(2) Neuroinflammatory and neuropeptide panel choice.
The variables analyzed are both quantitative/continuous and qualitative/nominal.
3.1. MODeLING.Vis: Development of A Protein Visualization Tool
| Are there categorical differences in the protein profiles matching our mental health strata? |
| Could an unsupervised learning analysis find corresponding electrophoretic signatures? |
| Could MODeLING.Vis cluster proteins with a high discriminative power? |
MODeLING.Vis is a GUI toolbox created to analyze electrophoretic data. MODeLING.Vis data input/output is based on local storage, nonetheless enhancing the reusability of our electrophoretic data. Respecting the FAIR principles [
31], we emphasize improving the ability of machines, in this case, a GUI toolbox, to automatically find and use the electrophoretic data, in addition to supporting its reuse by individuals. Henceforth, with the analysis proposed by this study, we supported data discovery through sound data management and maximized the added value by formal scholarly digital publishing.
Firstly, the full raw electropherogram of the Expected Protein Profiles of the 92 neurotypical young adults was obtained. Then Experion
TM Imaging software exported it to a comma-separated values file, “.csv”. This exported raw profile was treated with the same preliminary strategy as in the pipeline oral proteome study, with the raw data of the 22 control subjects (T-1). The pipeline oral proteome study is a preliminary exploratory study that had already been completed and published [
18] and justified the rationale for this work. This biomedical analysis methodology [
18] was conducted by SalivaTec laboratory and generated preliminary data with a sample size of 22 control subjects (T-1) and five preliminary subjects (T-1 (before and after the experimental procedure)), for which the total protein profiles were characterized by capillary electrophoresis. These raw data, a preliminary Expected Protein Profile workbook, were published as “EPPStrategyDataExport” (
https://0-doi-org.brum.beds.ac.uk/10.5281/zenodo.7054406, accessed on 28 November 2022) and can be consulted at:
https://tinyurl.com/EPPStrategyDataExport (accessed on 21 July 2022). This workbook consists of six worksheets demonstrating the preliminary strategy applied to the database—six stages were executed.
The first worksheet (first stage: Total) shows the total raw data of the 12 electrophoretic runs performed for all the samples of neurotypical young adults.
The second worksheet (second stage: Total Reviewed) reviewed the previous one, showing only the MW (shown in kDa) and Concentration (ng/µL) for each sample.
The third worksheet (third stage: Total Rounded) rounded the previous variables into decimals, as we did in the Pipeline Oral Proteome Study, and added the new variable “Order” to help sort the samples.
The fourth worksheet (fourth stage: Total Subgroups Sorted) added the following variables: Subphenome and molecular weight’s Color and Molecular Band. A Subphenome is a variable used to define which subgroup the sample belonged to ES = (i), the top phenome of the Experimental group; CS = (ii) the top phenome of the Control group; EI = (iii) the bottom phenome of the Experimental group; and CI = (iv) the bottom phenome of the Control group. Molecular weight’s Color is a variable showing the respective RGB color corresponding to each group. Molecular Bands is a variable that (i) sorted the rounded MW (shown in kDa) according to a crescent kDa and (ii) showed the respective RGB color correspondent, from which an electrophoretic run was executed.
In the fifth worksheet (fifth stage: Total Clustered), in the first part, the variable MolecularBands was repeated according to the number of electrophoretic runs detected (MolecularBandsRep). Then, in the second part, the variable MolecularBandsRep was colored according to the sample’s subgroup using the algorithm “Excel VLOOKUP Function”. This fifth stage originated the variable MolecularBandsSubgroups.
In the sixth worksheet (sixth stage, named “EPPStrategyDataForMOdeLINGVis”), the preliminary final database is shown, which is a triple-entry table. This worksheet used the previously acquired variables (Sample Number; Molecular Bands Subgroups; Concentration (ng/µL)) to create the final table.
Subsequently, this preliminary final database, the “EPPStrategyDataExport” database, was treated to be imported to MATLAB.
First, the section: “Present in the following subphenomes” was added, which comprised binary variables (present/absent) to identify in which subgroup the Molecular Bands were present. Secondly, the preliminary triple entry table was added to the Molecular Bands Summary for each Subject (ex: D01309).
This final full raw electropherogram was published as “ExportForMOdeLINGVis” (
https://0-doi-org.brum.beds.ac.uk/10.5281/zenodo.7054551, accessed on 26 November 2022) and can be consulted at:
https://tinyurl.com/ExportForMOdeLINGVis (accessed on 8 August 2022). Lastly, the database was implemented in the MODeLING.Vis toolbox and the variables were imported into arrays. Those arrays indexed a linear matrix of the variables: Molecular Bands Subgroups (kDa) and Concentration (ng/µL) of each sample (subject).
One of the limitations of exploring the data with Experion Imaging software (Biorad
®, Hercules, CA, USA) was its incapacity for generating MW intervals and clustering the subjects according to them. Therefore, we developed a toolbox for unsupervised/supervised machine learning, MODeLING.Vis, and assigned it a
https://0-doi-org.brum.beds.ac.uk/10.5281/zenodo.7041477 (accessed on 24 November 2022). The GUI MATLAB code, used in our toolbox, is accessible online (
https://www.limmit.org/uploads/2/6/8/4/26841837/modeling.vis.zip (accessed on 8 August 2022)), in the LIMMIT laboratory, Faculty of Medicine, University of Lisbon website, as a free and open-source MATLAB toolbox.
To start the GUI MATLAB code, follow the instructions provided by the video tutorial (
https://0-doi-org.brum.beds.ac.uk/10.5281/zenodo.7337428, accessed on 30 November 2022) and use the provided electrophoretic dataset “ExportForMOdeLINGVis”, i.e., protLabled.xls on the video tutorial.
