
Analysis of Homozygous-by-Descent (HBD) Segments for Purebred and Crossbred Pigs in Russia

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Data Processing and Data Analyses
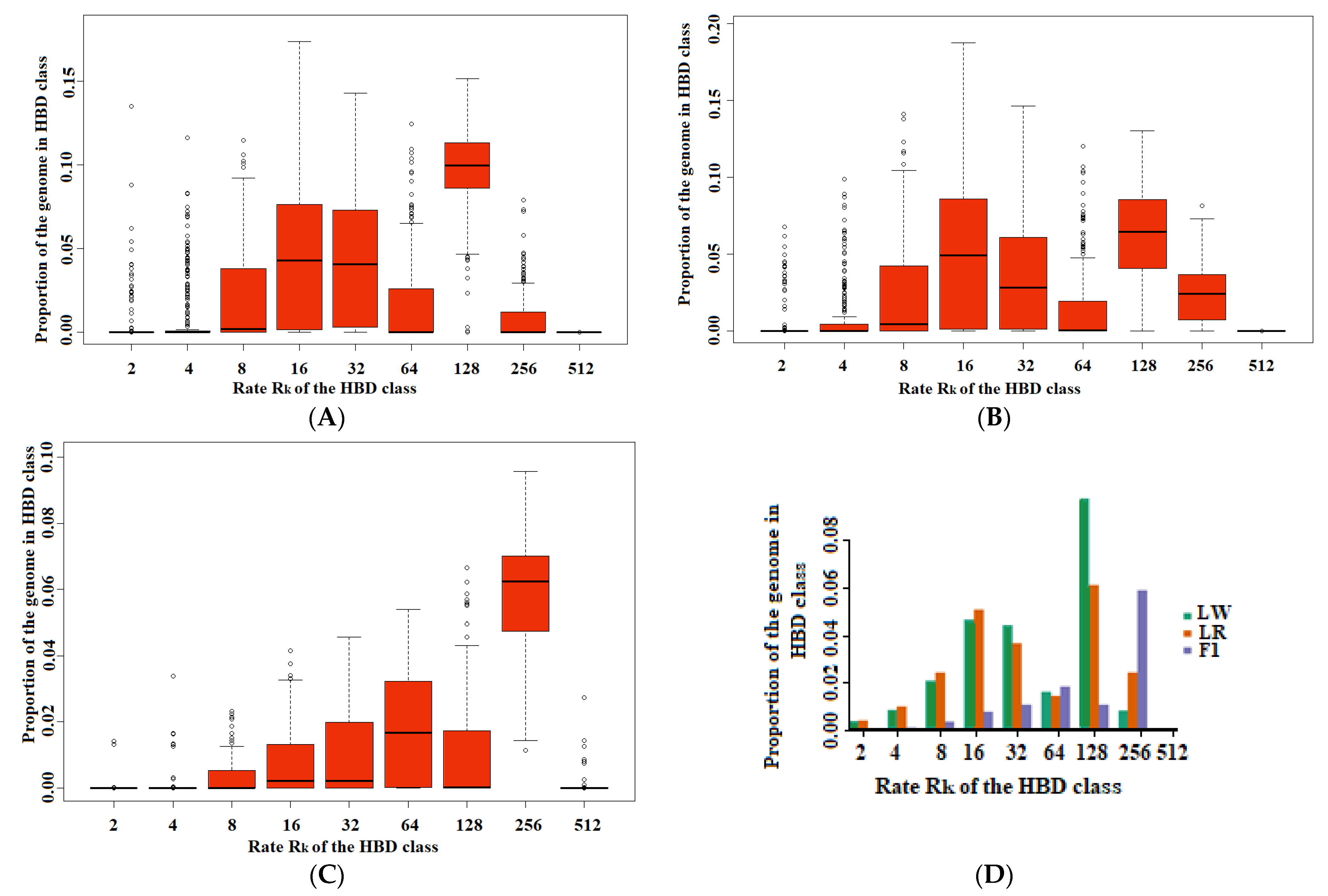
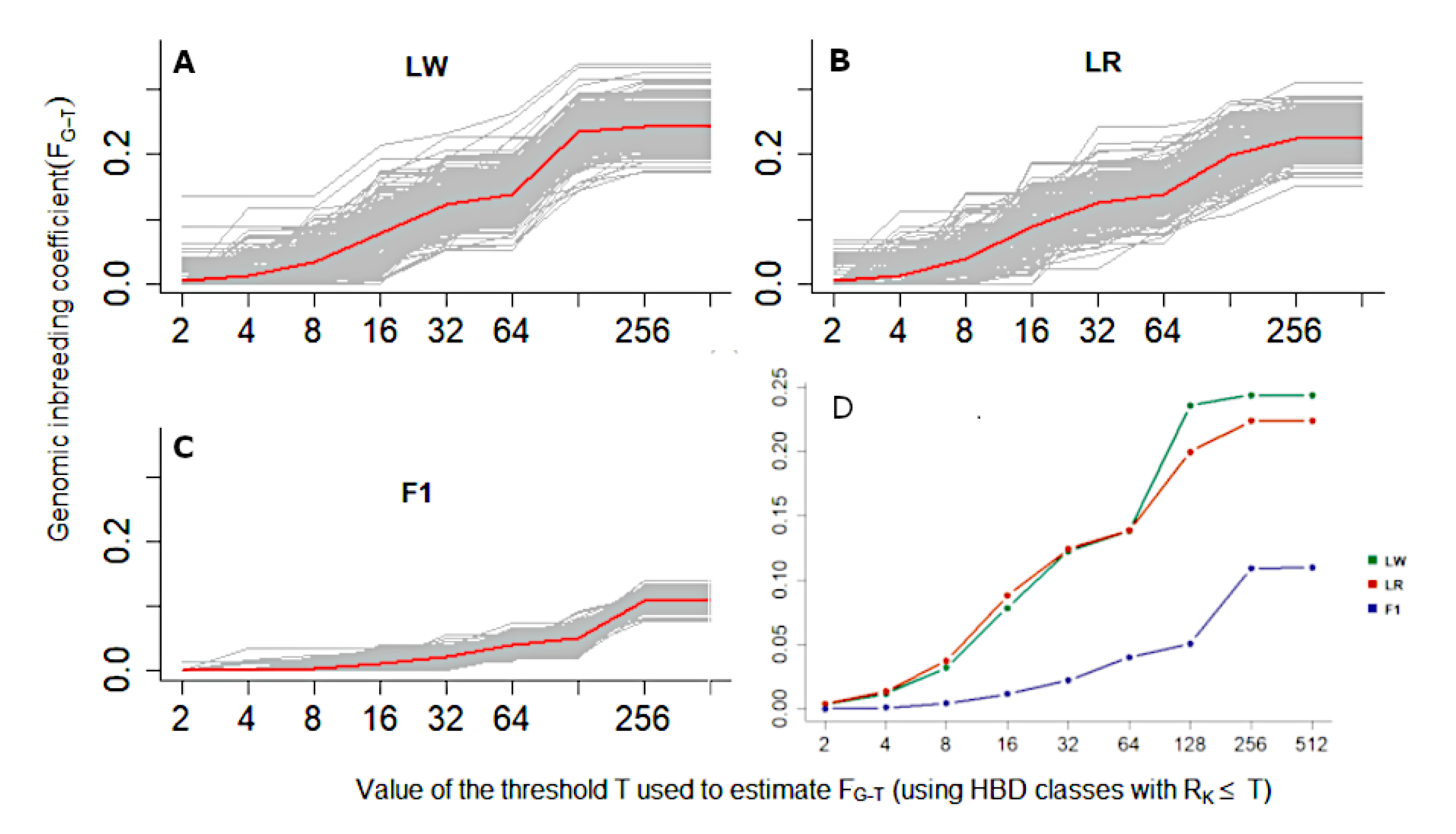
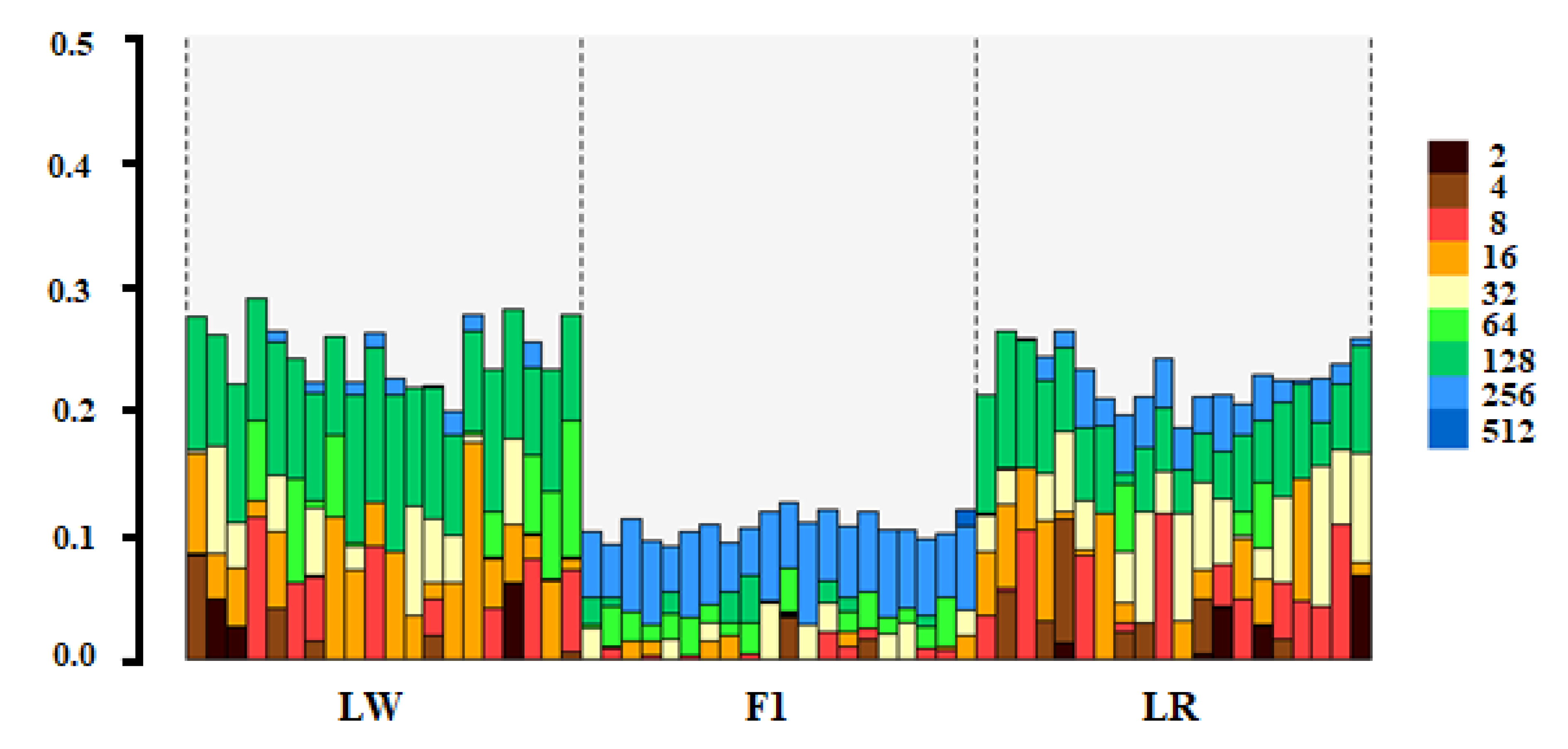
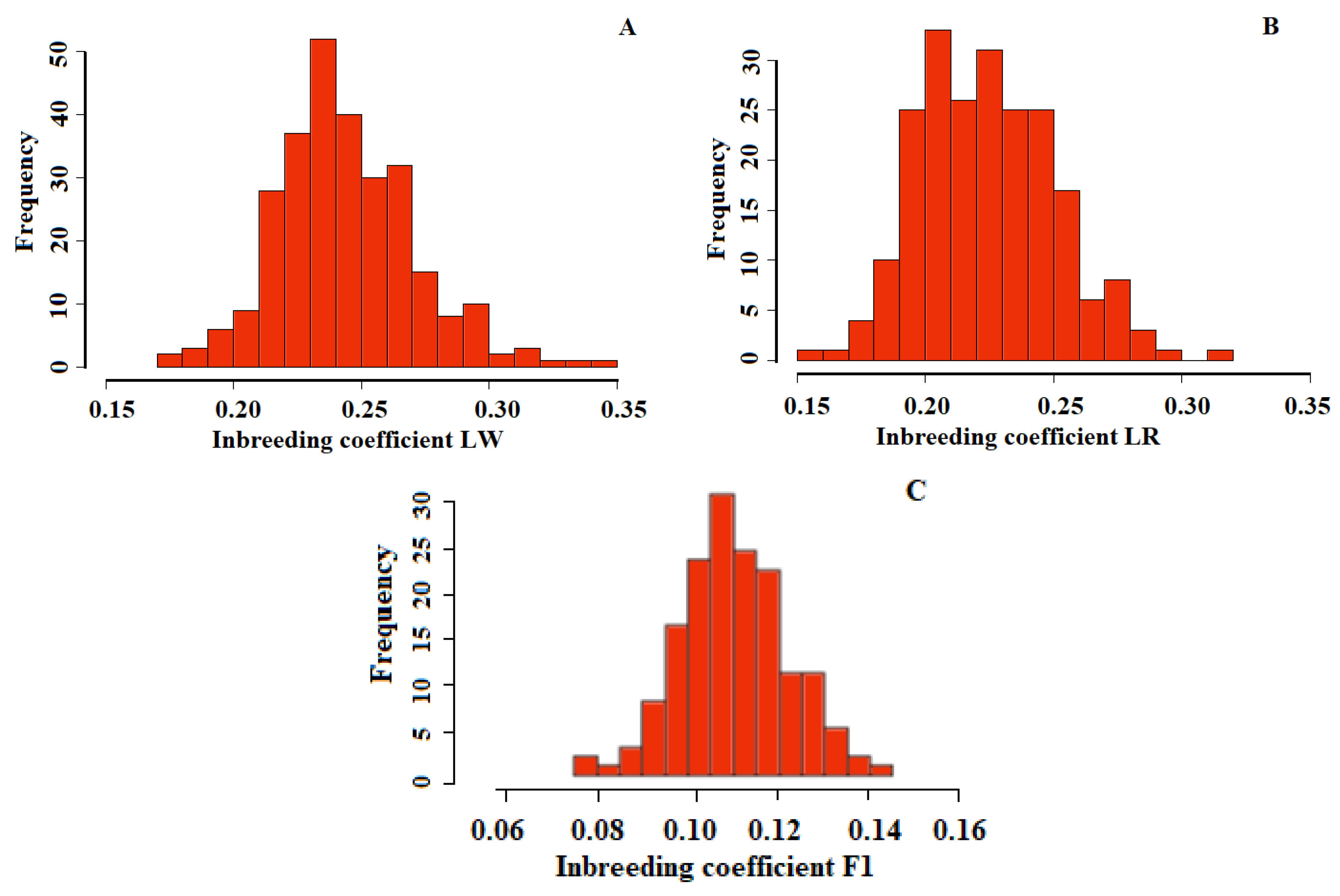
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Auton, A.; Bryc, K.; Boyko, A.R.; Lohmueller, K.E.; Novembre, J.; Reynolds, A.; Indap, A.; Wright, M.H.; Degenhardt, J.D.; Gutenkunst, R.N.; et al. Global distribution of genomic diversity underscores rich complex history of continental human populations. Genome. Res. 2009, 19, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.Q.; Xue, J.; Lian, M.J.; Yang, R.F.; Liu, T.F.; Zeng, Z.Y.; Jiang, A.A.; Jiang, Y.Z.; Zhu, L.; Bai, L.; et al. Inbreeding and genetic diversity in three imported Swine breeds in china using pedigree data. Asian-Australas J. Anim. Sci. 2013, 26, 755–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szpiech, Z.A.; Xu, J.; Pemberton, T.J.; Peng, W.; Zöllner, S.; Rosenberg, N.A.; Li, J.Z. Long runs of homozygosity are enriched for deleterious variation. Am. J. Hum. Genet. 2013, 93, 90–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltecca, C.; Tiezzi, F.; Cole, J.B.; Baes, C. Symposium review: Exploiting homozygosity in the era of genomics—Selection, inbreeding, and mating programs. J. Dairy Sci. 2020, 103, 5302–5313. [Google Scholar] [CrossRef] [PubMed]
- Saura, M.; Fernández, A.; Rodríguez, M.C.; Toro, M.A.; Barragán, C.; Fernández, A.I.; Villanueva, B. Genome-wide estimates of coancestry and inbreeding in a closed herd of ancient Iberian pigs. PLoS ONE 2013, 8, e78314. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sørensen, P.; Janss, L.; Ostersen, T.; Edwards, D. Genome-wide and local pattern of linkage disequilibrium and persistence of phase for 3 Danish pig breeds. BMC Genet. 2013, 14, 115. [Google Scholar] [CrossRef] [Green Version]
