
Astrocytes in Neurodegenerative Diseases: A Perspective from Tauopathy and α-Synucleinopathy

Laboratory of Molecular Biology, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA
*
Author to whom correspondence should be addressed.
Life 2021, 11(9), 938; https://0-doi-org.brum.beds.ac.uk/10.3390/life11090938
Submission received: 18 August 2021
/
Revised: 6 September 2021
/
Accepted: 6 September 2021
/
Published: 9 September 2021
(This article belongs to the Special Issue Alpha-Synuclein and Non-Motor Symptoms of Parkinson’s Disease)
Abstract
:Neurodegenerative diseases are aging-associated chronic pathological conditions affecting primarily neurons in humans. Inclusion bodies containing misfolded proteins have emerged as a common pathologic feature for these diseases. In many cases, misfolded proteins produced by a neuron can be transmitted to another neuron or a non-neuronal cell, leading to the propagation of disease-associated pathology. While undergoing intercellular transmission, misfolded proteins released from donor cells can often change the physiological state of recipient cells. Accumulating evidence suggests that astrocytes are highly sensitive to neuron-originated proteotoxic insults, which convert them into an active inflammatory state. Conversely, activated astrocytes can release a plethora of factors to impact neuronal functions. This review summarizes our current understanding of the complex molecular interplays between astrocyte and neuron, emphasizing on Tau and α-synuclein (α-syn), the disease-driving proteins for Alzheimer’s and Parkinson’s diseases, respectively.
1. Introduction
Neurodegeneration refers to the progressive loss of structure and function of neurons in pathological conditions. Depending on the type and location of the affected neurons, neurodegenerative diseases can display heterogeneous clinical and pathological expressions [1]. Although research in the past has long been ‘neurocentric’, recent studies have started to shift the paradigm as new roles by glial cells in neurodegenerative diseases are being revealed.
Glial cells were first reported in 1856 by a pathologist named Rudolf Virchow in the book ’Cellular Pathology’. Derived from the ancient Greek word “glía” (meaning “glue” in English), the name “Glia” suggests these cells as “glue” that holds neurons together. However, this view has changed significantly in recent years as more and more neuronal supporting functions were identified for glial cells.
Glial cells are historically categorized into two main groups: macroglia and microglia. The former includes astrocytes, oligodendrocytes, NG2-glia, and ependymal cells, while microglia are resident phagocytes of the central nervous system (CNS). Among these cell types, astrocytes have drawn significant attention recently due to their unique neuron-safeguarding functions. As the most abundant non-neuronal cells in the CNS, astrocytes are capable of responding to many neurodegeneration-associated events such as metabolic fluctuation, molecular damage, and energy and ion homeostasis disruption [2]. Additionally, as immune-responding cells, astrocytes also participate in neuroinflammation [3]. These functions are all tightly regulated during ageing and ageing-associated neurodegeneration. Here, we review the emerging roles of astrocytes in two major pathological conditions, tauopathies and α-synucleinopathies.
2. Astrocytes in Tauopathies
2.1. Tau and Tauopathies
Intracellular neurofibrillary tangles (NFTs) formed by hyperphosphorylated Tau are a pathological hallmark of a broad spectrum of neurodegenerative disorders collectively referred to as tauopathies [4,5,6]. Tauopathies are conventionally classified into two groups. Primary tauopathies, which include progressive supranuclear palsy (PSP), frontotemporal dementia parkinsonism linked to chromosome17 (FTDP-17), Pick’s disease (PiD), corticobasal degeneration (CBD), chronic traumatic encephalopathy (CTE), and argyrophilic grain disease (AGD), refer to disease conditions in which Tau deposit is the predominant pathology [4,7]. By contrast, secondary tauopathies involve other pathogenic drivers in addition to Tau deposition. For example, Alzheimer’s disease (AD), the most prevalent cause of dementia, is a secondary tauopathy because it also involves extracellular deposition of amyloid-β (Aβ) plaques [8,9].