On the MATLAB prompt, write:
>> cd C:\...\code (i.e., where the code is unzipped)
>> addpath(genpath(‘./’))
>> limmitGui
MODeLING.Vis includes three separate phases: (i) Data Visualization, (ii) Data Exploration, and (iii) Data Mining.
The first objective of (i) Data Visualization is to transform the independent continuous variable of MWs into molecular intervals through an algorithm based on the EM scheme to fit a Gaussian mixture model to the data in a maximum likelihood framework. The soundness of this computational method acknowledges various bioinformatics applications, with specific reference under the hypothesis of hidden variables underlying the observed features [
32].
The algorithm comprises not only the EM component but a definition of other functions to set concentration (ng/µL) thresholds and the quantity of MW intervals (kernels) of interest.
The number of kernels can be set to the number of isolated local maxima from visual data inspection. MODeLING.Vis permits overlaying the GMM fitting curve with the data distribution. If a local maximum has not been captured by a Gaussian component, then a new kernel can be added to the mixture. The mixture can be so defined within a few trials as part of the interactive data processing capabilities of MODeLING.Vis. It can also be possible to terminate the inclusion of new Gaussians once the data likelihood reaches a saturation point. It is, however, noted that the mixture definition through the visual identification of local maxima has been found very effective in the scope of the present work.
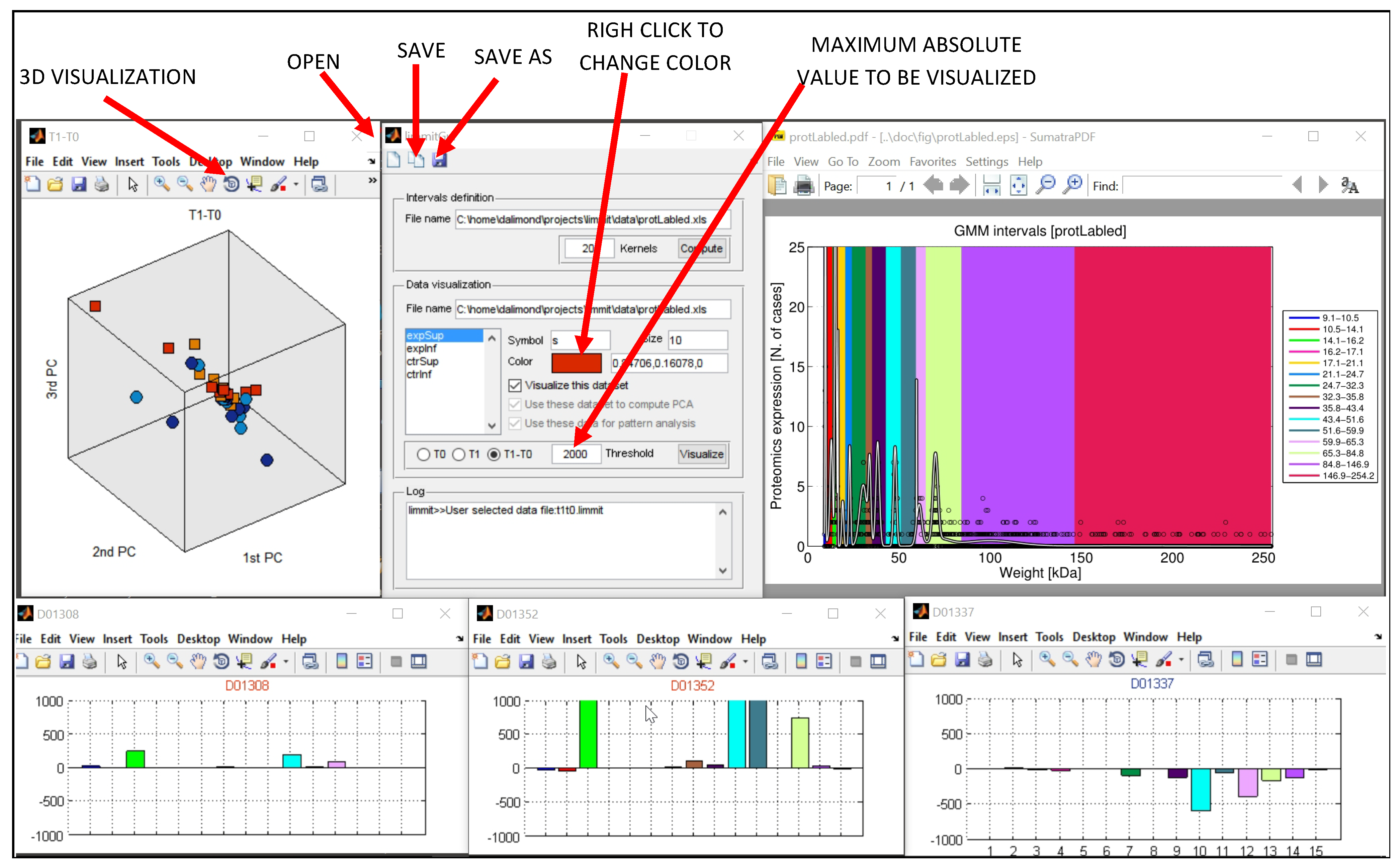
As shown in
Figure 1, the number of kernels (Gaussian components) was set to 13 because it was the best algorithm to treat our protein profile and the dispersion in our MW. Furthermore, this application was designed to import from other databases other than human salivary electropherograms and had already been positively tested.
The possibility of defining the number of kernels gives the researcher control over the data exploration of his specific dataset. It does not limit it to the constraints of restricted unsupervised machine learning.
