Using Reservoir Geology and Petrographic Observations to Improve CO2 Mineralization Estimates: Examples from the Johansen Formation, North Sea, Norway


Abstract
:1. Introduction
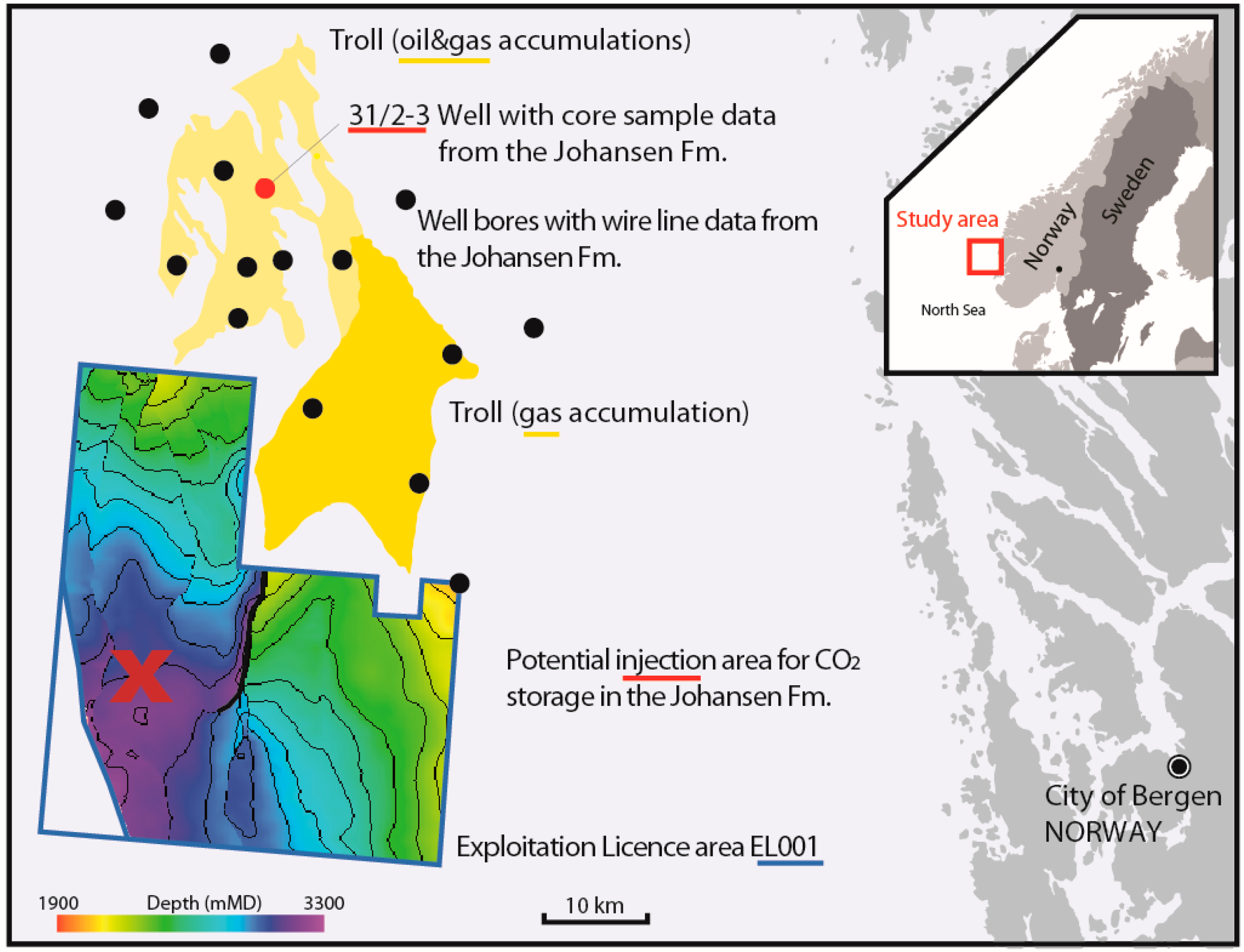
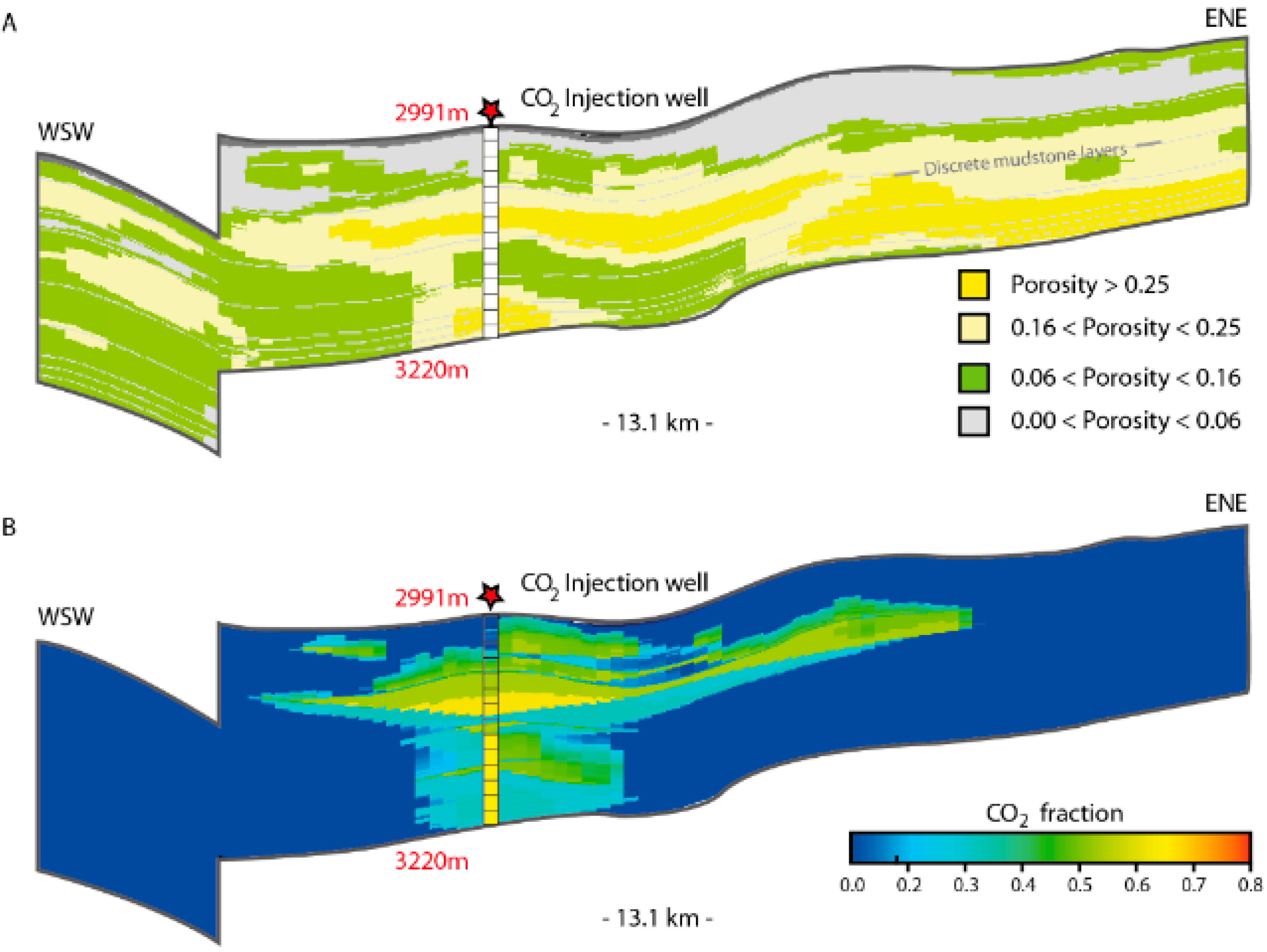
Case Study: The Johansen Formation, North Sea (NORWAY)
2. Estimating CO2 Mineralization Potential (I): Model Parameterization
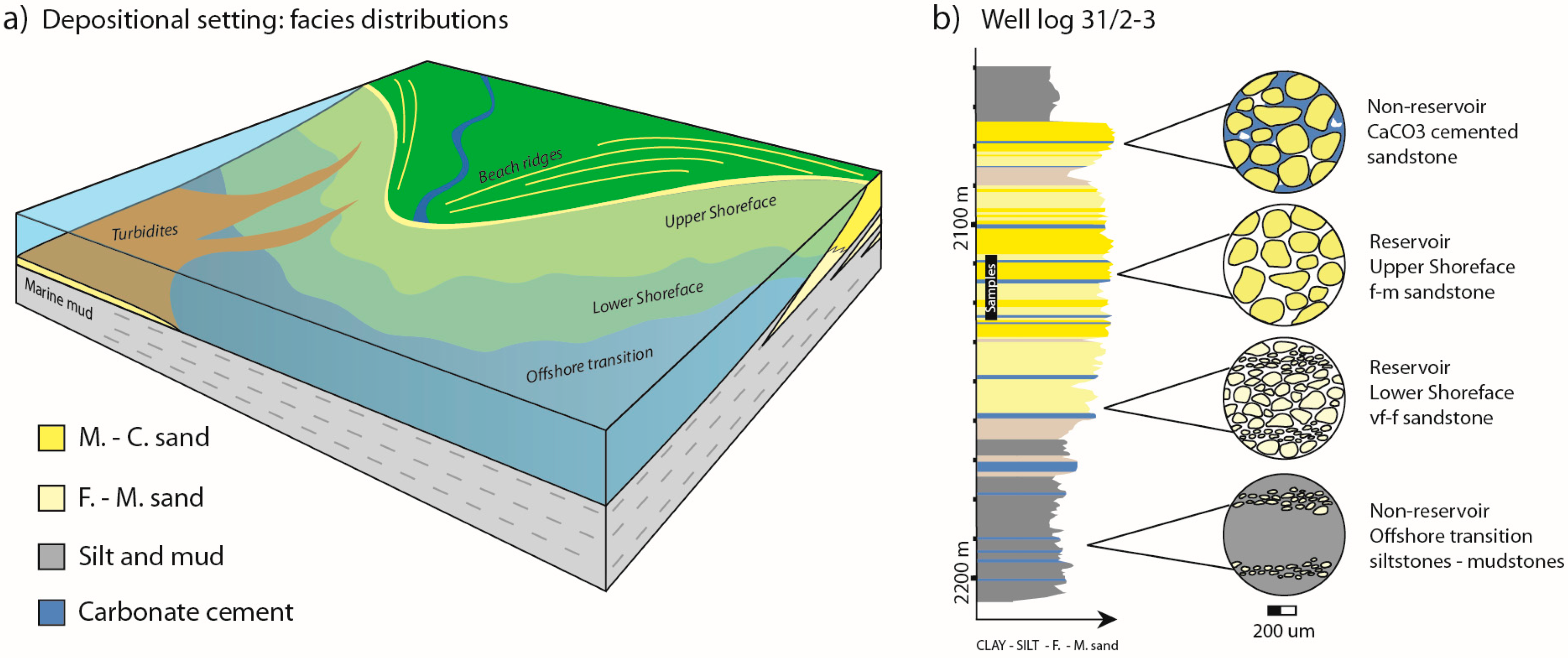
2.1. Qualitative and Quantitative Reservoir Mineralogy
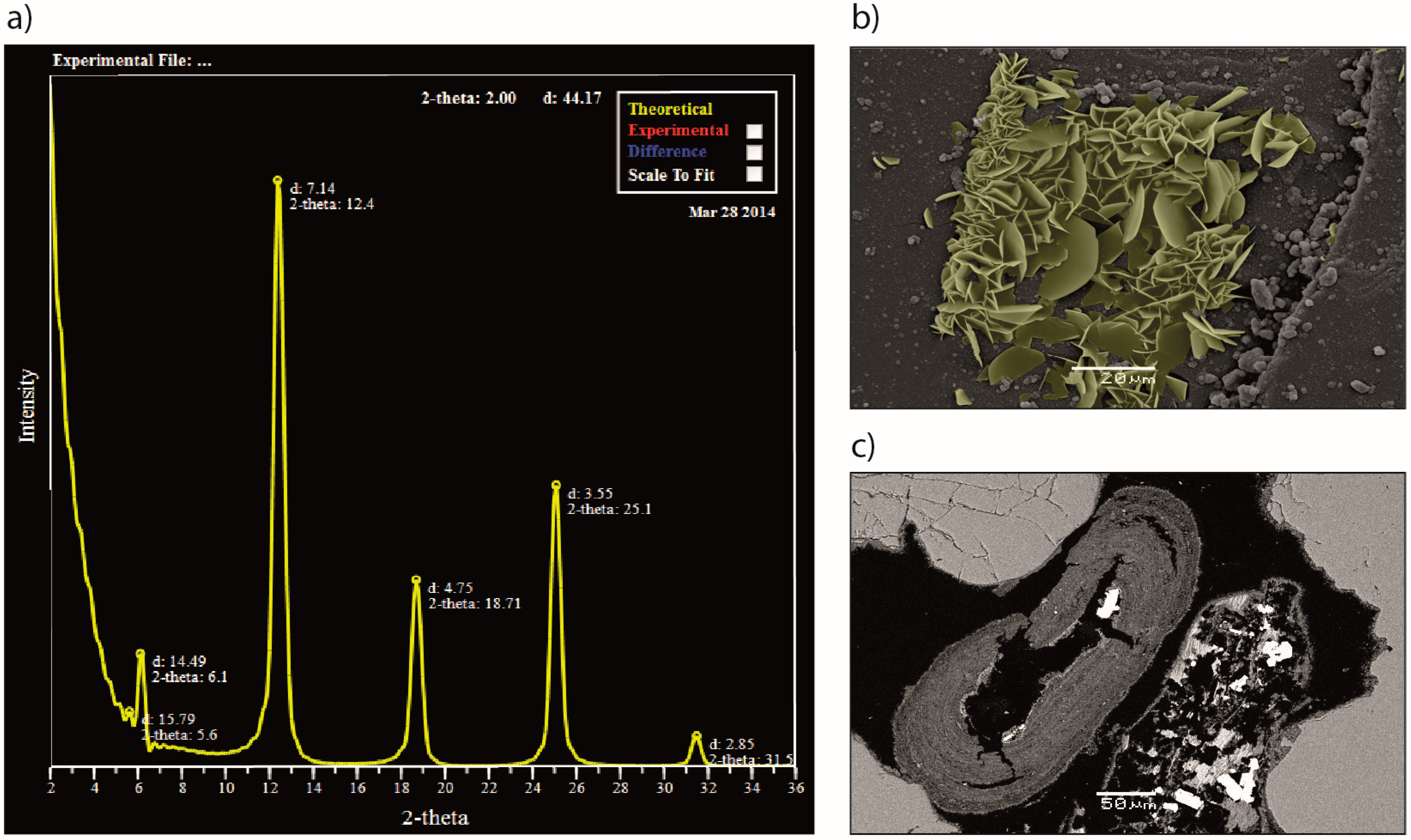
2.1.1. Characterization of Chlorites
2.1.2. Characterization of Feldspars
2.2. Reaction Potential
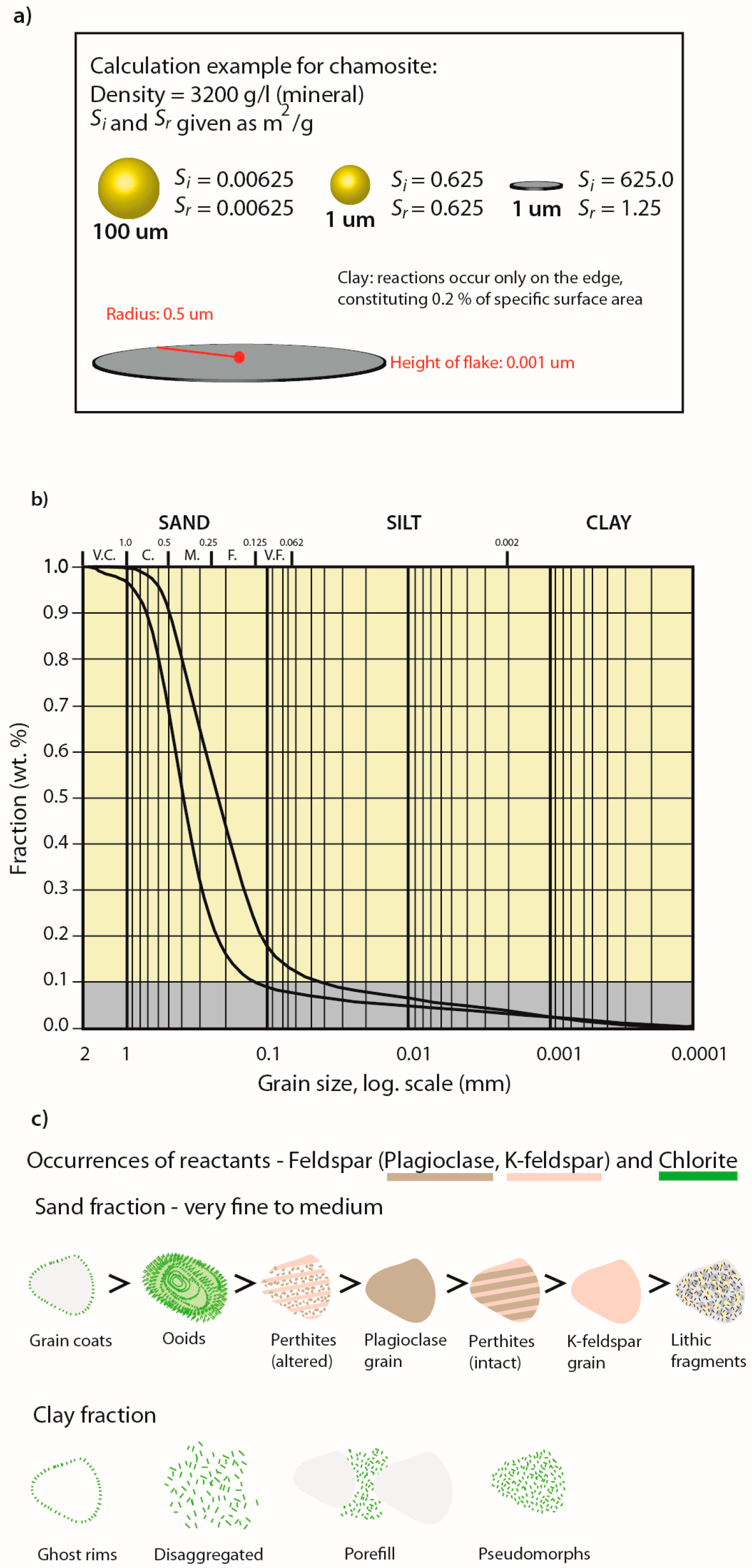
2.3. Reactive Surface Areas
3. Estimating CO2 Mineralization Potential (II): Example Modelling
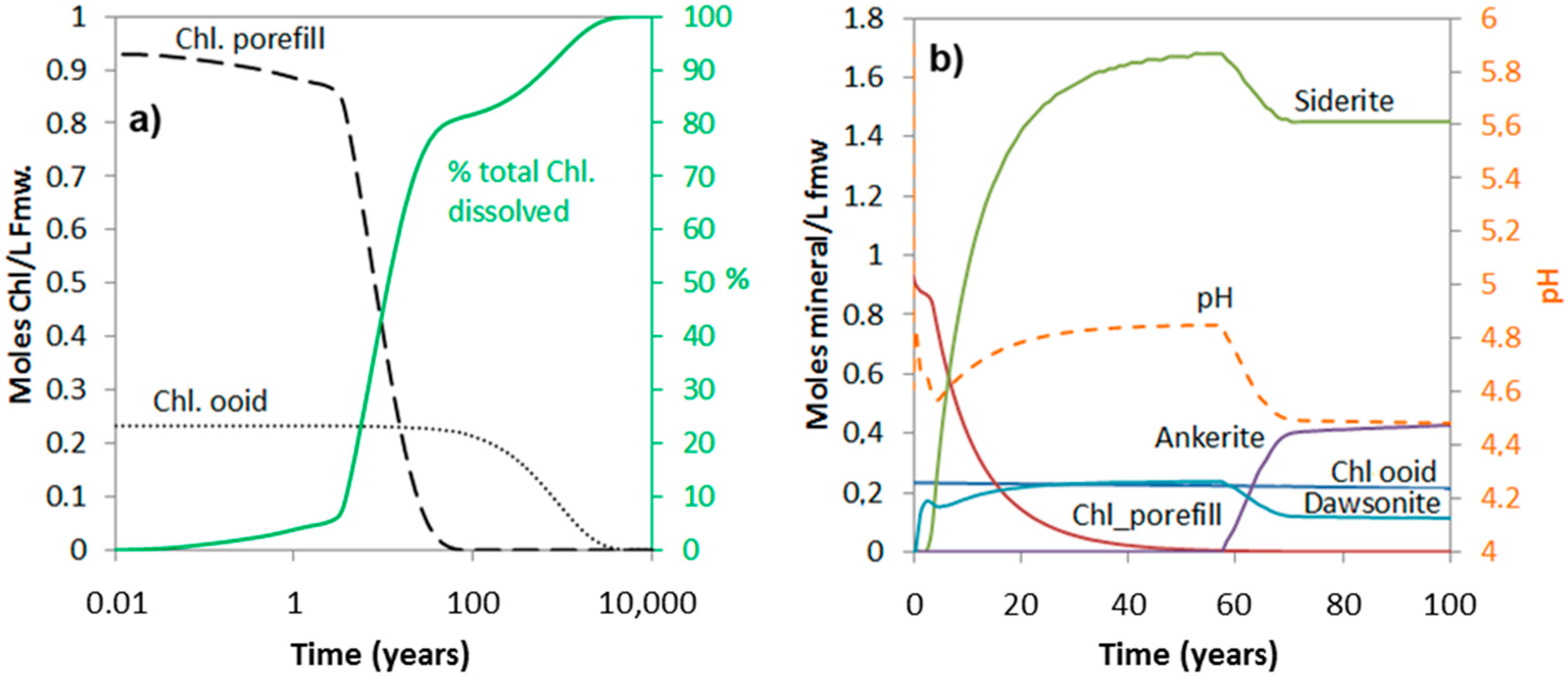
3.1. Carbonatization of Chlorite
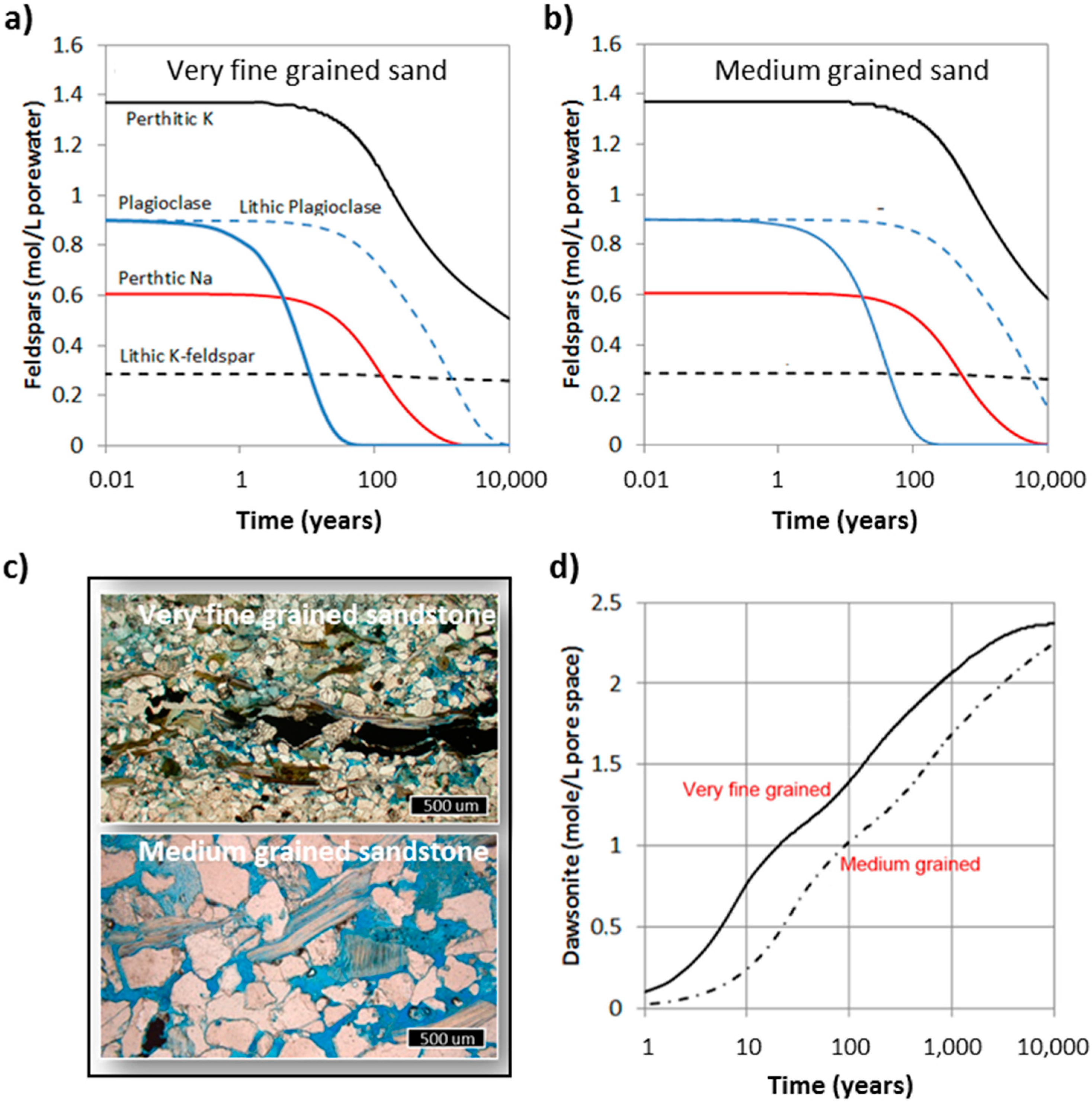
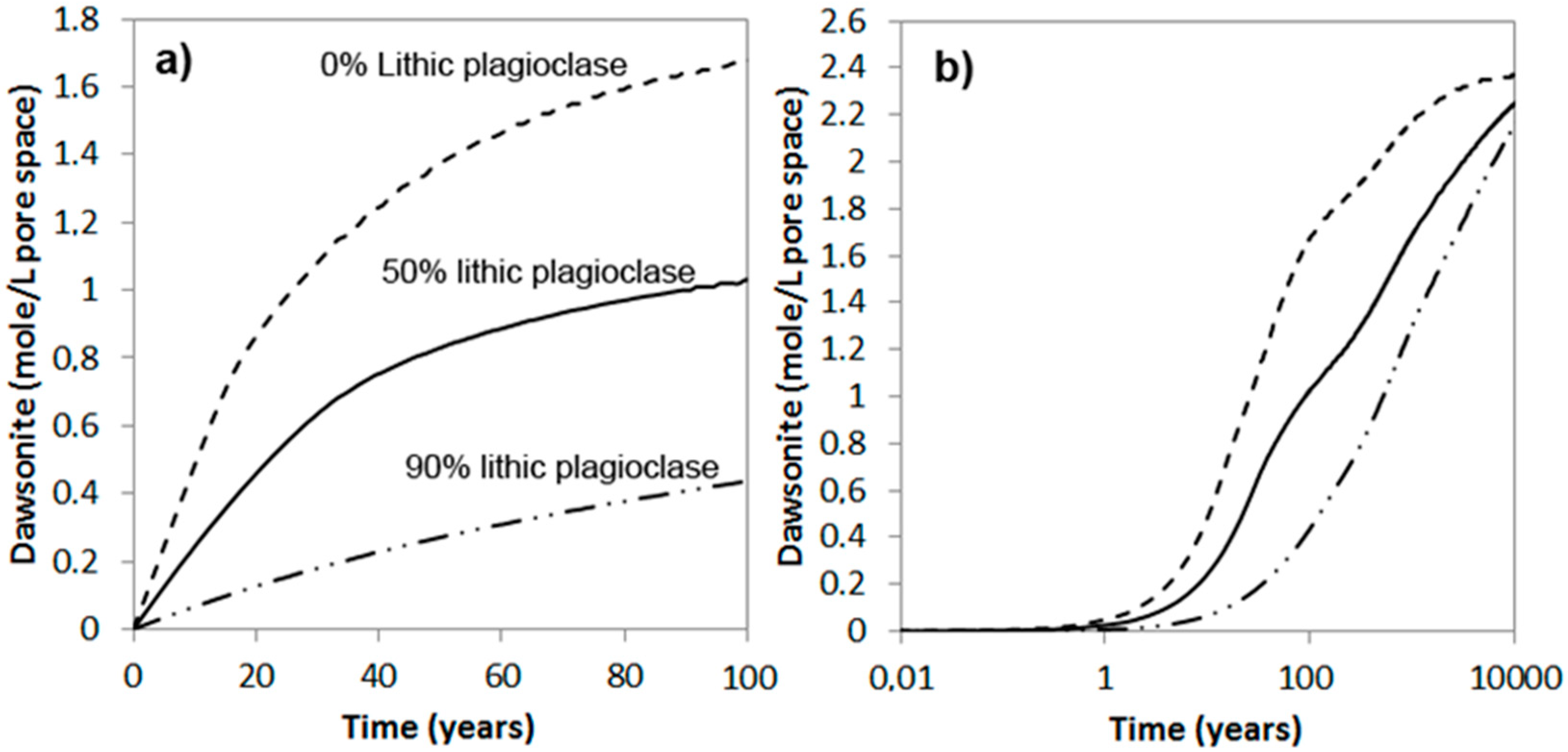
3.2. Carbonatization of Feldspars
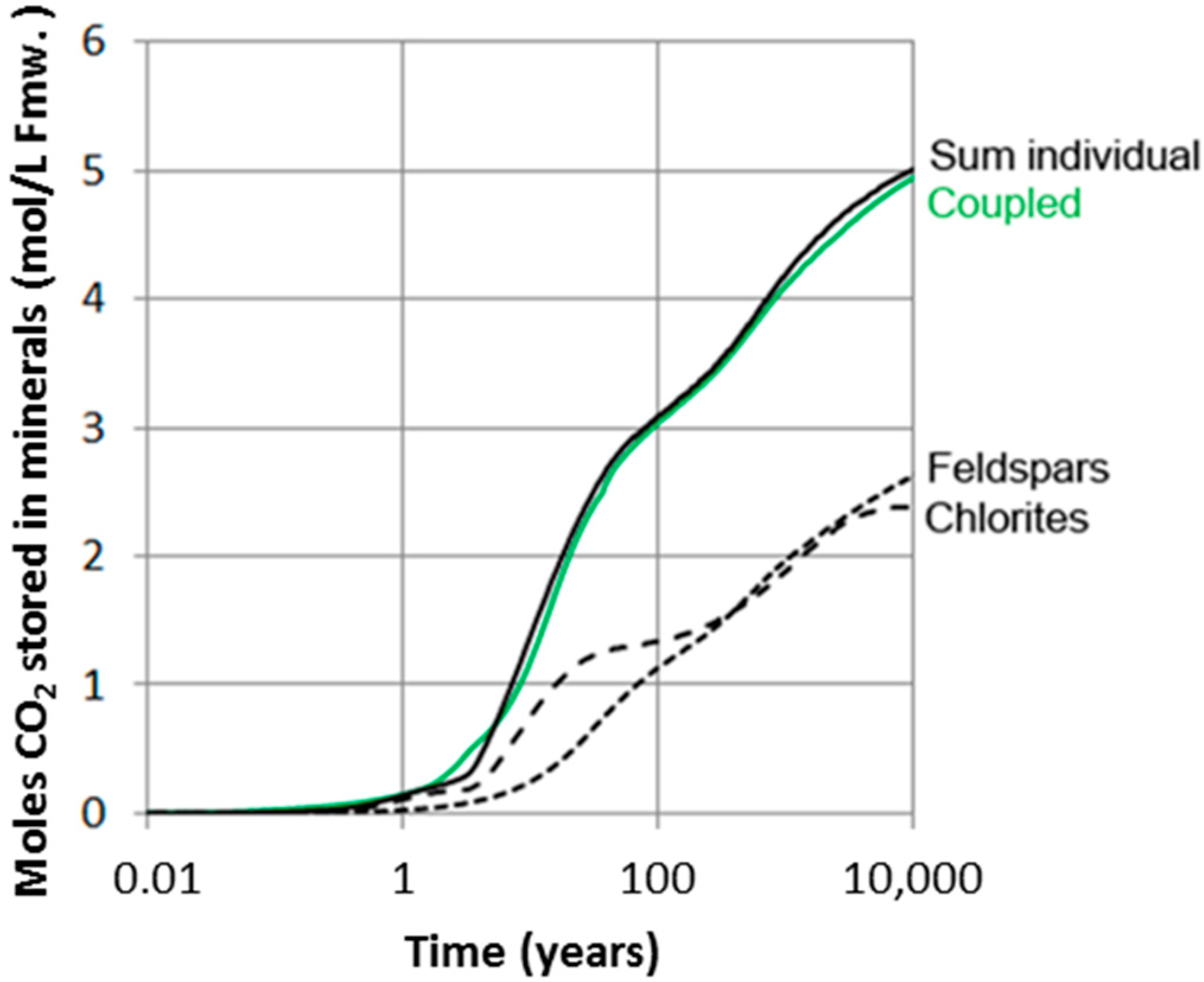
4. Discussion
4.1. Workflow
4.2. Input Data
4.3. Upscaling
4.4. Hydrogeochemical Trapping
