
Bacterial Diversity and Community Structure of a Municipal Solid Waste Landfill: A Source of Lignocellulolytic Potential




Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sites and Sampling
2.2. Physicochemical Properties
2.3. DNA Extraction and Sequencing
2.4. PCR Amplification and Sequencing
2.5. Next-Generation Sequencing and Data Analysis
3. Results
3.1. Physicochemical Characteristics
3.2. Bacterial Analysis
3.3. Bacterial Taxonomy Composition
3.4. Correlation between Bacterial Communities and Soil Physicochemical Properties
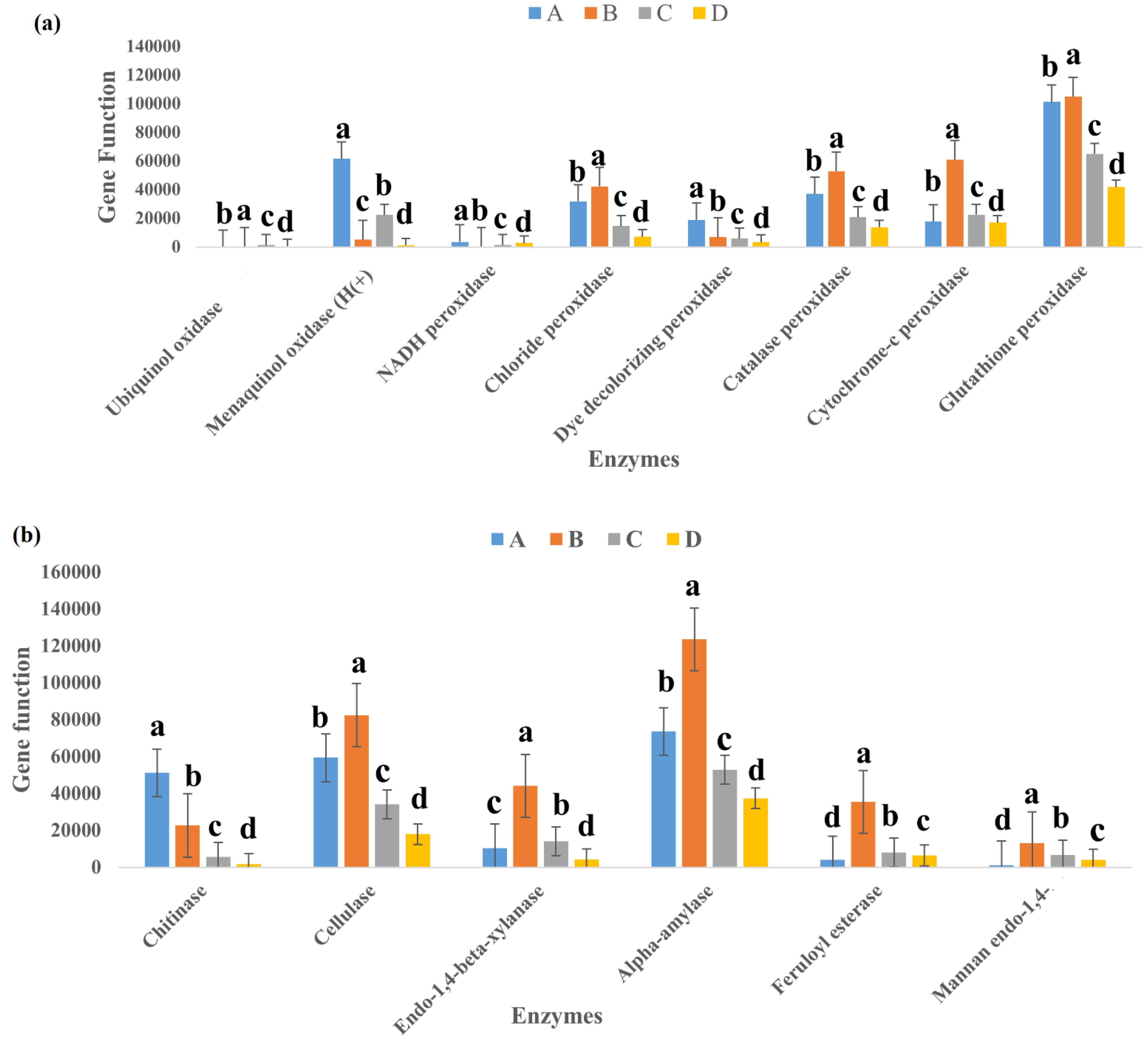
3.5. Function Prediction of Bacterial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghilamicael, A.M.; Budambula, N.L.M.; Anami, S.E.; Mehari, T.; Boga, H.I. Evaluation of prokaryotic diversity of five hot springs in Eritrea. BMC Microbiol. 2017, 17, 203. [Google Scholar] [CrossRef] [Green Version]
- Dornau, A.; Robson, J.F.; Thomas, G.H.; McQueen-Mason, S.J. Robust microorganisms for biofuel and chemical production from municipal solid waste. Microb. Cell Fact. 2020, 19, 68. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Shafy, H.I.; Mansour, M.S.M. Solid waste issue: Sources, composition, disposal, recycling, and valorization. Egypt. J. Pet. 2018, 27, 1275–1290. [Google Scholar] [CrossRef]
- Wang, X.; Cao, A.; Zhao, G.; Zhou, C.; Xu, R. Microbial community structure and diversity in a municipal solid waste landfill. Waste Manag. 2017, 66, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Ransom-Jones, E.; McCarthy, A.J.; Haldenby, S.; Doonan, J.; McDonald, J.E. Lignocellulose-Degrading Microbial Communities in Landfill Sites Represent a Repository of Unexplored Biomass-Degrading Diversity. mSphere 2017, 2, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Marriott, P.E.; Gómez, L.D.; Mcqueen-Mason, S.J. Unlocking the potential of lignocellulosic biomass through plant science. New Phytol. 2016, 209, 1366–1381. [Google Scholar] [CrossRef] [Green Version]
- DeAngelis, K.M.; Sharma, D.; Varney, R.; Simmons, B.; Isern, N.G.; Markilllie, L.M.; Nicora, C.; Norbeck, A.D.; Taylor, R.C.; Aldrich, J.T.; et al. Evidence supporting dissimilatory and assimilatory lignin degradation in Enterobacter lignolyticus SCF1. Front. Microbiol. 2013, 4, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosnow, J.J.; Anderson, L.N.; Nair, R.N.; Baker, E.S.; Wright, A.T. Profiling microbial lignocellulose degradation and utilization by emergent omics technologies. Crit. Rev. Biotechnol. 2017, 37, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Shah, G.; Sahota, S.; Singh, L.; Vijay, V.K. Biogas production from waste: Technical overview, progress, and challenges. In Bioreactors; Elsevier: Amsterdam, The Netherlands, 2020; pp. 89–104. [Google Scholar]
- Malgas, S.; Thoresen, M.; van Dyk, J.S.; Pletschke, B.I. Time dependence of enzyme synergism during the degradation of model and natural lignocellulosic substrates. Enzyme Microb. Technol. 2017, 103, 1–11. [Google Scholar] [CrossRef]
