
e).
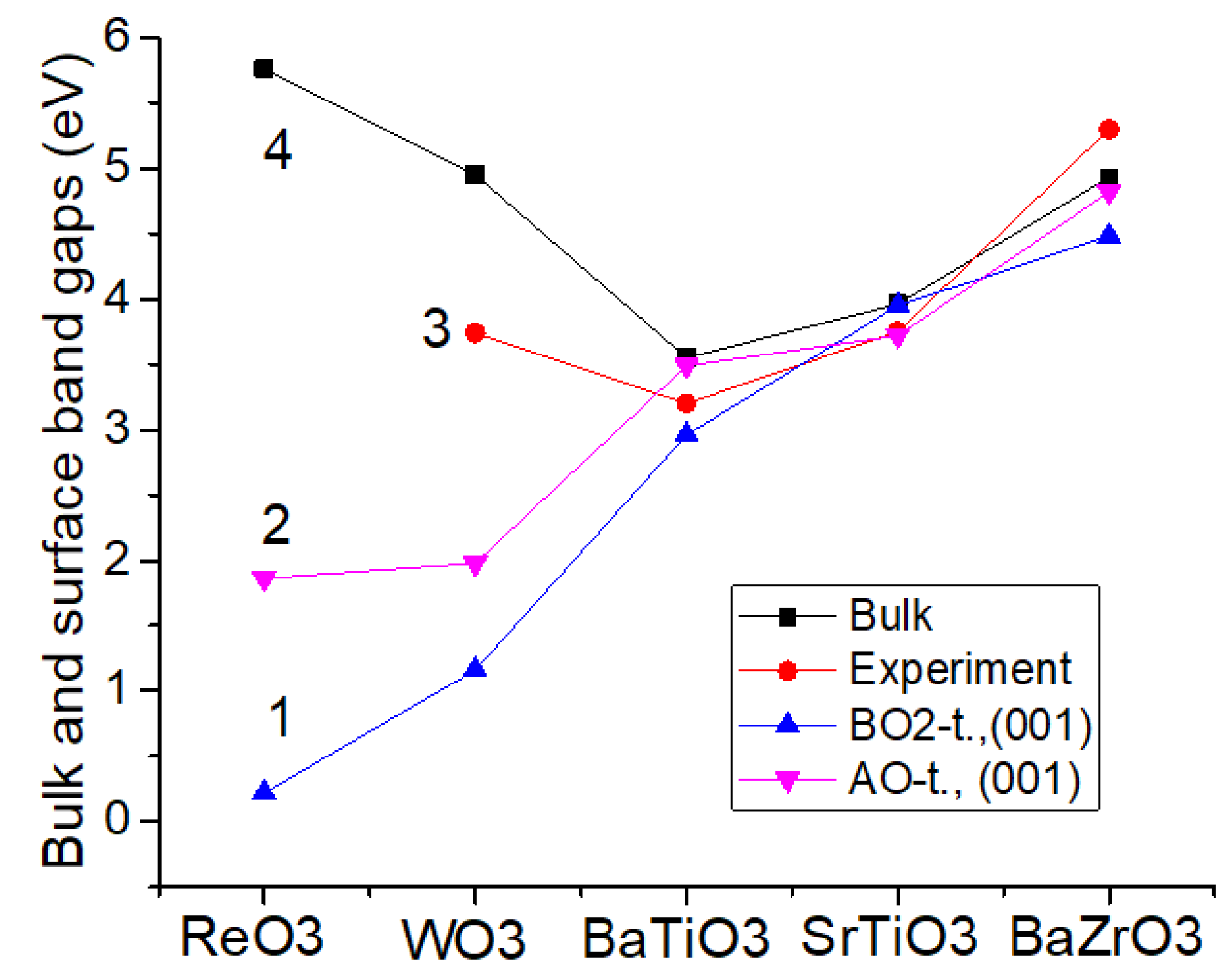
According to our ab initio computations, the Γ-Γ bandgaps near the BO
2, AO, or O-terminated ReO
3, WO
3, BaTiO
3, SrTiO
3 and BaZrO
3 (001) surfaces are always reduced with respect to their relevant bulk Γ-Γ bandgaps (
Table 9 and
Figure 8). Experimental data, where available, for the bulk Γ-Γ bandgaps are listed for comparison purposes (
Figure 8 and
Table 9). As we can see from
Table 9 and
Figure 8, for ReO
3 and WO
3 crystals, our ab initio computed Γ-Γ bandgaps near their O and especially ReO
2 and WO
2-terminated (001) surfaces are much more strongly reduced with respect to their bulk Γ-Γ bandgap values, than near the BO
2 and AO-terminated BaTiO
3, SrTiO
3 and BaZrO
3 perovskite (001) surfaces. For example, TiO
2 (3.95 eV) and SrO-terminated (3.72 eV) SrTiO
3 (001) surface Γ-Γ bandgaps are reduced only by (0.01 and 0.24 eV, respectively) regarding their bulk Γ-Γ bandgap value of 3.96 eV (
Table 9 and
Figure 8).
5. Conclusions
According to our ab initio computation results for BO2, AO and O-terminated ReO3, WO3, SrTiO3, BaTiO3 and BaZrO3 (001) surfaces, in most cases, the upper surface layer atoms relax inwards towards the bulk. The second surface layer atoms, again, in most cases, relax upwards, while the third layer atoms, again, relax inwards.
Our ab initio computation results for SrO-terminated SrTiO
3 (001) in most cases are in a fair agreement with previous calculation results and available experimental data. It is worth noting that our ab initio calculated interlayer distance Δ
d12 disagrees with the RHEED experiment (
Table 7) with respect to the sign. Nevertheless, since the RHEED experiment also disagrees with the LEED and SXRD experiments, we can probably not take this available RHEED experiment [
97] too seriously (
Table 7).
According to our ab initio computations, the Γ-Γ bandgaps near the BO2, AO or O-terminated ReO3, WO3, BaTiO3, SrTiO3 and BaZrO3 (001) surfaces are always reduced with respect to their relevant bulk Γ-Γ bandgaps.
Our ab initio computed B-O atom chemical bond populations in the BaTiO3, SrTiO3 and BaZrO3 perovskite bulk are always smaller than near their BO2-terminated (001) surfaces. Just opposite, the Re-O and W-O chemical bond populations in the ReO3 (0.212e) and WO3 (0.142e) bulk are slightly larger than near the ReO2 and WO2-terminated ReO3 as well as WO3 (001) surfaces (0.170e and 0.108e, respectively). Nevertheless, the largest chemical bond populations in the ReO3 and WO3 matrixes are among the upper layer Re atom and the second layer O atom (0.262e) as well as among the upper layer W atom and the second layer O atom (0.278e).


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}