- Bosse, M.; Megens, H.J.; Derks, M.F.L.; de Cara, Á.M.R.; Groenen, M.A.M. Deleterious alleles in the context of domestication, inbreeding, and selection. Evol. Appl. 2018, 12, 6–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorssen, W.; Meyermans, R.; Janssens, S.; Buys, N. A publicly available repository of ROH islands reveals signatures of selection in different livestock and pet species. Genet. Sel. Evol. 2021, 53, 2. [Google Scholar] [CrossRef]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef] [Green Version]
- Marchesi, J.A.P.; Buzanskas, M.E.; Cantão, M.E.; Ibelli, A.M.G.; Peixoto, J.O.; Joaquim, L.B.; Moreira, G.C.M.; Godoy, T.F.; Sbardella, A.P.; Figueiredo, E.A.P.; et al. Relationship of runs of homozygosity with adaptive and production traits in a paternal broiler line. Animal 2018, 12, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, S.; Sardina, M.T.; Tolone, M.; Di Gerlando, R.; Sutera, A.M.; Fontanesi, L.; Portolano, B. Genome-wide identification of runs of homozygosity islands and associated genes in local dairy cattle breeds. Animal 2018, 12, 2480–2488. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.C.D.C.; Ventura, R.V.; Baldi, F.; da Silva, M.V.G.B. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Bovo, S.; Bertolini, F.; Dall’Olio, S.; Costa, L.N.; Tinarelli, S.; Gallo, M.; Fontanesi, L. Runs of homozygosity islands in Italian cosmopolitan and autochthonous pig breeds identify selection signatures in the porcine genome. Livest. Sci. 2020, 240, 104219. [Google Scholar] [CrossRef]
- Druet, T.; Gautier, M. A model-based approach to characterize individual inbreeding at both global and local genomic scales. Mol. Ecol. 2017, 26, 5820–5841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunhareang, S.; Zhou, H.; Hickford, J.G.H. Rapid DNA extraction of pig ear tissues. Meat Sci. 2010, 85, 589–590. [Google Scholar] [CrossRef] [PubMed]