We wanted to find and compare among the four subgroups for our specific data and the number of fixed intervals; thus, we defined it as 13 kernels. This decision provided us with the following significant (p < 0.05) intervals of MW:
(A) [9.1;9.8] kDa;
(B) [9.8;10.3] kDa;
(C) [10.3;13.7] kDa;
(D) [13.7;17.5] kDa;
(E) [17.5;21.1] kDa;
(F) [21.1;24.7] kDa;
(G) [24.7;36] kDa;
(H) [36;42.6] kDa;
(I) [42.6;51.5] kDa;
(J) [51.5;65] kDa,;
(K) [65;77] kDa;
(L) [77;149.7] kDa.
Similarly, these intervals are consistent with the ones discovered by the visual analysis of the capillary gels and quantitative electropherograms. Moreover, these intervals are equally compatible with those found in the preliminary study of the 22 control subjects.
Hence, (a) the major density of protein peak dispersion—[12;18] kDa and [43;66] kDa—was statistically and relevantly subdivided into:
(C) [10.3;13.7] kDa;
(D) [13.7;17.5] kDa;
(E) [17.5;21.1] kDa;
(I) [42.6;51.5] kDa;
(J) [51.5;65] kDa.
Similarly, (b) the minor density of protein peak dispersion—[20;40] kDa and [70;145] kDa—was statistically and relevantly subdivided into:
(F) [21.1;24.7] kDa;
(G) [24.7;36] kDa;
(H) [36;42.6] KDa;
(K) [65;77] kDa;
(L) [77;149.7] kDa.
From this analysis, a new range of density of protein peak dispersion was discovered in the lower molecular range, which offered significant relevance to our specific molecular data dispersion—(c) the lower MW density protein peaks:
(A) [9.1;9.8] kDa;
(B) [9.8;10.3] kDa.
All data visualization and analysis are offered as an easy access tool for the researcher, who may update his proteomic dataset and evaluate how the proposed solution reflects her/his data and hypothesis, as shown in
Figure 1.
Subsequently, our GUI provided us with (ii) Data Exploration for hypothesis setting and testing. The toolbox was designed to explore not only one type of dataset but also integrate other datasets acquired for the same sample of subjects. As it is, the researcher can feed multiple clinical and molecular datasets: e.g., clinical evaluations, genomic data, immune detection data, etc.
An additional feature of the toolbox allows for the following:
(a) Integration of multiple omics datasets;
(b) Visual access to explore all the subject information, in particular, and the whole sample, in general.
This approach (a) allows the researcher to conduct better her/his multimolecular approaches in datasets (which tend to be multiple) and (b) addresses a possible solution for the increasingly prominent data characteristics of omics methods.
