
NGLY1 Deficiency: A Rare Newly Described Condition with a Typical Presentation

 , , , , and
, , , , and 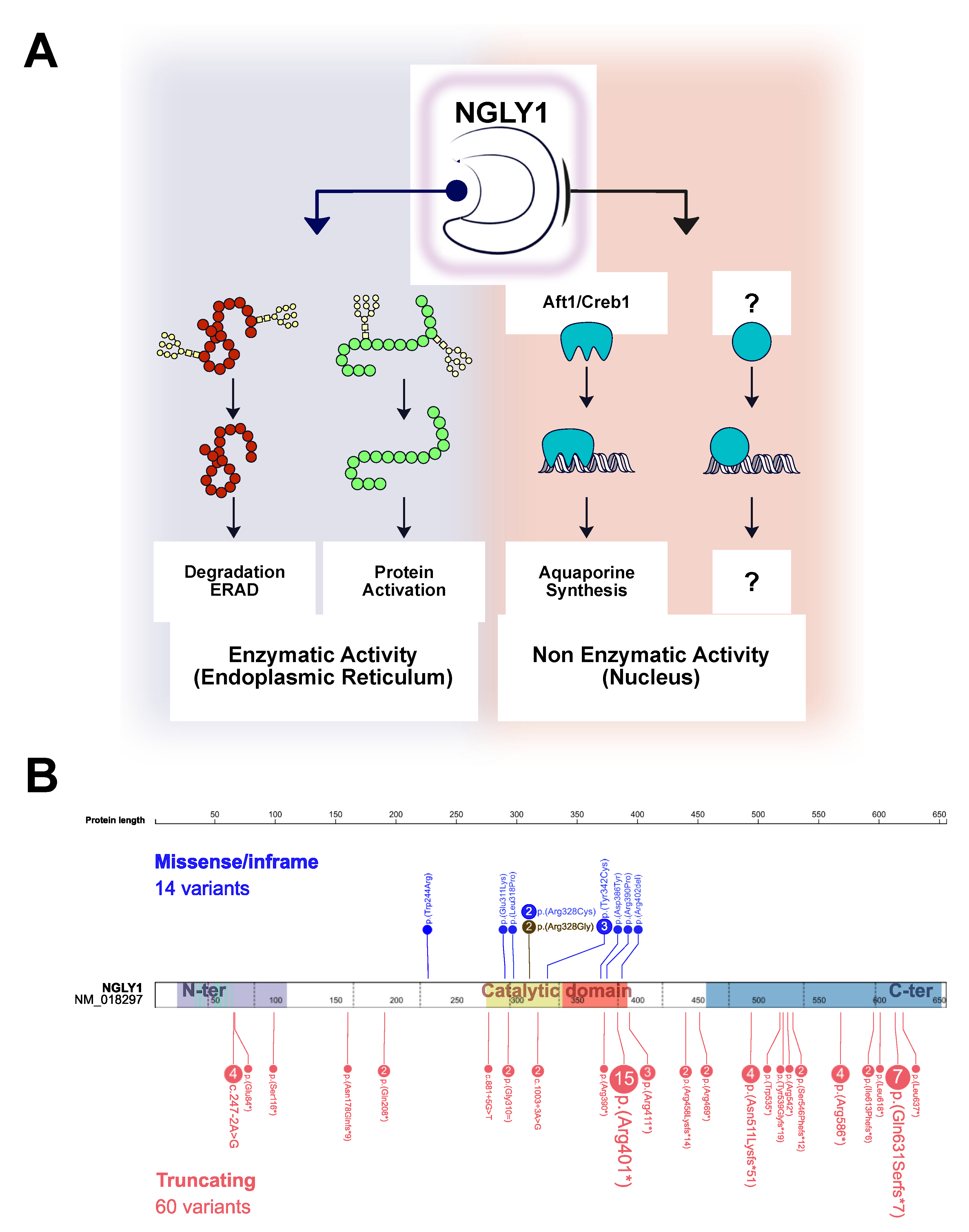{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Patients and Methods
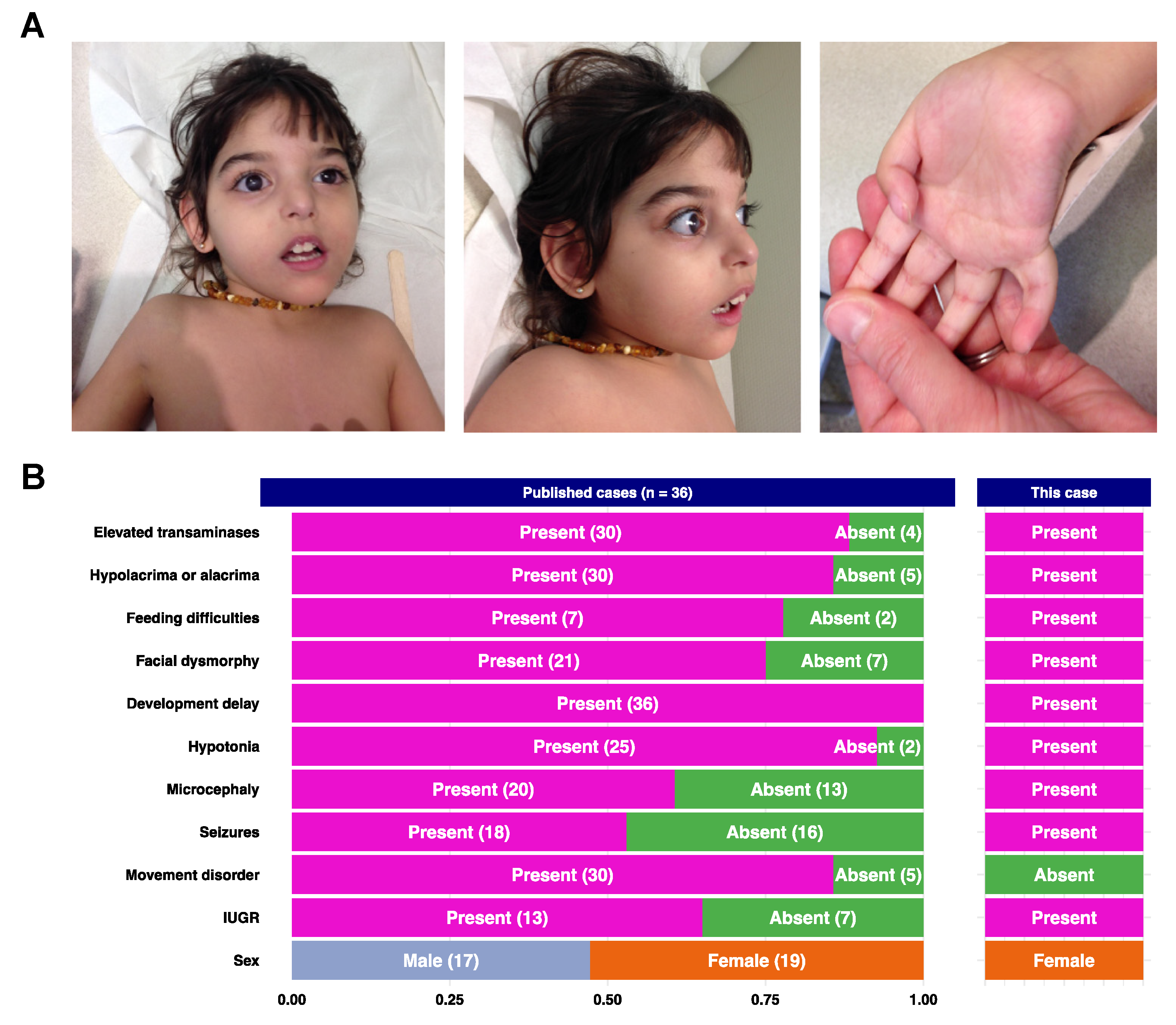
2.1. Case Description
2.2. Whole Exome Sequencing
2.3. Urine Oligosaccharide Screening
2.4. Literature Review
3. Results
3.1. Molecular and Biochemical Findings
3.2. Literature Review
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lam, C.; Ferreira, C.; Krasnewich, D.; Toro, C.; Latham, L.; Zein, W.M.; Lehky, T.; Brewer, C.; Baker, E.H.; Thurm, A.; et al. Prospective phenotyping of ngly1-cddg, the first congenital disorder of deglycosylation. Genet. Med. 2017, 19, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Park, H.; Hollingsworth, N.M.; Sternglanz, R.; Lennarz, W.J. Png1, a yeast gene encoding a highly conserved peptide:N-glycanase. J. Cell Biol. 2000, 149, 1039–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Need, A.C.; Shashi, V.; Hitomi, Y.; Schoch, K.; Shianna, K.V.; McDonald, M.T.; Meisler, M.H.; Goldstein, D.B. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 2012, 49, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Abuduxikuer, K.; Zou, L.; Wang, L.; Chen, L.; Wang, J.S. Novel ngly1 gene variants in chinese children with global developmental delay, microcephaly, hypotonia, hypertransaminasemia, alacrimia, and feeding difficulty. J. Hum. Genet. 2020, 65, 387–396. [Google Scholar] [CrossRef]
- Caglayan, A.O.; Comu, S.; Baranoski, J.F.; Parman, Y.; Kaymakçalan, H.; Akgumus, G.T.; Caglar, C.; Dolen, D.; Erson-Omay, E.Z.; Harmanci, A.S.; et al. Ngly1 mutation causes neuromotor impairment, intellectual disability, and neuropathy. Eur. J. Med. Genet. 2015, 58, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.A.; Wei, X.C.; Martin, S.R.; Sinasac, D.S.; Al-Hertani, W. Transiently elevated plasma methionine, s-adenosylmethionine and s-adenosylhomocysteine: Unreported laboratory findings in a patient with ngly1 deficiency, a congenital disorder of deglycosylation. JIMD Rep. 2019, 49, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Enns, G.M.; Shashi, V.; Bainbridge, M.; Gambello, M.J.; Zahir, F.R.; Bast, T.; Crimian, R.; Schoch, K.; Platt, J.; Cox, R.; et