Tau is a microtubule-binding protein predominantly expressed in neurons in the brain [10,11]. However, Tau deposits are prevalent in both neuronal and non-neuronal cells in tauopathies. Immunohistochemistry analyses of phosphorylated Tau revealed six distinct astroglial phenotypes associated with tauopathies including astrocytic plaques (AP), tufted astrocytes (TA), ramified astrocytes (RA), and globular astroglial inclusions (GAI) in primary tauopathies, and thorn shaped astrocytes (TSA) and granular/fuzzy astrocytes (GFA) in aging-related Tau astrogliopathy (ARTAG) [12,13,14,15].
The expression of Tau is regulated by alternative splicing of the Tau-encoding gene MAPT [16]. The resulting six isoforms contain either 3 or 4 microtubule-binding repeats (referred to as 3R and 4R, respectively) combined with zero to two amino-terminal insertions (NT). Healthy adults express approximately equal amounts of 3R- and 4R-Tau, and aggregates composed of either 3R or 4R Tau have been seen in different tauopathies. However, sporadic tauopathies such as PSP, CBD, FTDP-17, and AGD feature NFT deposits exclusively composed of 4R-Tau [14].
Post-translational modifications (PTMs) of Tau such as phosphorylation, acetylation, ubiquitination, SUMOylation, methylation, and glycation have long been recognized as a critical contributing factor to tauopathies [17,18,19,20,21]. Tau PTMs may enable the formation of the highly ordered β-sheet structures, which facilitates the formation of filamentous Tau inclusions, as indicated by a recent study that reported a role of Tau ubiquitination in filament formation and strain specification [22]. PTMs may also control Tau stability, and thus influence Tau pathology, as exemplified by the implication of ubiquitin ligase and deubiquitinase (DUB) in Tau stability regulation [23,24,25]. Among reported PTMs, Tau hyperphosphorylation is thought to be the most significant driving force of tauopathy, possibly because this modification changes the affinity of Tau to microtubule, and thus its aggregation propensity. Noticeably, Tau phosphorylation was also seen in astrocytes, implying a potential role in reactive astrogliosis [26].
2.2. Astrocytes as a Modulator of AD and Tauopathies
Although most tauopathies including late-onset AD-associated tauopathies arise sporadically within the population, genome-wide association study (GWAS) have identified many tauopathy-associated single-nucleotide polymorphism (SNP) markers [27,28,29]. Intriguingly, many genes associated with increased risk of neurodegeneration are glial genes (Table 1).
ApoE is the strongest genetic risk locus for AD. ApoE E4 carriers have enhanced AD pathology, accelerated cognitive decline and worsened memory performance compared to noncarriers [30]. As a secreted lipid transport protein that moves lipids between organs, ApoE is expressed primarily in a subset of astrocytes in the CNS [31,32]. The mechanism by which ApoE variants alter AD pathology is complex, which is likely linked to the deposition and clearance of Aβ in the brain [33,34,35,36,37].
Given the tight link between AD and tauopathy, the role of ApoE in tauopathies has also been examined. By crossing the P301S Tau transgenic mice to those bearing a human ApoE knock-in allele or lacking ApoE completely, Shi et al. showed that P301S/ApoE E4 mice had significantly higher levels of intracellular Tau, more microglia activation and reactive astrogliosis compared to P301S mice bearing other ApoE variants, while the P301S mice lacking ApoE completely had the least tauopathy [38]. More recently, the same group found that astrocyte specific removal of ApoE E4 allele markedly decreased phosphorylated Tau and Tau-associated neurodegeneration, which suggested that astrocyte-derived ApoE4 is a major regulator of tauopathies [61]. However, another study suggested that neuronal ApoE expression is linked to MHC-I upregulation, which causes tauopathy and selective neurodegeneration [62].
CLU gene variants (encoding ApoJ/Clusterin) are another strong genetic risk factor for late-onset AD, as established by GWAS [40,41]. Like ApoE, CLU is an apolipoprotein predominantly expressed in astrocytes in the brain [63]. As an extracellular chaperone, CLU secreted by astrocytes can bind to Aβ to prevent Aβ aggregation [64,65,66]. Accordingly, it has been proposed that increased CLU in glia may be protective in AD and tauopathies [67].