4.5. Kinetic Rate Uncertainties for Chlorite
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bachu, S. Sequestration of CO2 in geological media: Criteria and approach for site selection in response to climate change. Energy Convers. Manag. 2000, 41, 953–970. [Google Scholar] [CrossRef]
- Metz, B.; Davidson, O.; De Coninck, H.; Loos, M.; Meyer, L. IPCC Special Report on Carbon Dioxide Capture and Storage; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Michael, K.; Golab, A.; Shulakova, V.; Ennis-King, J.; Allinson, G.; Sharma, S.; Aiken, T. Geological storage of CO2 in saline aquifers—A review of the experience from existing storage operations. Int. J. Greenh. Gas Control 2010, 4, 659–667. [Google Scholar] [CrossRef]
- Halland, E.K.; Johansen, W.T.; Riis, F. CO2 Storage Atlas—Norwegian North Sea; Norwegian Petroleum Directorate: Stavanger, Norway, 2011. [Google Scholar]
- Bachu, S. Screening and ranking of sedimentary basins for sequestration of CO2 in geological media in response to climate change. Environ. Geol. 2003, 44, 277–289. [Google Scholar] [CrossRef]
- Benson, S.M.; Cole, D.R. CO2 sequestration in deep sedimentary formations. Elements 2008, 4, 325–331. [Google Scholar] [CrossRef]
- Xu, T.; Apps, J.A.; Pruess, K. Mineral sequestration of carbon dioxide in a sandstone–shale system. Chem. Geol. 2005, 217, 295–318. [Google Scholar] [CrossRef]
- André, L.; Audigane, P.; Azaroual, M.; Menjoz, A. Numerical modeling of fluid-rock chemical interactions at the supercritical CO2-liquid interface during CO2 injection into a carbonate reservoir, the Dogger aquifer (Paris Basin, France). Energy Convers. Manag. 2007, 48, 1782–1797. [Google Scholar] [CrossRef]
- Audigane, P.; Gaus, I.; Czernichowski-Lauriol, I.; Pruess, K.; Xu, T. Two-dimensional reactive transport modeling of CO2 injection in a saline aquifer at the Sleipner site, North Sea. Am. J. Sci. 2007, 307, 974–1008. [Google Scholar] [CrossRef]
- Pham, V.T.H.; Lu, P.; Aagaard, P.; Zhu, C.; Hellevang, H. On the potential of CO2–water–rock interactions for CO2 storage using a modified kinetic model. Int. J. Greenh. Gas Control 2011, 5, 1002–1015. [Google Scholar] [CrossRef]