- Rabiner, L.R. A tutorial on Hidden Markov Models and selected applications in speech recognition. Proc. IEEE 1989, 77, 257–286. [Google Scholar] [CrossRef]
- Bertrand, A.R.; Kadri, N.K.; Flori, L.; Gautier, M.; Druet, T. RZooRoH: An R package to characterize individual genomic autozygosity and identify homozygous-by-descent segments. Methods Ecol. Evol. 2019, 10, 860–866. [Google Scholar] [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef]
- Porter, V. Pigs. A Handbook to the Breeds of the World; Helm Information, Ltd: Mountfield, UK, 1993. [Google Scholar]
- Wilkinson, S.; Lu, Z.H.; Megens, H.J.; Archibald, A.L.; Haley, C.; Jackson, I.J.; Groenen, M.A.; Crooijmans, R.P.; Ogden, R.; Wiener, P. Signatures of diversifying selection in European pig breeds. PLoS Genet. 2013, 9, e1003453. [Google Scholar] [CrossRef] [Green Version]
- Getmantseva, L.; Bakoev, S.; Bakoev, N.; Karpushkina, T.; Kostyunina, O. Mitochondrial DNA Diversity in Large White Pigs in Russia. Animals 2020, 10, 1365. [Google Scholar] [CrossRef]
- Falkenberg, H.; Hammer, H. Zur Geschichte und Kultur der Schweinezüchtung und-haltung 4. Mitt.: Schweinezucht und-produktion in Europa zwischen 1900 und 1945. Züchtungskunde 2008, 80, 315–333. [Google Scholar]
- Bakoev, S.; Getmantseva, L.; Bakoev, F.; Kolosova, M.; Gabova, V.; Kolosov, A.; Kostyunina, O. Survey of SNPs Associated with Total Number Born and Total Number Born Alive in Pig. Genes 2020, 11, 491. [Google Scholar] [CrossRef] [PubMed]
- Getmantseva, L.; Kolosova, M.; Bakoev, F.; Zimina, A.; Bakoev, S. Genomic Regions and Candidate Genes Linked to Capped Hock in Pig. Life. 2021, 11, 510. [Google Scholar] [CrossRef]