Moreover, as shown in
Figure 2, the researcher can define the following:
(a) Colors;
(b) Type of symbol;
(c) Size for the clustering of subgroups.
This configuration eases the identification of specific clusters and makes hypothesis testing more visible. In our study, we created a solution to import T0, T1, and Δ (T1 − T0) datasets and defined the supervised search of the four subgroups. In this part of the data mining, we wanted to feed the algorithm with a specific classification to learn and recognize the four specified labels, which are our subgroups. Moreover, we created the threshold variable for the independent variable (in the case of the electrophoretic data: concentration in ng/µL). The threshold allows the researcher to define how many subjects she or he wants to plot according to the T0, T1, and Δ (T1 x2212; T0) intergroup variability or effect size.
Intergroup statistical testing is performed by simple principal component analysis (PCA), as the data that is routinely fed into the toolbox and its algorithms need an orthogonal linear transformation, which projects the data into a new coordinate system with a reduced number of dimensions, hence allowing for the visualization and interpretation of the data. In
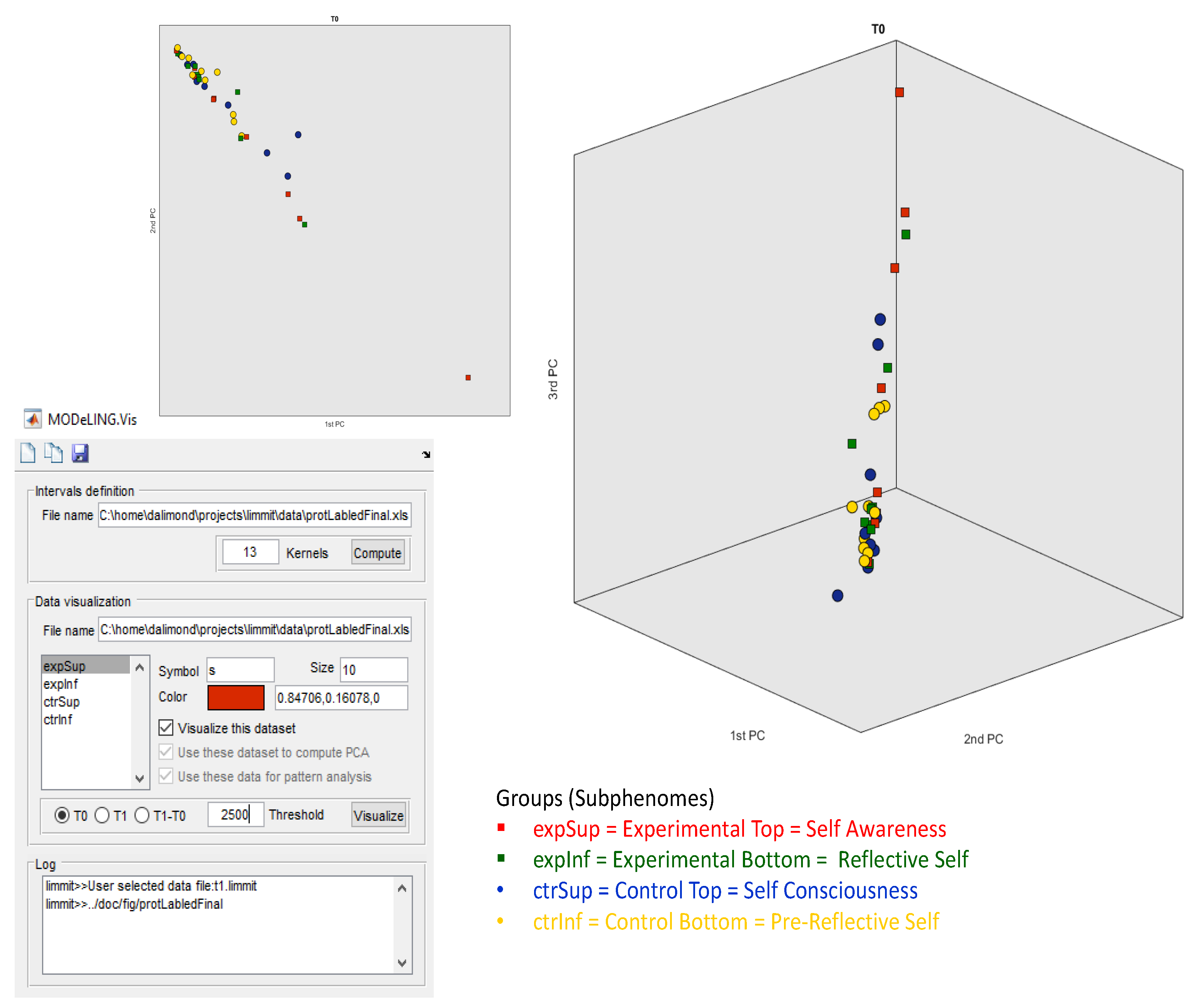
Figure 2, the data exploration of our electrophoretic dataset for T0 in MODeLING.Vis is shown. As mentioned, we had the option to define the threshold to 2500 ng/µL because it better fits our data. Then, we set the analysis to T0 and chose the Data Visualization of our electrophoretic data (“ExportForMOdeLINGVis”) and the interval definition acquired before. Finally, we defined colors and symbols for our subgroups. This analysis shows data clusters in only two PCA components and a small, but not very significant, separation of the Experimental Top (red square) and Control Bottom (yellow circle) subgroups.
In
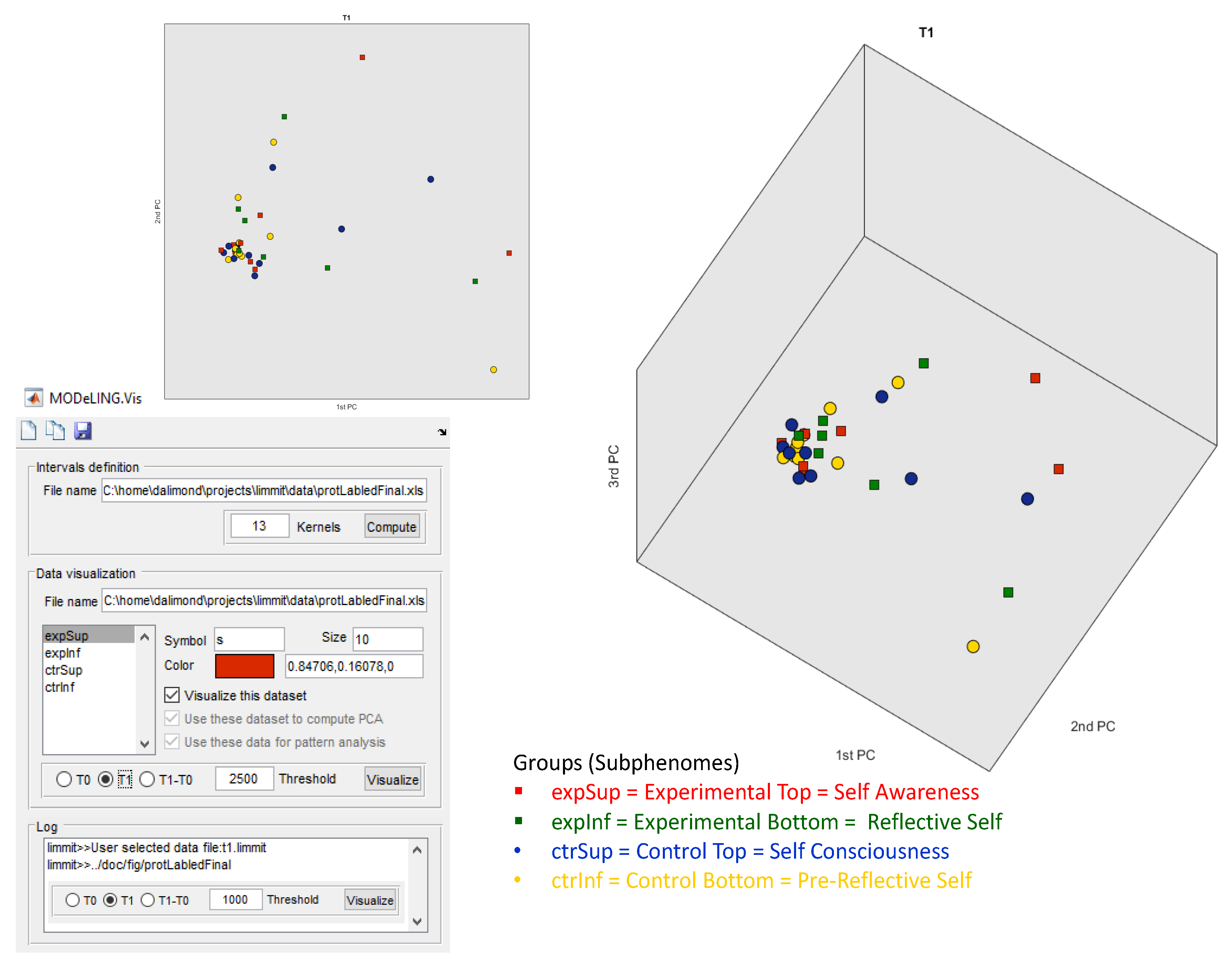
Figure 3, the data exploration of our electrophoretic dataset for T1 in MODeLING.Vis is shown. The same parameters were set. This data exploration presents data clustering in three PCA components, and there is a more relevant separation of the Experimental Top (red square) and Control Bottom (yellow circle) subgroups (when compared to T0), which is not visually perceived.