al. Mutations in ngly1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet. Med. 2014, 16, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Haijes, H.A.; de Sain-van der Velden, M.G.M.; Prinsen, H.; Willems, A.P.; van der Ham, M.; Gerrits, J.; Couse, M.H.; Friedman, J.M.; van Karnebeek, C.D.M.; Selby, K.A.; et al. Aspartylglycosamine is a biomarker for ngly1-cddg, a congenital disorder of deglycosylation. Mol. Genet. Metab. 2019, 127, 368–372. [Google Scholar] [CrossRef]
- Lipari Pinto, P.; Machado, C.; Janeiro, P.; Dupont, J.; Quintas, S.; Sousa, A.B.; Gaspar, A. Ngly1 deficiency-a rare congenital disorder of deglycosylation. JIMD Rep. 2020, 53, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Rios-Flores, I.M.; Bonal-Pérez, M.; Castellanos-González, A.; Velez-Gómez, E.; Bertoli-Avella, A.M.; Bobadilla-Morales, L.; Peña-Padilla, C.; Appendini-Andrade, V.; Corona-Rivera, A.; Romero-Valenzuela, I.; et al. Acute liver failure in a male patient with ngly1-congenital disorder of deglycosylation. Eur. J. Med. Genet. 2020, 63, 103952. [Google Scholar] [CrossRef]
- van Keulen, B.J.; Rotteveel, J.; Finken, M.J.J. Unexplained death in patients with ngly1 mutations may be explained by adrenal insufficiency. Physiol. Rep. 2019, 7, e13979. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T. The cytoplasmic peptide:N-glycanase (ngly1)-basic science encounters a human genetic disorder. J. Biochem. 2015, 157, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Peng, M.; Ostrovsky, J.; Kwon, Y.J.; Oretsky, O.; McCormick, E.M.; He, M.; Argon, Y.; Falk, M.J. Mitochondrial function requires ngly1. Mitochondrion 2018, 38, 6–16. [Google Scholar] [CrossRef]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordoñez-Rueda, D.; et al. Inhibition of ngly1 inactivates the transcription factor nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Huang, R.; Fujihira, H.; Suzuki, T.; Yan, N. N-glycanase ngly1 regulates mitochondrial homeostasis and inflammation through nrf1. J. Exp. Med. 2018, 215, 2600–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tambe, M.A.; Ng, B.G.; Freeze, H.H. N-glycanase 1 transcriptionally regulates aquaporins independent of its enzymatic activity. Cell Rep. 2019, 29, 4620–4631.e4. [Google Scholar] [CrossRef] [Green Version]