- Hulsegge, I.; Calus, M.; Hoving-Bolink, R.; Lopes, M.; Megens, H.-J.; Oldenbroek, K. Impact of merging commercial breeding lines on the genetic diversity of Landrace pigs. Genet. Sel Evol. 2019, 51, 60. [Google Scholar] [CrossRef] [Green Version]
- Norring, M.; Valros, A.; Bergman, P.; Marchant-Forde, J.N.; Heinonen, M. Body condition, live weight and success in agonistic encounters in mixed parity groups of sows during gestation. Animal 2019, 13, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Rekiel, A.; Bartosik, J.; Więcek, J.; Batorska, M.; Kuczyńska, B.; Łojek, A. Effect of Piglet Birth Weight on Selected Characteristics of Pork. Ann. Anim. Sci. 2014, 14, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Traspov, A.A.; Kostyunina, O.V.; Belous, A.A.; Karpushkina, T.V.; Svezhentseva, N.A.; Zinovieva, N.A. Whole-genome association studies of distribution of developmental abnormalities and other breeding-valuable qualitative traits in offspring of the Russian large-white boars. Vavilovskii Zhurnal Genet. Selektsii 2020, 24, 185–190. (In Russian) [Google Scholar]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Orlov, Y.L.; Galieva, A.G.; Orlova, N.G.; Ivanova, E.N.; Mozyleva, Y.A.; Anashkina, A.A. Rekonstruktsiia gennoĭ seti bolezni Parkinsona dlia poiska genov-misheneĭ [Reconstruction of gene network associated with Parkinson disease for gene targets search]. Biomed Khim 2021, 67, 222–230. (In Russian) [Google Scholar] [CrossRef]
- Wakeling, M.N.; Laver, T.W.; Wright, C.F.; Franco, E.D.; Stals, K.L.; Patch, A.-M.; Hattersley, A.T.; Flanagan, S.E.; Ellard, S. Homozygosity mapping provides supporting evidence of pathogenicity in recessive Mendelian disease. Genet. Med. 2019, 21, 982–986. [Google Scholar] [CrossRef]