However, this separation is more evident in
Figure 4, which shows the data exploration of our electrophoretic dataset for (T1 − T0).
Likewise, the same parameters were set. Nevertheless, in this analysis, data clusters in three PCA components and a significant separation of our cluster of subjects, i.e., clustering in subgroups.
The square symbols (Experimental subgroups), distributed along the top 2PCA and 3PCA axis, are separated from the circle symbols (Control subgroups), spread along the bottom 2PCA and bottom 1PCA axis, with statistical relevance. This approach offers confident consistency for an electrophoretic profile intergroup separation in between the Experimental and Control groups.
Additionally, but not so significantly, there is a separation of the red squares (Experimental Top group) and green squares (Experimental Bottom group), alongside PCA1, and of the blue circles (Control Top group) and yellow circles (Control Bottom group), also alongside PCA1. This offers some consistency for the possibility of an electrophoretic profile separation in between the intra-Experimental electropherograms (Experimental subgroups) and the intra-Control electropherograms (Control subgroups). However, this electrophoretic profile separation is not statistically relevant for a defined threshold Δ (T1 − T0) and effect size of 2500 ng/µL.
More significant is the separation and clustering, both alongside the 3PCA components, between (i) the red squares (Experimental Top group) vs. the blue circles (Control Top group) and (ii) the green squares (Experimental Bottom subgroup) vs. the yellow circles (Control Bottom subgroup).
This data exploration offers some consistency for a possible electrophoretic profile separation between (i) the Experimental Top and Control Subphenomes and (ii) the Experimental Bottom and Control Subphenomes.
This clustering lacks proper hypothesis testing to evaluate the exact concentration (ng/µL) of the Δ (T1 − T0), which is the main limitation of this analysis.
Notwithstanding, it may offer the opportunity to define a consequent hypothesis, i.e., to better profile and stratify substrata in our total electrophoretic data. Therefore, the subsequent data analysis was executed as a reasonable solution for this limitation.
The toolbox has the objective of (iii) Data Mining the individual molecular profile (subject to subject/sample to sample) and comparing it to the whole sample (neurotypical young adults).
As referred, it was designed to integrate multiple clinical and molecular datasets.
As such, in
Figure 4, we present an example of the comparison of four subjects after the phases of the GUI toolbox:
(i) Data Visualization;
(ii) Data Exploration;
(iii) Data Mining.
We show, fittingly, subject D01383 (Experimental Top subgroup (1)), subject D01371 (Experimental Bottom subgroup (3)), subject D01337 (Control Top subgroup (2)), and subject D01319 (Control Bottom subgroup (4)).
These four subjects (with the same colors) are the most significant subjects of each subgroup and represent the specific and characterizing stratum of the electrophoretic profile of their subgroup.
From the molecular intervals found, those which are more relevant are the red (No. 2) and the pink (No. 4) ones, which correspond to (B) [9.8;10.3] kDa and (D) [13.7;17.5] kDa in the lighter MW range (
Figure 1).
Additionally, with a correspondent relevance are the purple (No. 9) and light blue (No. 10) ones, which correspond to (I) [42.6;51.5] kDa and (J) [51.5;65] kDa in the heavier MW range (
Figure 1).
The Δ (T1 − T0) ng/µL of the (B) [9.8;10.3] kDa and (D) [13.7;17.5] kDa molecular weight range is for:
(1) Subject D01383 (representing the Experimental Top group) ≅ 50 ng/µL and −900 ng/µL;
(2) Subject D01337 (representing the Control Top group) ≅ −10 ng/µL and −30 ng/µL;
(3) Subject D01371 (representing the Experimental Bottom group) ≅ 600 ng/µL and 800 ng/µL;
(4) Subject D01319 (representing the Control Bottom group) ≅ 0 ng/µL and 2000 ng/µL.
Those MW ranges [(B) [9.8;10.3] kDa and (D) [13.7;17.5] kDa] are characteristic of molecules that have been documented to cross the blood-brain barrier [
33] (Banks, 2009). Please note that an error variable should be considered and correspond to the lack of accuracy offered by Experion
TM analysis and the identified MW ranges. This consideration should take this inaccuracy into account, but also the process of protein degradation observed and well documented in saliva.
Different molecular characteristics are associated with the capacity to cross the blood-brain barrier, a significant field of study in neuropharmacology [
34,
35].
However, in the interest of molecular biology, it is essential to understand those small molecules’ physiology and biological function.
Banks [
36] has described the biological characteristics of those small peptides crossing the blood-brain barrier and correlated them to the neuropeptide response. Likewise, this light MW [(B) and (D)] range was earlier associated with neuroinflammatory response [
37]. Still, more recently, Erickson and Banks [
38] described it as part of the neuroimmune axes of the blood-brain barriers and blood-brain interfaces.