- Ishii, N. Evaluation of endo-β-n-acetylglucosaminidase activity based on a fret-quenching system. Trends Glycosci. Glycotechnol. 2020, 32, E105–E108. [Google Scholar] [CrossRef]
- Bi, Y.; Might, M.; Vankayalapati, H.; Kuberan, B. Repurposing of proton pump inhibitors as first identified small molecule inhibitors of endo-β-n-acetylglucosaminidase (engase) for the treatment of ngly1 deficiency, a rare genetic disease. Bioorganic Med. Chem. Lett. 2017, 27, 2962–2966. [Google Scholar] [CrossRef] [PubMed]
- Marguet, F.; Laquerrière, A.; Goldenberg, A.; Guerrot, A.M.; Quenez, O.; Flahaut, P.; Vanhulle, C.; Dumant-Forest, C.; Charbonnier, F.; Vezain, M.; et al. Clinical and pathologic features of aicardi-goutières syndrome due to an ifih1 mutation: A pediatric case report. Am. J. Med. Genet. A 2016, 170, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Grangeon, L.; Wallon, D.; Charbonnier, C.; Quenez, O.; Richard, A.-C.; Rousseau, S.; Budowski, C.; Lebouvier, T.; Corbille, A.-G.; Vidailhet, M. Biallelic myorg mutation carriers exhibit primary brain calcification with a distinct phenotype. Brain 2019, 142, 1573–1586. [Google Scholar] [CrossRef]
- Quenez, O.; Cassinari, K.; Coutant, S.; Lecoquierre, F.; Le Guennec, K.; Rousseau, S.; Richard, A.-C.; Vasseur, S.; Bouvignies, E.; Bou, J. Detection of copy-number variations from ngs data using read depth information: A diagnostic performance evaluation. Eur. J. Hum. Genet. 2020, 29, 99–109. [Google Scholar] [CrossRef]
- Xia, B.; Asif, G.; Arthur, L.; Pervaiz, M.A.; Li, X.; Liu, R.; Cummings, R.D.; He, M. Oligosaccharide analysis in urine by maldi-tof mass spectrometry for the diagnosis of lysosomal storage diseases. Clin. Chem. 2013, 59, 1357–1368. [Google Scholar] [CrossRef] [Green Version]
- Hall, P.L.; Lam, C.; Alexander, J.J.; Asif, G.; Berry, G.T.; Ferreira, C.; Freeze, H.H.; Gahl, W.A.; Nickander, K.K.; Sharer, J.D.; et al. Urine oligosaccharide screening by maldi-tof for the identification of ngly1 deficiency. Mol. Genet. Metab. 2018, 124, 82–86. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ge, H.; Wu, Q.; Lu, H.; Huang, Y.; Zhou, T.; Tan, D.; Zhong, Q. Two novel compound heterozygous mutations in ngly1as a cause of congenital disorder of deglycosylation: A case presentation. BMC Med. Genet. 2020, 21, 135. [Google Scholar] [CrossRef]