- Nielsen, V.; Bendixen, C.; Arnbjerg, J.; Sørensen, C.; Jensen, H.; Shukri, N.M.; Thomsen, B. Abnormal growth plate function in pigs carrying a dominant mutation in type X collagen. Mamm. Genome 2000, 11, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Zhang, Y.; Jia, Q.; Wang, X.; Zheng, Q.; Zhang, H.; Song, R.; Li, Y.; Luo, A.; Hong, Q.; et al. An exonic splicing enhancer mutation in DUOX2 causes aberrant alternative splicing and severe congenital hypothyroidism in Bama pigs. Dis. Model. Mech. 2019, 12, dmm036616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Guo, W.; Ren, L.; Yang, M.; Zhao, Y.; Guo, Z.; Yi, H.; Li, M.; Hu, Y.; Long, X.; et al. A de novo silencer causes elimination of MITF-M expression and profound hearing loss in pigs. BMC Biol. 2016, 14, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Chen, J.; Cao, W.; Yang, L.; Chen, Q.; He, J.; Yi, Q.; Huang, H.; Zhang, E.; Cai, Z. The many substrates and functions of NEDD4-1. Cell Death Dis. 2019, 10, 904. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.; Trevers, K.E.; Reyes-Corral, M.; Antinucci, P.; Hindges, R. Expression and roles of teneurins in zebrafish. Front. Neurosci. 2019, 13, 158. [Google Scholar] [CrossRef] [Green Version]
- Feldman, G.; Kappes, D.; Mookerjee-Basu, J.; Freeman, T.; Fertala, A.; Parvizi, J. Novel mutation in Teneurin 3 found to co-segregate in all affecteds in a multi-generation family with developmental dysplasia of the hip. J. Orthop. Res. 2019, 37, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Srivastava, P.; Phadke, S.R. Sequence variations in TENM3 gene causing eye anomalies with intellectual disability: Expanding the phenotypic spectrum. Eur. J. Med. Genet. 2019, 62, 61–64. [Google Scholar] [CrossRef]
- Schröder, R.; van der Ven, P.F.; Warlo, I.; Schumann, H.; Fürst, D.O.; Blümcke, I.; Schmidt, M.C.; Hatzfeld, M. p0071, a member of the armadillo multigene family, is a constituent of sarcomeric I-bands in human skeletal muscle. J. Muscle Res. Cell Motil. 2000, 21, 577–586. [Google Scholar] [CrossRef]