Please note that uncertainties should be considered, which correspond to the lack of accuracy offered by ExperionTM analysis and the identified MW ranges. In addition to this inaccuracy, the process of protein degradation observed and well-documented in saliva should be considered.
Hence, the importance of these small peptides, detected by capillary electrophoresis in this light MW [(B) and (D)] range, for the physiological and pathological regulation of neurotypical/atypical subjects.
The Δ (T1 − T0) ng/µL of the (I) [42.6;51.5] kDa and (J) [51.5;65] kDa molecular weight range is for:
(1) Subject D01383 (representing the Experimental Top group) ≅ −100 ng/µL and 0 ng/µL;
(2) Subject D01337 (representing the Control Top group) ≅ −600 ng/µL and −400 ng/µL;
(3) Subject D01371 (representing the Experimental Bottom group) ≅ 2200 ng/µL and 2000 ng/µL;
(4) Subject D01319 (representing the Control Bottom group) ≅ 900 ng/µL and 800 ng/µL.
Those MWs are characteristic of a group of larger systemic molecules, which have not been documented to cross the blood-brain barrier [
33] (Banks, 2009).
Hence, they are not directly relevant to our study as they are not brain-produced proteins but indirectly important as systemic protein expression. In another oriented study design, they could be interesting for heavier protein molecular profiling of the subjects with systemic-produced proteins.
Specifically, this heavier MW range is essential for comprehending the role of larger proteins and protein complexes in non-neuropsychiatric diseases.
As an example, proteins, such as alpha-1-antitrypsin, 47 kDa [
39], pyruvate kinase PKM, 58 kDa [
40], and serum albumin, 69 kDa [
41], are essential markers for hereditary, metabolic, and cardiovascular diseases, respectively.
As a hypothesis for better conduction of our study and better statistical generalization power, it is essential to quantify those lighter MW ranges, i.e., (B) [9.8;10.3] kDa and (D) [13.7;17.5] kDa with more accurate sensibility and sensitivity.
The quantification and identification of those lighter MW ranges are imperative to understand better what is affecting this electrophoretic profile.
However, acquiring data with the ExperionTM automated electrophoresis system (Biorad®) offers a low capacity to discriminate which proteins reflect that stratum.
This low capacity is explained because electrophoretic patterns refer to a conjunction of proteins that migrate to the same MW and not a specific and single protein migration, and an error correspondent to the lack of accuracy offered by ExperionTM analysis.
A better acquisition method, with higher sensitivity, sensibility, and discrimination, is necessary to explain which proteins are changing.
Identifying those specific proteins can improve our understanding of how they influence the total protein profile in those light molecular ranges: [9.1;30] kDa.
This specific molecular range reflects the whole spectrum of peptides, peptide complexes, and small proteins that migrate in electrophoresis in (c) the lower MW density protein peaks.
For that purpose, simultaneous immune detection, with specific antibodies for specific peptides in those MW ranges, is mandatory for adequate quantification and discrimination.
Moreover, this quantification offers a suitable possibility for multivariate hypothesis testing of the identified peptides and proteins by immune detection.
Following the work of Banks [
33,
36] and the objective of our study design, immune detection of the peptides and the small proteins implicated in the neuropeptide and the neuroinflammatory response should be addressed.
This identification is essential for better characterization of the protein strata in this light MW range and understanding of how they affect neurotypical young adults.
3.2. Neuroinflammatory and Neuropeptide Panel Choice
A MODeLING.Vis analysis helped us understand which proteins are responsible for the changes observed in the MW range [9.1;30] kDa.
The [9.1;30] kDa MW interval corresponds to small proteins like the ones already identified in saliva by Rosa and colleagues [
42] and listed in the OralCard by Arrais and colleagues [
43]:
Histatin-1, 7 kDa;
Submaxillary gland androgen-regulated protein 3B, 8 kDa;
Acyl-CoA-binding protein, 10 kDa;
Protein S100-A8, 11 kDa;
Cystatin-A, 11 kDa;
Protein S100-A9, 13 kDa;
Profilin-1, 15 kDa;
Fatty acid-binding protein, 15 kDa;
Cystatin-SA, 16kDa;
Cystatin-SN, 16 kDa;
Cystatin-S, 16 kDa;
Cystatin-C, 16 kDa;
CALML3, 17 kDa;
PIP, 17 kDa;
PRH1, 17 kDa;
Interleukin-1 receptor antagonist protein, 20 kDa;
Glutathione S-transf P, 23 kDa;
HSP β-1, 23 kDa;
ZG16 homol β, 23 kDa;
BPI fold-containing family A member, 27 kDa;
14-3-3 protein sigma, 28 kDa;
Kallikrein-1, 29 kDa.
The listed proteins are small enough either (i) to pass the blood-brain barrier or (ii) to be detected in saliva.
Those proteins have not only well-known neurological functions, for instance, Cystatin-C in amyotrophic lateral sclerosis [
44], but may also be altered in neurodevelopmental conditions, for instance, Interleukin-1 receptor antagonist protein (part of the neuroimmune system) in intellectual disability [
45].
The best four subjects of the (1) Experimental Top, (2) Control Top, (3) Experimental Bottom, and (4) Control Bottom subgroups are plotted.
These four subjects represent the molecular profile with more significant intergroup variability and intragroup homogeneity.
Therefore, they are the subjects more characteristic of each group and have a more representative molecular profile.
Henceforth, we chose those four subjects of each four subgroups to perform the following analysis. Additionally, we selected one control subject for each group.
As the objective of the following analysis was (i) to study the MW range considered for particles passing the blood-brain barrier and (ii) to probe into the neuroinflammatory and neuropeptide system, we chose one control subject of each subgroup with an inflammatory disease, undergoing the same cognitive load and task.