- Heeley, J.; Shinawi, M. Multi-systemic involvement in ngly1-related disorder caused by two novel mutations. Am. J. Med. Genet. A 2015, 167, 816–820. [Google Scholar] [CrossRef]
- Lipiński, P.; Bogdańska, A.; Różdżyńska-Świątkowska, A.; Wierzbicka-Rucińska, A.; Tylki-Szymańska, A. Ngly1 deficiency: Novel patient, review of the literature and diagnostic algorithm. JIMD Rep. 2020, 51, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Panneman, D.M.; Wortmann, S.B.; Haaxma, C.A.; van Hasselt, P.M.; Wolf, N.I.; Hendriks, Y.; Küsters, B.; van Emst-de Vries, S.; van de Westerlo, E.; Koopman, W.J.H.; et al. Variants in ngly1 lead to intellectual disability, myoclonus epilepsy, sensorimotor axonal polyneuropathy and mitochondrial dysfunction. Clin. Genet. 2020, 97, 556–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahan, E.M.; Frick, S.L. Orthopaedic phenotyping of ngly1 deficiency using an international, family-led disease registry. Orphanet J. Rare Dis. 2019, 14, 148. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabaj, I.; Sudrié-Arnaud, B.; Lecoquierre, F.; Raymond, K.; Ducatez, F.; Guerrot, A.-M.; Snanoudj, S.; Coutant, S.; Saugier-Veber, P.; Marret, S.; et al. NGLY1 Deficiency: A Rare Newly Described Condition with a Typical Presentation. Life 2021, 11, 187. https://0-doi-org.brum.beds.ac.uk/10.3390/life11030187
Dabaj I, Sudrié-Arnaud B, Lecoquierre F, Raymond K, Ducatez F, Guerrot A-M, Snanoudj S, Coutant S, Saugier-Veber P, Marret S, et al. NGLY1 Deficiency: A Rare Newly Described Condition with a Typical Presentation. Life. 2021; 11(3):187. https://0-doi-org.brum.beds.ac.uk/10.3390/life11030187
Chicago/Turabian StyleDabaj, Ivana, Bénédicte Sudrié-Arnaud, François Lecoquierre, Kimiyo Raymond, Franklin Ducatez, Anne-Marie Guerrot, Sarah Snanoudj, Sophie Coutant, Pascale Saugier-Veber, Stéphane Marret, and et al. 2021. "NGLY1 Deficiency: A Rare Newly Described Condition with a Typical Presentation" Life 11, no. 3: 187. https://0-doi-org.brum.beds.ac.uk/10.3390/life11030187