- Thakur, K.; Chownk, M.; Kumar, V.; Purohit, A.; Vashisht, A.; Kumar, V.; Yadav, S.K. Bioprospecting potential of microbial communities in solid waste landfills for novel enzymes through metagenomic approach. World J. Microbiol. Biotechnol. 2020, 36, 34. [Google Scholar] [CrossRef]
- Chukwuma, O.B.; Rafatullah, M.; Tajarudin, H.A.; Ismail, N. Lignocellulolytic Enzymes in Biotechnological and Industrial Processes: A Review. Sustainability 2020, 12, 7282. [Google Scholar] [CrossRef]
- Sang, N.N.; Soda, S.; Ishigaki, T.; Ike, M. Microorganisms in landfill bioreactors for accelerated stabilization of solid wastes. J. Biosci. Bioeng. 2012, 114, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Lei, L.; Duan, Y.; Zhang, K.Q.; Yang, J. Culture-independent methods for studying environmental microorganisms: Methods, application, and perspective. Appl. Microbiol. Biotechnol. 2012, 93, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, R.C.; Singh, R.; Eltis, L.D.; Mohn, W.W. Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J. 2019, 13, 413–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Li, Y.; Ge, X.; Yang, L.; Li, Y. Anaerobic digestion of food waste—Challenges and opportunities. Bioresour. Technol. 2018, 247, 1047–1058. [Google Scholar] [CrossRef]
- He, Y.; Xie, K.; Xu, P.; Huang, X.; Gu, W.; Zhang, F.; Tang, S. Evolution of microbial community diversity and enzymatic activity during composting. Res. Microbiol. 2013, 164, 189–198. [Google Scholar] [CrossRef]
- Westlake, K.; Archer, D.B.; Boone, D.R. Diversity of cellulolytic bacteria in landfill. J. Appl. Bacteriol. 1995, 79, 73–78. [Google Scholar] [CrossRef]
- Kamaruddin, M.A.; Yusoff, M.S.; Aziz, H.A.; Alrozi, R. Current status of Pulau Burung Sanitary Landfill leachate treatment, Penang, Malaysia. AIP Conf. Proc. 2016, 1774, 030014. [Google Scholar]
- Sinclair, L.; Osman, O.A.; Bertilsson, S.; Eiler, A. Microbial community composition and diversity via 16S rRNA gene amplicons: Evaluating the illumina platform. PLoS ONE 2015, 10, e0116955. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Wongwilaiwalin, S.; Mhuantong, W.; Champreda, V.; Tangphatsornruang, S.; Panichnumsin, P.; Ratanakhanokchai, K.; Tachaapaikoon, C. Structural and metabolic adaptation of cellulolytic microcosm in co-digested Napier grass-swine manure and its application in enhancing thermophilic biogas production. RSC Adv. 2018, 8, 29806–29815. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Principles of Enzymes☆. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Riesenfeld, C.S.; Schloss, P.D.; Handelsman, J. Metagenomics: Genomic analysis of microbial communities. Annu. Rev. Genet. 2004, 38, 525–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Lu, W.; Liu, Y.Y.; Ming, Z.; Liu, Y.Y.; Meng, R.; Wang, H. Structure and diversity of bacterial communities in two large sanitary landfills in China as revealed by high-throughput sequencing (MiSeq). Waste Manag. 2017, 63, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Rahman, M.S.; Hasan, M.M.; Paul, N.; Sajib, A.A. Microbial degradation of lignocellulosic biomass: Discovery of novel natural lignocellulolytic bacteria. Biotechnologia 2018, 99, 137–146. [Google Scholar] [CrossRef]