The subjects followed the analysis already discussed in (ii) the Expected Protein Profile Results.
The ExperionTM Automated Electrophoresis System (Biorad®) analysis was repeated, but in this case, for those five subjects (4 best + 1 control) of each group. The final objective of this analysis was to evaluate if those five subjects should advance for simultaneous immune detection and quantification.
We want to warn about the limitations of conducting such an analysis and hypothesis testing.
This nonblind analysis lacks the statistical power for generalization for the researcher, and it would be a type 1 statistical error to act as such.
However, such an exploration is valid as an exploratory study aiming at the sole understanding of the protein expression in this small MW range.
Therefore, the most significant protein profiles were selected. This (c) lower MW range [9.1;30] kDa was one of the protein density peaks with more intergroup concentration (ng/µL) difference and intragroup curve similarity.
This [9.1;30] kDa interval is known as characterizing neuroinflammatory response [
46], as well as neuropeptide response, as published in the NeuroPep database [
47].
The cytokines, interleukins, and neuropeptides are small proteins that migrate in the electrophoresis in this molecular range.
Hence, the vital role that those small neuroimmune molecules [
48] and small neuropeptides [
49] may have in neurodevelopmental conditions; e.g., autism spectrum disorder [
50].
In the following pictures, two molecular ranges should be separated.
From the [9.1;30] kDa range studied, interval I. [9.1;17] kDa, corresponding to the electropherogram’s first peaks, is associated with the smallest molecules of neuropeptide response.
Complementarily, interval II. [17;30] kDa corresponds to slightly larger molecules associated with the neuroinflammatory response.
For a more accessible display, on the x-axis, we added two markers indicated in the figures as Bioplex Th17 (Start and End). From the beginning of the x-axis to Bioplex Th17 Start, the interval is associated with the neuropeptide response.
The interval is associated with the neuroinflammatory response from the Bioplex Th17 [Start; End].
We named the markers indicatively and referred to a possible Bioplex Th17 immunodetection panel, which would be a good panel for understanding the peptides involved in this MW range.
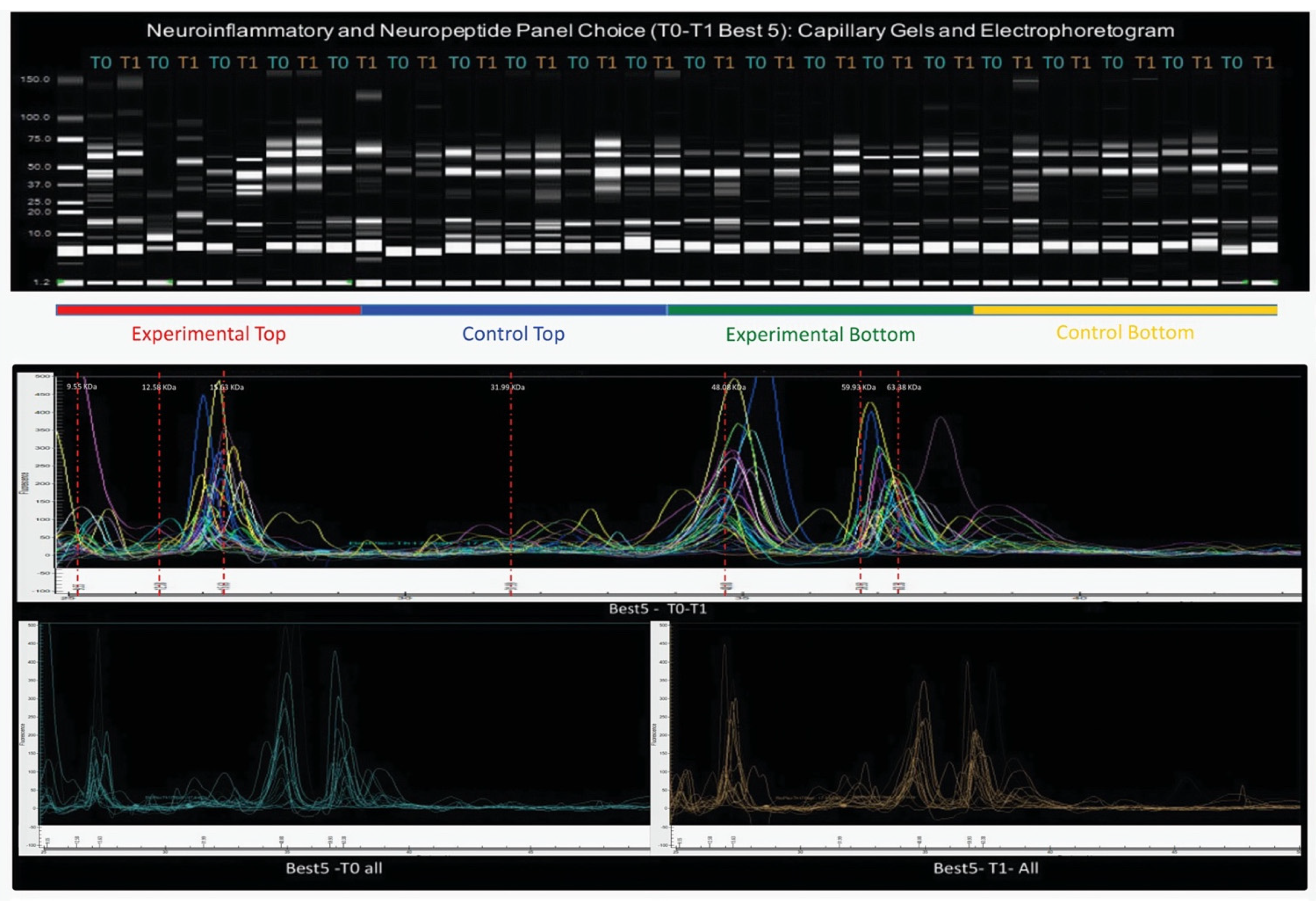
Figure 5 shows all four subgroups’ capillary gels and electropherograms of the five chosen subjects for the neuroinflammatory and neuropeptide panel.