- Balashov, V.N.; Guthrie, G.D.; Hakala, J.A.; Lopano, C.L.; Rimstidt, J.D.; Brantley, S.L. Predictive modeling of CO2 sequestration in deep saline sandstone reservoirs: Impacts of geochemical kinetics. Appl. Geochem. 2013, 30, 41–56. [Google Scholar] [CrossRef]
- Kampman, N.; Bickle, M.; Wigley, M.; Dubacq, B. Fluid flow and CO2-fluid-mineral interactions during CO2-storage in sedimentary basins. Chem. Geol. 2014, 369, 22–50. [Google Scholar] [CrossRef]
- White, A.F.; Brantley, S.L. The effect of time on the weathering of silicate minerals: Why do weathering rates differ in the laboratory and field? Chem. Geol. 2003, 202, 479–506. [Google Scholar] [CrossRef]
- White, A.F.; Peterson, M.L. Role of reactive-surface-area characterization in geochemical kinetic models. In Chemical modeling of aqueous systems II. ACS Symp. Ser. 1999, 416, 461–475. [Google Scholar]
- Lüttge, A. Etch pit coalescence, surface area, and overall mineral dissolution rates. Am. Miner. 2005, 90, 1776–1783. [Google Scholar] [CrossRef]
- Bolourinejad, P.; Omrani, P.S.; Herber, R. Effect of reactive surface area of minerals on mineralization and carbon dioxide trapping in a depleted gas reservoir. Int. J. Greenh. Gas Control 2014, 21, 11–22. [Google Scholar] [CrossRef]
- Hellevang, H.; Aagaard, P. Can the long-term potential for carbonatization and safe long-term CO2 storage in sedimentary formations be predicted? Appl. Geochem. 2013, 39, 108–118. [Google Scholar] [CrossRef]
- Hellevang, H.; Pham, V.T.H.; Aagaard, P. Kinetic modelling of CO2-water-rock interactions. Int. J. Greenh. Gas Control 2013, 15, 3–15. [Google Scholar] [CrossRef]
- Gunter, W.D.; Perkins, E.H.; McCann, T.J. Aquifer disposal of CO2-rich gases: Reaction design for added capacity. Energy Convers. Manag. 1993, 34, 941–948. [Google Scholar] [CrossRef]
- Perkins, E.H.; Gunter, W.D. Mineral traps for carbon dioxide. In Aquifer Disposal of Carbon Dioxide, B.; Hitchon, Geoscience Publishing: Alberta, Canada, 1996; pp. 93–113. [Google Scholar]
- Knauss, K.G.; Johnson, J.W.; Steefel, C.I. Evaluation of the impact of CO2, co-contaminant gas, aqueous fluid and reservoir rock interactions on the geologic sequestration of CO2. Chem. Geol. 2005, 217, 339–350. [Google Scholar] [CrossRef]
- Peters, C.A. Accessibilities of reactive minerals in consolidated sedimentary rock: An imaging study of three sandstones. Chem. Geol. 2009, 265, 198–208. [Google Scholar] [CrossRef]
- Landrot, G.; Ajo-Franklin, J.B.; Yang, L.; Cabrini, S.; Steefel, C.I. Measurement of accessible reactive surface area in a sandstone, with application to CO2 mineralization. Chem. Geol. 2018, 318, 113–125. [Google Scholar] [CrossRef]
- Park, J.; Baek, K.; Lee, M.; Chung, C.W.; Wang, S. The use of the surface roughness value to quantify the extent of supercritical CO2 involved geochemical reaction at a CO2 sequestration site. Appl. Sci. 2017, 7, 572. [Google Scholar] [CrossRef]
- Ringrose, P.; Bentley, M. Reservoir Model Design: A Practitioner’s Guide; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Northern Lights: A European CO2 Transport and Storage Project. Available online: https://www.slideshare.net/globalccs/northern-lights-a-european-CO2-transport-and-storage-project (accessed on 28 October 2019).