- Blumenthal, J.; Behar, L.; Elliott, E.; Ginzburg, I. Dcp1a phosphorylation along neuronal development and stress. FEBS Lett. 2009, 583, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Bai, R.-Y.; Koester, C.; Ouyang, T.; Hahn, S.A.; Hammerschmidt, M.; Peschel, C.; Duyster, J. SMIF, a Smad4-interacting protein that functions as a co-activator in TGF-beta signalling. Nat. Cell Biol. 2002, 4, 181–190. [Google Scholar] [CrossRef]
- Noormohammadi, A.; Khodakarami, A.; Gutierrez-Garcia, R.; Lee, H.J.; Koyuncu, S.; König, T.; Schindler, C.; Saez, I.; Fatima, A.; Dieterich, C.; et al. Somatic increase of CCT8 mimics proteostasis of human pluripotent stem cells and extends C. elegans lifespan. Nat. Commun. 2016, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LW | LR | F1 | |
|---|---|---|---|
| Min | 0.17 | 0.15 | 0.08 |
| 1st Qu. | 0.27 | 0.20 | 0.10 |
| Median | 0.24 | 0.22 | 0.11 |
| Mean | 0.24 | 0.22 | 0.11 |
| 3rd Qu. | 0.26 | 0.24 | 0.12 |
| Max. | 0.34 | 0.31 | 0.14 |
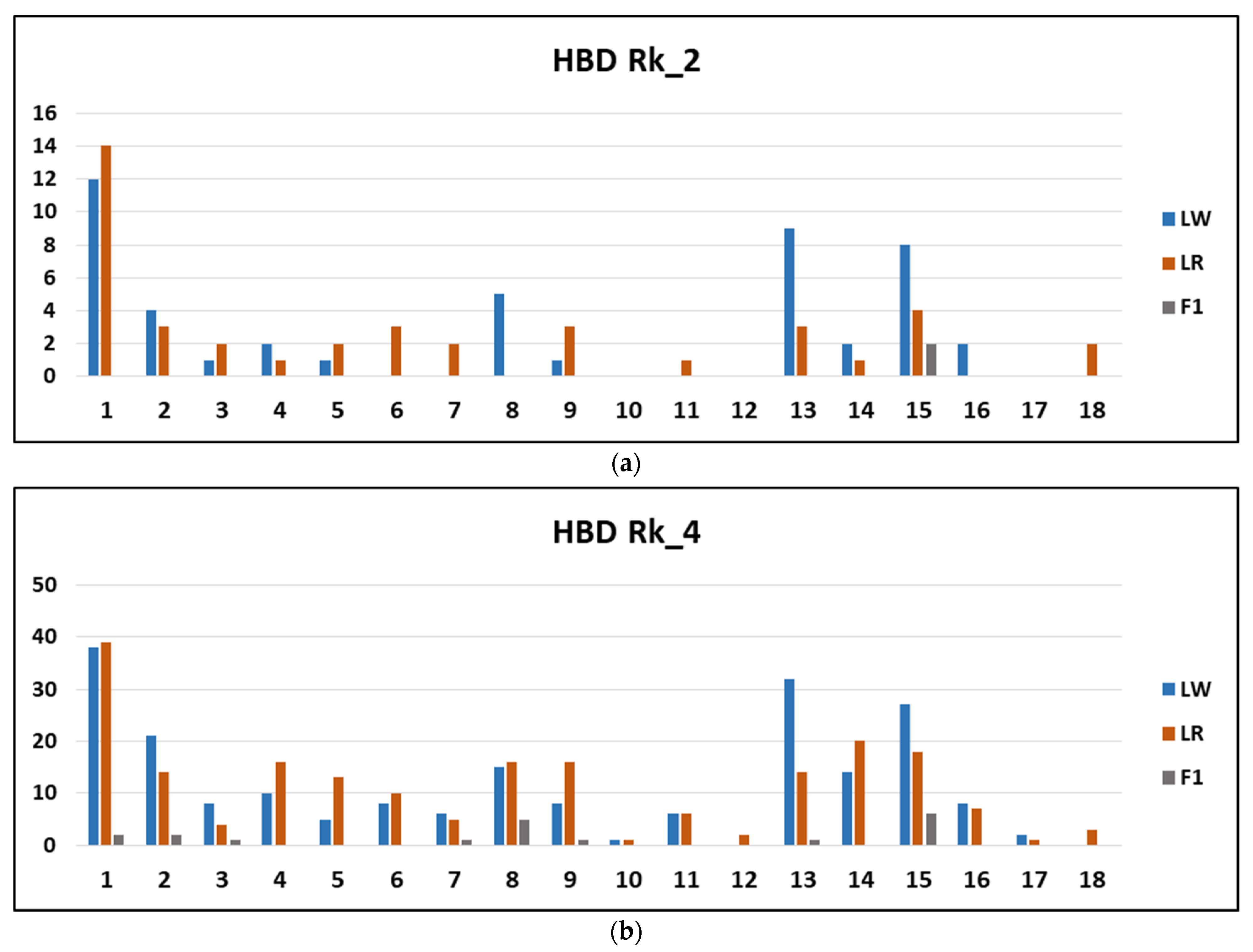
| Rk | LW | LR | F1 | |||
|---|---|---|---|---|---|---|
| Number | Length | Number | Length | Number | Length | |
| 2 | 47 | 76.286 ± 5.120 | 41 | 73.447 ± 5.486 | 2 | 63.836 ± 1.797 |
| 4 | 209 | 41.175 ± 1.463 | 205 | 38.593 ± 1.491 | 19 | 32.509 ± 3.605 |
| 8 | 1042 | 20.225 ± 0.362 | 864 | 20.204 ± 0.414 | 128 | 19.471 ± 0.715 |
| 16 | 4589 | 9.576 ± 0.089 | 3910 | 9.088 ± 0.096 | 446 | 10.497 ± 0.303 |
| 32 | 5820 | 5.356 ± 0.032 | 3873 | 4.725 ± 0.040 | 1362 | 4.837 ± 0.072 |
| 64 | 2349 | 3.068 ± 0.026 | 2176 | 2.697 ± 0.027 | 2679 | 3.030 ± 0.024 |
| 128 | 35,254 | 1.283 ± 0.004 | 17,145 | 1.292 ± 0.006 | 1543 | 1.537 ± 0.019 |
| 256 | 1114 | 0.554 ± 0.009 | 5372 | 0.572 ± 0.004 | 14,889 | 0.732 ± 0.003 |
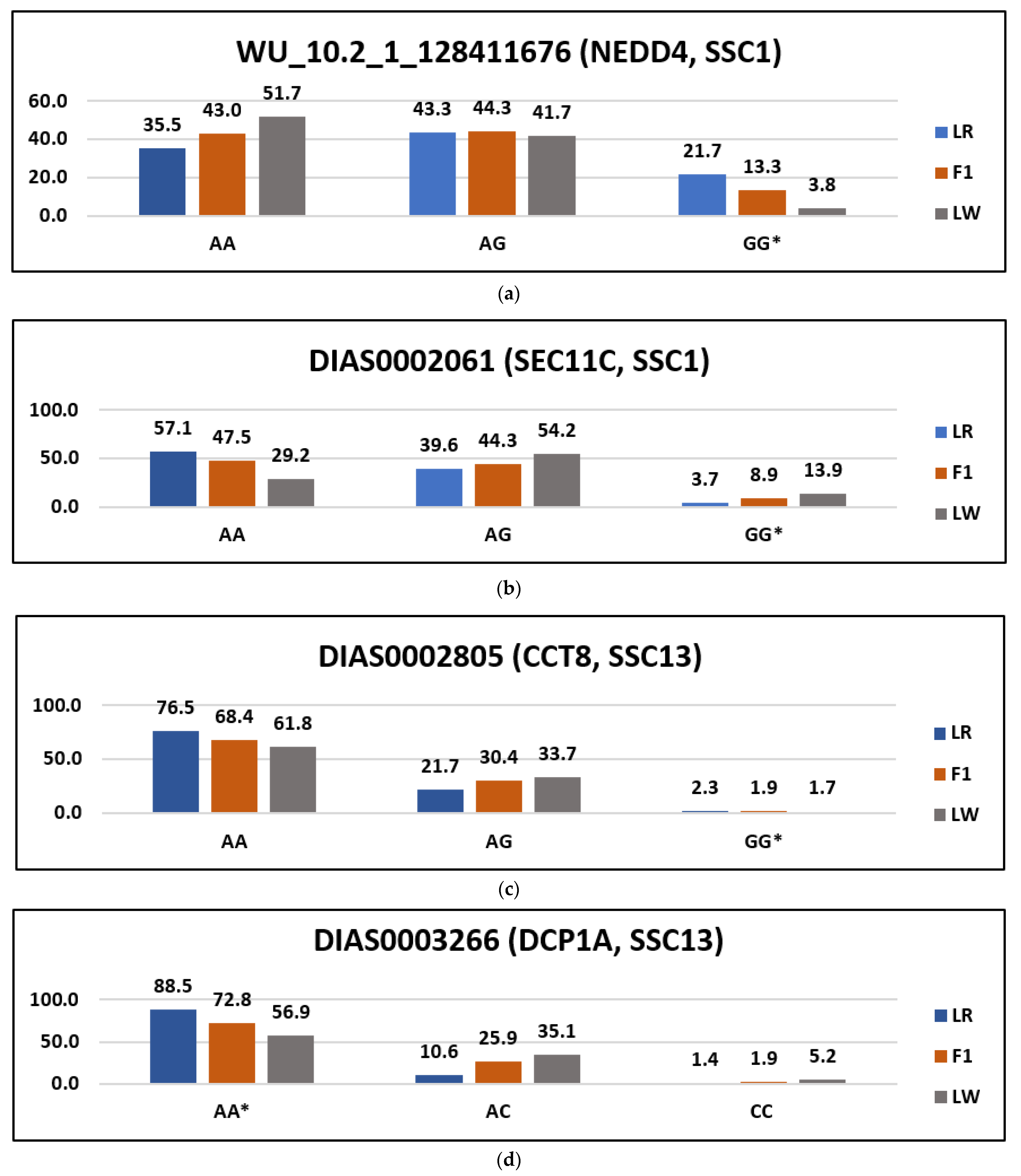
| Uploaded_Variation | Position | Allele | Symbol | Amino_Acids | Codons | Variation | SIFT |