- Sekhohola-Dlamini, L.; Tekere, M. Microbiology of municipal solid waste landfills: A review of microbial dynamics and ecological influences in waste bioprocessing. Biodegradation 2020, 31, 1–21. [Google Scholar] [CrossRef]
- Wong, J.T.F.; Chen, X.; Deng, W.; Chai, Y.; Ng, C.W.W.; Wong, M.H. Effects of biochar on bacterial communities in a newly established landfill cover topsoil. J. Environ. Manage. 2019, 236, 667–673. [Google Scholar] [CrossRef]
- Thomas, F.; Hehemann, J.-H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and Gut Bacteroidetes: The Food Connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef] [Green Version]
- Nunoura, T.; Hirai, M.; Miyazaki, M.; Kazama, H.; Makita, H.; Hirayama, H.; Furushima, Y.; Yamamoto, H.; Imachi, H.; Takai, K. Isolation and Characterization of a Thermophilic, Obligately Anaerobic and Heterotrophic Marine Chloroflexi Bacterium from a Chloroflexi-dominated Microbial Community Associated with a Japanese Shallow Hydrothermal System, and Proposal for Thermomarinilin. Microbes Environ. 2013, 28, 228–235. [Google Scholar] [CrossRef] [Green Version]
- de Jonge, N.; Davidsson, Å.; la Cour Jansen, J.; Nielsen, J.L. Characterisation of microbial communities for improved management of anaerobic digestion of food waste. Waste Manag. 2020, 117, 124–135. [Google Scholar] [CrossRef]
- Wang, X.; Pecoraro, L. Analysis of Soil Fungal and Bacterial Communities in Tianchi Volcano Crater, Northeast China. Life 2021, 11, 280. [Google Scholar] [CrossRef] [PubMed]
- Bredon, M.; Dittmer, J.; Noël, C.; Moumen, B.; Bouchon, D. Lignocellulose degradation at the holobiont level: Teamwork in a keystone soil invertebrate 06 Biological Sciences 0605 Microbiology. Microbiome 2018, 6, 162. [Google Scholar] [CrossRef] [Green Version]
- Ventorino, V.; Aliberti, A.; Faraco, V.; Robertiello, A.; Giacobbe, S.; Ercolini, D.; Amore, A.; Fagnano, M.; Pepe, O. Exploring the microbiota dynamics related to vegetable biomasses degradation and study of lignocellulose-degrading bacteria for industrial biotechnological application. Sci. Rep. 2015, 5, 8161. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Dong, D.; Wang, H.; Müller, K.; Qin, Y.; Wang, H.; Wu, W. Metagenomic analysis of microbial consortia enriched from compost: New insights into the role of Actinobacteria in lignocellulose decomposition. Biotechnol. Biofuels 2016, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zainudin, M.H.M.; Hassan, M.A.; Md Shah, U.K.; Abdullah, N.; Tokura, M.; Yasueda, H.; Shirai, Y.; Sakai, K.; Baharuddin, A.S. Bacterial community structure and biochemical changes associated with composting of lignocellulosic oil palm empty fruit bunch. BioResources 2014, 9, 316–335. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Song, L. Succession of bacterial community structure and metabolic function during solid waste decomposition. Bioresour. Technol. 2019, 291, 121865. [Google Scholar] [CrossRef] [PubMed]