- Vollset, J.; Doré, A.G. A Revised Triassic and Jurassic lithostratigraphic nomenclature for the Norwegian North Sea. Nor. Pet. Dir. Bull. 1984, 3, 1–33. [Google Scholar]
- Sundal, A.; Miri, R.; Ravn, T.; Aagaard, P. Modelling CO2 migration in aquifers; considering 3D seismic property data and the effect of site-typical depositional heterogeneities. Int. J. Greenh. Gas Control 2015, 39, 349–365. [Google Scholar] [CrossRef]
- Sundal, A.; Nystuen, J.P.; Rørvik, K.L.; Dypvik, H.; Aagaard, P. The Lower Jurassic Johansen Formation, northern North Sea–Depositional model and reservoir characterization for CO2 storage. Mar. Pet. Geol. 2016, 77, 1376–1401. [Google Scholar] [CrossRef]
- Marjanac, T.; Steel, R.J. Dunlin Group sequence stratigraphy in the northern North sea: A model for Cook Sandstone deposition. AAPG bulletin 1997, 81, 276–292. [Google Scholar]
- Dreyer, T.; Whitaker, M.; Dexter, J.; Flesche, H.; Larsen, E. From spit system to tide-dominated delta: Integrated reservoir model of the Upper Jurassic Sognefjord Formation on the Troll West Field. In Proceeding of the Petroleum Geology Conference series, London, UK, 1 January 2005; pp. 423–448. [Google Scholar]
- Holgate, N.E.; Jackson, C.A.L.; Hampson, G.J.; Dreyer, T. Sedimentology and sequence stratigraphy of the middle–upper Jurassic Krossfjord and Fensfjord formations, Troll Field, northern North Sea. Pet. Geosci. 2013, 19, 237–258. [Google Scholar] [CrossRef]
- Aagaard, P.; Jahren, J.S.; Harstad, A.O.; Nilsen, O.; Ramm, M. Formation of grain-coating chlorite in sandstones. Laboratory synthesized vs. natural occurrences. Clay Miner. 2000, 35, 261–269. [Google Scholar] [CrossRef]
- Ehrenberg, S.N. Preservation of anomalously high porosity in deeply buried sandstones by grain-coating chlorite: Examples from the Norwegian continental shelf. AAPG Bulletin 1993, 77, 1260–1286. [Google Scholar]
- Olivarius, M.; Sundal, A.; Weibel, R.; Gregersen, U.; Baig, I.; Thomsen, T.B.; Kristensen, L.; Hellevang, H.; Nielsen, L.H. Provenance and sediment maturity as controls on CO2 mineral sequestration potential of the Gassum Formation in Skagerrak. Front. Sediment. Res. 2019. in review. [Google Scholar]
- Van Wagoner, J.; Posamentier, H.; Mitchum, R.; Vail, P.; Sarg, J.; Loutit, T.; Hardenbol, J. An overview of sequence stratigraphy and key definitions. In Sea Level Changes—An Integrated Approach; Wilgus, C., Hastings, B., Kendall, C.G.S.C., Posamentier, H., Ross, C., Van Wagoner, J., Eds.; SEPM Special Publication: Tulsa, OK, USA, 1988; pp. 39–45. [Google Scholar]
- Van Wagoner, J.C.; Mitchum, R.; Campion, K.; Rahmanian, V. Siliciclastic sequence stratigraphy in well logs, cores, and outcrops: Concepts for high-resolution correlation of time and facies. In Siliciclastic Sequence Stratigraphy in Well Logs, Cores, and Outcrops: Concepts for High-Resolution Correlation of Time and Facies; American Association of Petroleum Geologists: Tulsa, OK, USA, 1990; pp. 3–55. [Google Scholar]
- Hillier, S. Accurate quantitative analysis of clay and other minerals in sandstones by XRD: Comparison of a Rietveld and a reference intensity ratio (RIR) method and the importance of sample preparation. Clay Miner. 2000, 35, 291–302. [Google Scholar] [CrossRef]
- Johnson, J.W.; Nitao, J.J.; Knauss, K.G. Reactive transport modeling of CO2 storage in saline aquifers to elucidate fundamental processes, trapping mechanisms and sequestration partitioning. In Geological Storage of Carbon Dioxide; Bains, S.J., Worden, R.H., Eds.; Geological Society Special Publications: London, UK, 2004; pp. 107–128. [Google Scholar]
- Deer, W.A.; Howie, R.A.; Zussman, J. Rock Forming Minerals: Layered Silicates Excluding Micas and Clay Minerals; Geological Society: London, UK, 2009. [Google Scholar]
- Hillier, S. Pore-lining chlorites in siliciclastic reservoir sandstones: Electron microprobe, SEM and XRD data, and implications for their origin. Clay Miner. 1994, 29, 665–680. [Google Scholar] [CrossRef]
- Hillier, S.; Fallick, A.; Matter, A. Origin of pore-lining chlorite in the aeolian Rotliegend of northern Germany. Clay Miner. 1996, 31, 153–171. [Google Scholar] [CrossRef]
- Jahren, J.; Aagaard, P. Compositional variations in diagenetic chlorites and illites, and relationships with formation-water chemistry. Clay Miner. 1989, 24, 157–170. [Google Scholar] [CrossRef]
- Maast, T.E. Reservoir Quality of Deeply Buried Sandstones—A Study of Burial Diagenesis from the North Sea. Ph.D. Thesis, University of Oslo, Oslo, Norway, 2013. [Google Scholar]
- Jahren, J.; Aagaard, P. Diagenetic illite-chlorite assemblages in arenites. I. Chemical evolution. Clays Clay Miner. 1992, 40, 540. [Google Scholar] [CrossRef]
- Palandri, J.L.; Kharaka, Y.K. A Compilation of Rate Parameters of Water-Mineral Interaction Kinetics for Application to Geochemical Modeling; OPEN-FILE-2004-1068; US Geological Survey: Menlo Park, CA, USA, 2004.
- Critelli, T.; Marini, L.; Schott, J.; Mavromatis, V.; Apollaro, C.; Rinder, T.; De Rosa, R.; Oelkers, E.H. Dissolution rates of actinolite and chlorite from a whole-rock experimental study of metabasalt dissolution from 2 ≤ pH ≤ 12 at 25 °C. Chem. Geol. 2014, 390, 100–108. [Google Scholar] [CrossRef]
- Smith, M.M.; Wolery, T.J.; Carroll, S.A. Kinetics of chlorite dissolution at elevated temperatures and CO2 conditions. Chem. Geol. 2013, 347, 1–8. [Google Scholar] [CrossRef]
- Smith, M.M.; Carroll, S.A. Chlorite dissolution kinetics at pH 3–10 and temperature to 275 °C. Chem. Geol. 2016, 421, 55–64. [Google Scholar] [CrossRef]
- Lowson, R.T.; Comarmond, M.-C.J.; Rajaratnam, G.; Brown, P.L. The kinetics of the dissolution of chlorite as a function of pH and at 25 °C. Geochim. Cosmochim. Acta 2005, 69, 1687–1699. [Google Scholar] [CrossRef]
- Bachu, S.; Bennion, B. Effects of in-situ conditions on relative permeability characteristics of CO2-brine systems. Environ. Geol. 2008, 54, 1707–1722. [Google Scholar] [CrossRef]
- Morad, S. Feldspars in sedimentary rocks. In Encyclopedia of Sediments and Sedimentary Rocks; Middleton, G.V., Church, M.J., Coniglio, M., Hardie, L.A., Longstaffe, F.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Aagaard, P.; Egeberg, P.K.; Saigal, G.C.; Morad, S.; Bjørlykke, K. Diagenetic albitization of detrital K-feldspars in Jurassic, Lower Cretaceous and Tertiary clastic reservoir rocks from offshore Norway; II, Formation water chemistry and kinetic considerations. J. Sediment. Res. 1990, 60, 575–581. [Google Scholar] [CrossRef]
- Warren, E.A.; Smalley, P.C. North Sea formation waters atlas. Oceanogr. Lit. Rev. 1995, 6, 471. [Google Scholar]
- Bachu, S.; Gunter, W.D.; Perkins, E.H. Aquifer disposal of CO2: Hydrodynamic and mineral trapping. Energy Convers. Manag. 1994, 35, 269–279. [Google Scholar] [CrossRef]
- Wawersik, W.R.; Rudnicki, J.W.; Dove, P.; Harris, J.; Logan, J.M.; Pyrak-Nolte, L.; Orr, F.M.; Ortoleva, P.J.; Richter, F.; Warpinski, N.R.; et al. Terrestrial sequestration of CO2: An assessment of research needs. Adv. Geophys. 2001, 43, 97–177. [Google Scholar]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3: A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; U.S. Geological Survey: Reston, VA, USA, 2013; pp. 1–497.