|---|---|---|---|---|---|---|---|
| WU_10.2_1_128411676 | 1:115993957 | G | NEDD4 | I/V | Ata/Gta | rs331958194 | tolerated_low_confidence (1) |
| DIAS0002234 | 1:29042621 | G | HBS1L | I/V | Att/Gtt | rs80787670 | Tolerated (1) |
| MARC0008887 | 1:95536583 | C | EPG5 | I/V | Atc/Gtc | rs81253573 | Tolerated (1) |
| MARC0005035 | 1:108340862 | G | HERC1 | N/S | aAc/aGc | rs80998094 | Tolerated (1) |
| WU_10.2_1_202674735 | 1:182404193 | T | STYX | S/L | tCg/tTg | rs321526744 | Tolerated (1) |
| DRGA0001262 | 1:82324692 | A | - | E/K | Gaa/Aaa | rs80984416 | Tolerated (0.71) |
| ALGA0002996 | 1:44498050 | G | ROS1 | D/G | gAt/gGt | rs80883735 | Tolerated (0.65) |
| DIAS0002722 | 1:25558722 | A | REPS1 | G/D | gGt/gAt | rs333867086 | Tolerated (0.64) |
| DIAS0002980 | 1:26316436 | C | PERP | T/P | Act/Cct | rs55618815 | Tolerated (0.39) |
| ASGA0004152 | 1:106877209 | G | FECH | V/A | gTg/gCg | rs81216562 | Tolerated (0.22) |
| DIAS0003245 | 1:179469474 | A | LRR1 | E/K | Gaa/Aaa | rs332693293 | Tolerated (0.13) |
| DIAS0002061 | 1:161757995 | G | SEC11C | C/R | Tgt/Cgt | rs80807772 | deleterious_low_confidence (0) |
| M1GA0025601 | 13:34117528 | A | - | V/I | Gta/Ata | rs81478482 | Tolerated (1) |
| DIAS0004147 | 13:106612102 | A | SERPINI1 | L/I | Ctt/Att | rs322745111 | Tolerated (1) |
| WU_10.2_13_42333381 | 13:38487829 | C | CCDC66 | I/T | aTa/aCa | rs335407407 | Tolerated (0.52) |
| DIAS0001169 | 13:13969030 | G | SLC4A7 | T/P | Act/Cct | rs339676777 | Tolerated (0.5) |
| INRA0040732 | 13:89489538 | G | - | I/T | aTc/aCc | rs345909418 | Tolerated (0.5) |
| M1GA0025255 | 13:29119930 | T | FYCO1 | D/N | Gac/Aac | rs81478691 | Tolerated (0.47) |
| DIAS0003138 | 13:22199433 | A | GOLGA4 | E/K | Gaa/Aaa | rs328179266 | Tolerated (0.31) |
| DIAS0003446 | 13:24649204 | A | ENTPD3 | A/T | Gca/Aca | rs81216415 | Tolerated (0.25) |