- López-Mondéjar, R.; Algora, C.; Baldrian, P. Lignocellulolytic systems of soil bacteria: A vast and diverse toolbox for biotechnological conversion processes. Biotechnol. Adv. 2019, 37, 107374. [Google Scholar] [CrossRef]
- Song, L.; Wang, Y.; Zhao, H.; Long, D.T. Composition of bacterial and archaeal communities during landfill refuse decomposition processes. Microbiol. Res. 2015, 181, 105–111. [Google Scholar] [CrossRef]
- Jing, T.Z.; Qi, F.H.; Wang, Z.Y. Most dominant roles of insect gut bacteria: Digestion, detoxification, or essential nutrient provision? Microbiome 2020, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, M.; Su, J.; Hu, H.; Yang, M.; Huang, Z.; Chen, D.; Wu, J.; Feng, Z. Overcoming biomass recalcitrance by synergistic pretreatment of mechanical activation and metal salt for enhancing enzymatic conversion of lignocellulose. Biotechnol. Biofuels 2019, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.M.; Yannarell, A.C.; Sims, G.K.; Cadavid-Restrepo, G.; Moreno Herrera, C.X. Characterization of bacterial diversity at different depths in the Moravia Hill landfill site at Medellín, Colombia. Soil. Biol. Biochem. 2011, 43, 1275–1284. [Google Scholar] [CrossRef]
- Heo, Y.; Park, J.; Lim, S.I.; Hur, H.G.; Kim, D.; Park, K. Size-resolved culturable airborne bacteria sampled in rice field, sanitary landfill, and waste incineration sites. J. Environ. Monit. 2010, 12, 1619–1624. [Google Scholar] [CrossRef]
- Puentes-Téllez, P.E.; Falcao Salles, J. Construction of Effective Minimal Active Microbial Consortia for Lignocellulose Degradation. Microb. Ecol. 2018, 76, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spasic, J.; Mandic, M.; Djokic, L.; Nikodinovic-Runic, J. Streptomyces spp. in the biocatalysis toolbox. Appl. Microbiol. Biotechnol. 2018, 102, 3513–3536. [Google Scholar] [CrossRef]
- Jaramillo, P.A.P.; Borda-Molina, D.; Montaña, J.S.; Baena, S. Isolation of lipolytic bacteria from Colombian Andean soils: A target for bioprospecting. J. Microbiol. Biotechnol. Food Sci. 2017, 6, 1250–1256. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Kazy, S.K. Microbial diversity, community composition and metabolic potential in hydrocarbon contaminated oily sludge: Prospects for in situ bioremediation. Environ. Sci. Pollut. Res. 2014, 21, 7369–7389. [Google Scholar] [CrossRef]