- Hellevang, H.; Declercq, J.; Kvamme, B.; Aagaard, P. The dissolution rates of dawsonite at pH 0.9 to 5 and temperatures of 22, 60 and 77 °C. Appl. Geochem. 2010, 25, 1575–1586. [Google Scholar] [CrossRef]
- Hellevang, H.; Aagaard, P.; Jahren, J. Will dawsonite form during CO2 storage? Greenh. Gases: Sci. Technol. 2014, 4, 191–199. [Google Scholar] [CrossRef]
- Tambach, T.J.; Hellevang, H. Discussion on the paper titled “Effect of reactive surface area of minerals on mineralization and carbon dioxide trapping in a depleted gas reservoir” by Bolourinejad et al. (2014). Int. J. Greenh. Gas Control 2015, 100, 141–143. [Google Scholar] [CrossRef]
- Sundal, A.; Hellevang, H.; Miri, R.; Dypvik, H.; Nystuen, J.P.; Aagaard, P. Variations in mineralization potential for CO2 related to sedimentary facies and burial depth–a comparative study from the North Sea. Energy Procedia 2014, 63, 5063–5070. [Google Scholar] [CrossRef]
- Förster, A.; Schöner, R.; Förster, H.J.; Norden, B.; Blaschke, A.W.; Luckert, J.; Beutler, G.; Gaupp, R.; Rhede, D. Reservoir characterization of a CO2 storage aquifer: The Upper Triassic Stuttgart Formation in the Northeast German Basin. Mar. Pet. Geol. 2010, 27, 2156–2172. [Google Scholar] [CrossRef]
- Miri, R.; Aagaard, P.; Hellevang, H. Examination of CO2-SO2 solubility in water by SAFT1. Implications for CO2 transport and storage. J. Phys. Chem. 2014, 118, 10214–10223. [Google Scholar] [CrossRef]
- Hovorka, S.D.; Doughty, C.; Benson, S.M.; Pruess, K.; Knox, P.R. The impact of geological heterogeneity on CO2 storage in brine formations: A case study from the Texas Gulf Coast. Geol. Soc. 2004, 233, 147–163. [Google Scholar] [CrossRef]
- Gaus, I. Role and impact of CO2-rock interactions during CO2 storage in sedimentary rocks. Int. J. Greenh. Gas Control 2010, 4, 73–89. [Google Scholar] [CrossRef]
- Bachu, S. Drainage and Imbibition CO2/Brine Relative Permeability Curves at in Situ Conditions for Sandstone Formations in Western Canada. Energy Procedia 2013, 37, 4428–4436. [Google Scholar] [CrossRef]
- Lowson, R.T.; Brown, P.L.; Comarmond, M.C.J.; Rajaratnam, G. 2007. The kinetics of chlorite dissolution. Geochim. Cosmochim. Acta 2007, 71, 1431–1447. [Google Scholar] [CrossRef]
- Black, J.R.; Haese, R.R. Chlorite dissolution rates under CO2 saturated conditions from 50 to 120 °C and 120 to 200 bar CO2. Geochim. Cosmochim. Acta 2014, 125, 225–240. [Google Scholar] [CrossRef]
- Aagaard, P.; Oelkers, E.H.; Schott, J. Glauconite dissolution kinetics and application to CO2 storage in the subsurface. Geochim. Cosmochim. Acta 2004, 68, A143. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T | pH | Na | K | Ca | Mg | Fe | Al | Cl | Alk | Si | O | O2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| °C | ppm | ppm | ppm | ppm | ppm | ppm | ppm | meq | ppm | ppm | Log P | |
| 96 | 5.9 | 7544 | 113 | 890 | 53 | 0 | 1 × 10−8 | 13,187 | 293 | 1 × 10−4 | 1 × 10−3 | −50 |
| Mineral | log k+ T = 25 pH = 0 | Ea+ (1) | N (2) | log k- (3) | Ea- |
|---|---|---|---|---|---|
| Albite | −10.2 | 65.0 | 0.46 | log k+ −2 | Ea+ |
| Oligoclase | −9.7 | 65.0 | 0.46 | log k+ −2 | Ea+ |
| K-feldspar | −10.1 | 52.0 | 0.5 | log k+ −2 | Ea+ |
| Chamosite 7A | −9.8 | 22.0 | 0.53 | log k+ −2 | Ea+ |
| Quartz/Chalcedony | EQUIL | Ea+ | |||
| Calcite | EQUIL | Ea+ | |||
| Kaolinite | EQUIL | Ea+ | |||
| Dawsonite(4) | −4.5 | 63.8 | 0.98 | log k+ −2 | Ea+ |
| Siderite | −7.5 | 48.0 | 0.94 | log k+ −2 | Ea+ |
| Ankerite (5) | −3.2 | 56.7 | 0.5 | log k+ −2 | Ea+ |
| Study | CO2 (1) | pH | T | log k (2) | Ea | N (3) | log k | log k |
|---|---|---|---|---|---|---|---|---|
| range | °C | (kJ/mol) | at 37 °C, pH 5 | at 75 °C, pH 5 | ||||
| Clinochlore-14A | 0 | Acidic | – | –11.1 | 88.0 | 0.5 | –13.0 | –10.6 |
| Fe-rich (Mg/Fe = 1.4) | 0 | Acidic | 25–95 | –9.8 | 94.3 | 0.49 | –11.6 | –9.1 |
| Clinochlore-14A | 0.1–0.5 M | 3.0–5.7 | 100–275 | –9.9 | 25.1 | 0.49 | –12.2 | –11.5 |
| Clinochlore-14A | 120–200 M | 3.5–5.4 | 50–120 | –12.0 | 16.0 | 0.076 | –12.3 | –11.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundal, A.; Hellevang, H. Using Reservoir Geology and Petrographic Observations to Improve CO2 Mineralization Estimates: Examples from the Johansen Formation, North Sea, Norway. Minerals 2019, 9, 671. https://0-doi-org.brum.beds.ac.uk/10.3390/min9110671
Sundal A, Hellevang H. Using Reservoir Geology and Petrographic Observations to Improve CO2 Mineralization Estimates: Examples from the Johansen Formation, North Sea, Norway. Minerals. 2019; 9(11):671. https://0-doi-org.brum.beds.ac.uk/10.3390/min9110671
Chicago/Turabian StyleSundal, Anja, and Helge Hellevang. 2019. "Using Reservoir Geology and Petrographic Observations to Improve CO2 Mineralization Estimates: Examples from the Johansen Formation, North Sea, Norway" Minerals 9, no. 11: 671. https://0-doi-org.brum.beds.ac.uk/10.3390/min9110671