All subjects (from all the subgroups) in T0 + T1 are plotted together, and in T0 (before) and T1 (after), the Intervention Protocol. Likewise, the capillary gel from the total four subgroups is shown separately, in T0 + T1.
In the interval I. [9.1;17] kDa, the total four subgroups of the study showed two protein peaks of a considerably high heterogeneity, both in the MW range and in fluorescence (concentration (ng/µL)), which need further investigation. Likewise, in interval II. [17;30] kDa, the total four subgroups of the study showed one protein peak of considerably high heterogeneity, more in the MW range variable than in the concentration variable.
This heterogeneity can also be observed in the capillary gels.
In
Figure 6, it is possible to see the electropherograms, separately in T0 and T1, of the subjects belonging only to the (1) Experimental Top Subphenome.
Five subjects were plotted. Moreover, they were also charted together in T0 + T1 without the positive control for that group. The fourth graph shows T0 + T1 for the positive control, a subject with an ICD-10 classification: J30.1—Allergic rhinitis due to pollen and medicated with the antihistaminergic Zyrtec.
As is shown in the first two graphs, there is considerable variability from the T1 to the T0, specifically, a slight increase in the concentration (ng/µL) in the interval I. [9.1;17] kDa and a substantial decrease in the concentration (ng/µL) in the interval II. [17;30] kDa. These results confirm the hypotheses made previously in the MODeLING.Vis for the Δ (T1 − T0) ng/µL of the (B) [9.8;10.3] kDa and (D) [13.7;17.5] kDa MW range.
In the third graph, we can see the overall intragroup homogeneity of the concentration (ng/µL) in the [9.1;30] kDa (the (c) lower MW range), contrastingly to the positive control, plotted in the fourth graph, showing a significant increase in the concentration in this MW range.
Specifically, this augmentation is visible in interval I. [9.1;17] kDa; this augmentation is visible in interval I. [9.1;17] kDa.
In
Figure 7, the same electropherograms are equally shown, but for the five subjects belonging to (3) the Experimental Bottom Subphenome.
In this case, the fourth graph shows T0 + T1 for the positive control, a subject with an ICD 10: J30.9—Allergic Rhinitis, unspecified and nonmedicated.
As is shown in the first two graphs, there is considerable variability from T1 to T0, specifically a significant increase in the concentration (ng/µL) of both intervals I. [9.1;17] kDa and II. [17;30] kDa. In this electropherogram, the atypical protein profile is due to the positive control in the [9.1;17] kDa, and is better demonstrated in the fourth graph, showing a significant decrease in the concentration in this MW range.
These results also confirm the hypothesis made previously in the MODeLING.Vis for the Δ (T1 − T0) ng/µL of the (B) [9.8;10.3] kDa and (D) [13.7;17.5] kDa MW range.
In the third graph, we can also see the overall intragroup homogeneity of the concentration in the [9.1;30] kDa, contrasting with the positive control, plotted in the fourth graph.
Figure 8 shows the five subjects belonging to the (2) Control Top Subphenome. In this situation, the fourth graph shows T0 + T1 for the positive control, a subject with an ICD-10: J45—Asthma, nonmedicated.
As is shown in the first two graphs, there is considerable variability from T1 to T0, specifically a slight decrease in the concentration (ng/µL) in both intervals I. [9.1;17] kDa and II. [17;30] kDa.
These results also confirm the hypothesis made previously in the MODeLING.Vis for the Δ (T1 − T0) ng/µL of the (B) [9.8;10.3] kDa and (D) [13.7;17.5] kDa MW range. In the third graph, we can see the overall intragroup homogeneity of the concentration in the [9.1;30] kDa.
Additionally, the electropherogram of the positive control, plotted in the fourth graph, shows a baseline control concentration in interval II. [17;30] kDa, characteristic of the neuroinflammatory response.
Figure 9 shows the five subjects belonging to the (4) Control Bottom Subphenome. In this condition, the fourth graph shows T0 + T1 for the positive control, which was a subject with an ICD-10: J30.9—Allergic Rhinitis, unspecified, and medicated with a leukotriene receptor antagonist (Singulair
®), a corticosteroid (Pulmicort
®), and a long-acting β2-agonist (Simbicort
®).
As shown in the first two graphs, there is considerable variability from T1 to T0, specifically the significant concentration increase (ng/µL) in interval II. [17;30] kDa.
These results also confirm the hypothesis made previously in the MODeLING.Vis for the Δ (T1 − T0) ng/µL of the B) [9.8;10,3] kDa (which remains constant) and D) [13.7;17.5] kDa (which augments considerably) MW range. The third graph shows a slight overall intragroup heterogeneity in the [9.1;30] kDa MW range compared to the other subgroups.
This heterogeneity is due to the lower clinical score characterizing this subgroup [
17] and, therefore, lower specific molecular print in this MW range.
The electropherogram of the positive control, plotted in the fourth graph, shows a baseline control concentration in interval II. [17;30] kDa, which is also characteristic of the neuroinflammatory response.
After this analysis, we postulate that those five subjects should be chosen to advance for a sequential phase of our molecular screening: simultaneous immune detection.
The excerpt of the research outline (
Figure 10) systemizes the experimental study and helps the reader understand this paper’s sequence and integration in the overall experiment conducted by the authors.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