| DBMA0000259 | 13:122067534 | T | EIF2B5 | T/M | aCg/aTg | rs45435374 | Tolerated (0.22) |
| DIAS0002680 | 13:73754891 | A | ACAD11 | A/V | gCa/gTa | rs326329989 | Tolerated (0.14) |
| DIAS0003266 | 13:35402355 | T | DCP1A | A/D | gCc/gAc | rs81211881 | Deleterious (0.04) |
| DIAS0002805 | 13:192415347 | G | CCT8 | S/P | Tct/Cct | rs81214915 | Deleterious (0) |
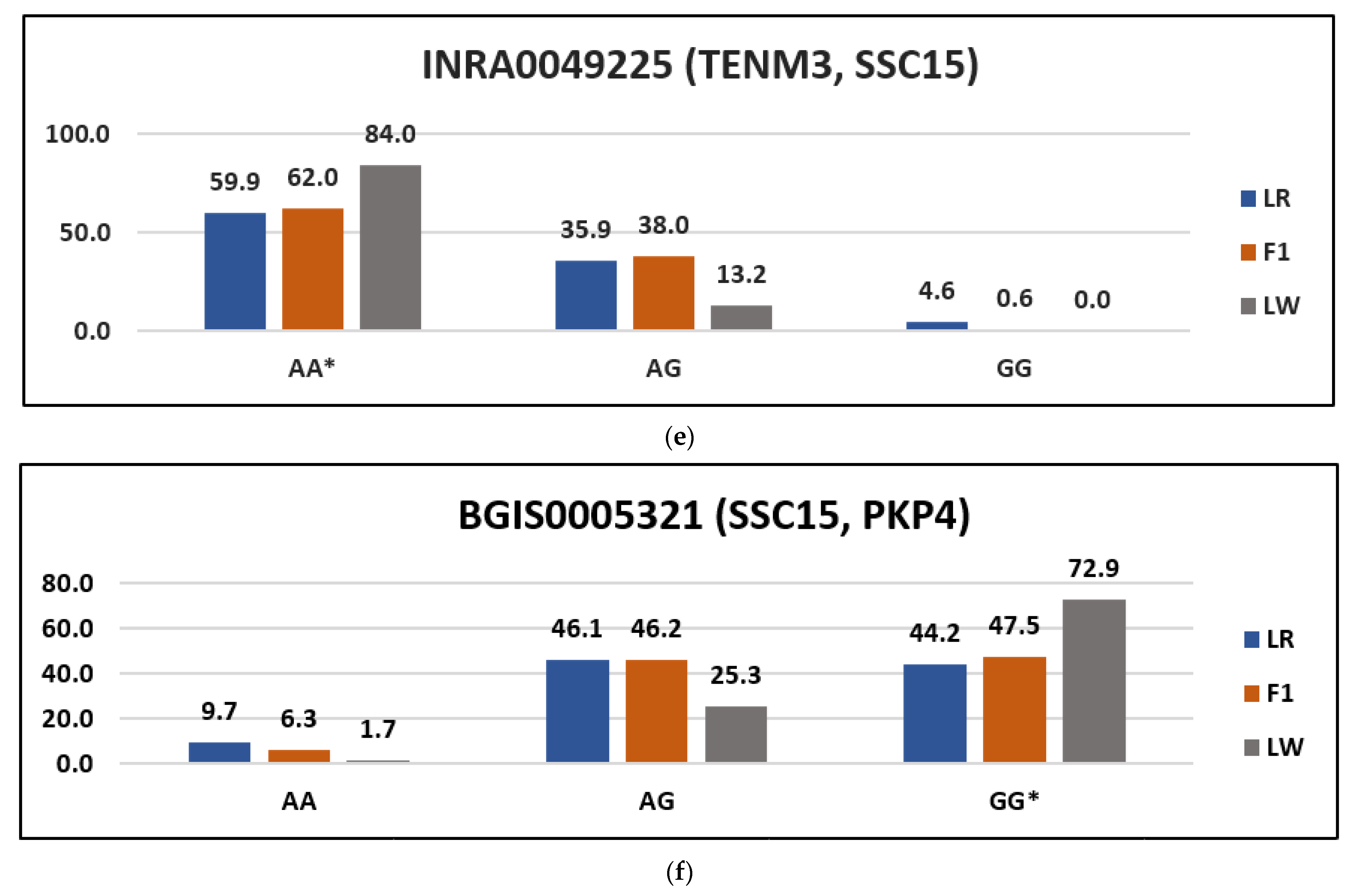
| BGIS0005321 | 15:65680055 | G | PKP4 | I/V | Ata/Gta | rs80939022 | tolerated_low_confidence (1) |
| DIAS0001114 | 15:72586615 | T | SCN1A | R/K | aGg/aAg | rs340033396 | Tolerated (1) |
| DIAS0000678 | 15:121561503 | A | OBSL1 | A/S | Gct/Tct | rs332398561 | Tolerated (0.79) |
| MARC0063762 | 15:106573135 | A | CYP20A1 | R/Q | cGg/cAg | rs81252138 | Tolerated (0.64) |
| MARC0035976 | 15:44768162 | T | WWC2 | P/S | Ccc/Tcc | rs81230064 | Tolerated (0.28) |
| INRA0049225 | 15:44344760 | A | TENM3 | V/M | Gtg/Atg | rs345636277 | Deleterious (0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakoev, S.; Kolosov, A.; Bakoev, F.; Kostyunina, O.; Bakoev, N.; Romanets, T.; Koshkina, O.; Getmantseva, L. Analysis of Homozygous-by-Descent (HBD) Segments for Purebred and Crossbred Pigs in Russia. Life 2021, 11, 861. https://0-doi-org.brum.beds.ac.uk/10.3390/life11080861
Bakoev S, Kolosov A, Bakoev F, Kostyunina O, Bakoev N, Romanets T, Koshkina O, Getmantseva L. Analysis of Homozygous-by-Descent (HBD) Segments for Purebred and Crossbred Pigs in Russia. Life. 2021; 11(8):861. https://0-doi-org.brum.beds.ac.uk/10.3390/life11080861
Chicago/Turabian StyleBakoev, Siroj, Anatoly Kolosov, Faridun Bakoev, Olga Kostyunina, Nekruz Bakoev, Timofey Romanets, Olga Koshkina, and Lyubov Getmantseva. 2021. "Analysis of Homozygous-by-Descent (HBD) Segments for Purebred and Crossbred Pigs in Russia" Life 11, no. 8: 861. https://0-doi-org.brum.beds.ac.uk/10.3390/life11080861