- Hayward, A.C.; Fegan, N.; Fegan, M.; Stirling, G.R. Stenotrophomonas and Lysobacter: Ubiquitous plant-associated gamma-proteobacteria of developing significance in applied microbiology. J. Appl. Microbiol. 2010, 108, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Nasipuri, P.; Herschend, J.; Brejnrod, A.D.; Madsen, J.S.; Espersen, R.; Svensson, B.; Burmølle, M.; Jacquiod, S.; Sørensen, S.J. Community-intrinsic properties enhance keratin degradation from bacterial consortia. PLoS ONE 2020, 15, e0228108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, V.M. Navneet Review on the current status of polymer degradation: A microbial approach. Bioresour. Bioprocess. 2017, 4, 15. [Google Scholar] [CrossRef]
- de Oliveira Gorgulho Silva, C.; Filho, E.X.F. A Review of Holocellulase Production Using Pretreated Lignocellulosic Substrates. Bioenergy Res. 2017, 10, 592–602. [Google Scholar] [CrossRef]
- Deng, Y.J.; Wang, S.Y. Synergistic growth in bacteria depends on substrate complexity. J. Microbiol. 2016, 54, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamps, B.W.; Lyles, C.N.; Suflita, J.M.; Masoner, J.R.; Cozzarelli, I.M.; Kolpin, D.W.; Stevenson, B.S. Municipal Solid Waste Landfills Harbor Distinct Microbiomes. Front. Microbiol. 2016, 7, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | pH | Temperature (°C) | Moisture Content (%) | C (%) | H (%) | N (%) |
|---|---|---|---|---|---|---|
| A | 6.80 ± 1.13 | 35.00 ± 1.01 | 17.07 ± 4.08 | 7.64 ± 0.18 | 0.88 ± 0.17 | 0.30 ± 0.06 |
| B | 6.82 ± 0.12 | 45.00 ± 0.86 | 3.41 ± 1.56 | 13.36 ± 5.29 | 1.88 ± 1.19 | 0.75 ± 0.47 |
| C | 8.44 ± 0.34 | 32.00 ± 0.08 | 7.66 ± 2.01 | 0.55 ± 0.14 | 0.44 ± 0.13 | 0.14 ± 0.02 |
| D | 7.98 ± 0.48 | 33.00 ± 1.87 | 37.26 ± 5.8 | 17.84 ± 7.51 | 2.25 ± 2.16 | 0.67 ± 0.55 |
| Sample | Observed OTUs | Chao1 | ACE | Shannon | Simpson |
|---|---|---|---|---|---|
| A | 539.79 ± 28.33 c | 570.02 ± 0.55 c | 570.84 ± 0.35 c | 3.52 ± 0.01 c | 0.92 ± 0.02 b |
| B | 512.00 ± 0.09 d | 538.52 ± 0.57 d | 543.32 ± 0.90 d | 4.16 ± 0.03 b | 0.95 ± 0.01 ab |
| C | 837.01 ± 0.14 b | 908.68 ± 0.56 b | 908.46 ± 0.48 b | 4.59 ± 0.07 a | 0.97 ± 0.02 a |
| D | 1253.63 ± 0.57 a | 1316.53 ± 0.49 a | 1314.50 ± 0.55 a | 4.50 ± 0.04 a | 0.91 ± 0.01 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chukwuma, O.B.; Rafatullah, M.; Tajarudin, H.A.; Ismail, N. Bacterial Diversity and Community Structure of a Municipal Solid Waste Landfill: A Source of Lignocellulolytic Potential. Life 2021, 11, 493. https://0-doi-org.brum.beds.ac.uk/10.3390/life11060493
Chukwuma OB, Rafatullah M, Tajarudin HA, Ismail N. Bacterial Diversity and Community Structure of a Municipal Solid Waste Landfill: A Source of Lignocellulolytic Potential. Life. 2021; 11(6):493. https://0-doi-org.brum.beds.ac.uk/10.3390/life11060493
Chicago/Turabian StyleChukwuma, Ogechukwu Bose, Mohd Rafatullah, Husnul Azan Tajarudin, and Norli Ismail. 2021. "Bacterial Diversity and Community Structure of a Municipal Solid Waste Landfill: A Source of Lignocellulolytic Potential" Life 11, no. 6: 493. https://0-doi-org.brum.beds.ac.uk/10.3390/life11060493