Other AD risk factors identified by GWAS include FERMT2 (encoding Kindlin-2) [43] and WWOX [29]. FERMT2 is mainly expressed in astrocytes [68] but can also be detected in human induced pluripotent stem cell (iPSC)-derived neurons [45]. It is localized to focal adhesions where it interacts with and activates β3 integrin [69]. The role of FERMT2 in AD and tauopathy is largely unknown, but a genome-wide siRNA screen suggested that FERMT2 may increase Aβ peptide production by elevating the levels of mature APP at the cell surface via membrane recycling [44]. Another candidate-based screening found that knockdown of FERMT2 led to a reduction of phosphorylated Tau [45]. WWOX, encoding a putative oxidoreductase, is expressed in both astrocytes and neurons [29]. WWOX regulates Aβ aggregation and also binds to Tau to influence Tau hyperphosphorylation and neurofibrillary formation [47,48]. Taken together, these genome-wide studies not only identified genetic risk factors for AD and related tauopathies, but also underscored a role for glial cells, especially astrocytes, in driving Aβ- and Tau-associated neuropathology.
2.3. Astrocyte in the Propagation of Tauopathies
An unusual characteristic of tauopathies is the prion-like propagation of Tau-containing fibrils, which correlates with cognition decline and disease progression. Braak and colleagues first reported the spatial and temporal dynamics of Tau-containing fibrils in AD brains. Specifically, NFTs, first uncovered in the transentorhinal region, appear to traverse along several anatomical paths to reach the hippocampus and eventually the neocortex region [70]. The progressive spreading of Tau inclusions was later recapitulated in mouse models [71,72,73,74]. There is now comprehensive evidence that supports the idea that pathogenic Tau species undergo cell-to-cell transmission with a prion-like property [75,76,77,78]. However, the ultimate spatial distribution of Tau NFTs is distinct among tauopathies due to strain distinctions. Additionally, external factors may also influence the spreading pattern of tauopathy. For example, in AD, genetic and clinical evidence indicates that Aβ plaque deposition can facilitate the spreading of tauopathy [79,80,81]. Moreover, Tau-containing aggregates accumulated in glial cells (both microglia and astrocytes) may also modulate Tau transmission (see below).
The intercellular transmission of Tau is likely initiated when neurons release Tau either in monomeric or small oligomerized forms. Indeed, Tau is readily detected in the interstitial fluid (ISF) of the brain under normal conditions [82]. Accumulating evidence suggests that Tau species can be released from neurons independent of cell death, and this process is modulated by neuronal activities [83,84,85]. The mechanisms underlying Tau release are controversial. Specifically, some studies showed that Tau is predominantly released in a free soluble form [86,87,88,89] but other studies suggested membrane-associated vesicles such as exosome as an extracellular Tau carrier [90,91]. It is possible that multiple mechanisms coexist to regulate Tau secretion.
Once in the cell exterior, Tau may be taken up by cells via endocytosis [92], micropinocytosis [93] or other forms of cargo internalization [94]. One study suggests that healthy neurons efficiently take up both normal and aggregated Tau by distinct but overlapping mechanisms, which indicates the existence of multiple Tau receptors for internalization [95]. Not only neurons, but other cell types in the brain such as microglia and astrocytes can also engulf Tau proteins [39,93,96]. In certain immortalized cells, endocytosis of Tau preformed fibrils (PFFs) is initiated when Tau binds to the cell surface heparan sulfate proteoglycans (HSPGs) [94,97,98], which cooperate with a membrane receptor to mediate Tau internalization [99]. However, HSPG does not play a major role in Tau uptake in primary astrocytes [99,100]. We recently used a spatially resolved proteomic mapping strategy to identify the integrin αV/β1 complex as a receptor that binds human Tau fibrils to mediate their entry into astrocytes [39]. When inside the astrocyte, Tau may be cleared by lysosomal degradation or the recently reported astrocytic glymphatic system [101].
Although Tau aggregates have been observed in various cell types in the brain, most attention in the field has been given to intraneuronal or extracellular Tau deposits, while the glial involvement was rarely considered. This deficiency may significantly hinder our understanding of the mechanisms underlying the transmission of tauopathy. To better understand the role of glial Tau deposits in tauopathy, the following questions need to be carefully addressed. (i) Which glial cell type accumulates the most pathological Tau in tauopathies? (ii) Which Tau species is propagated in each tauopathy and how is their distribution in the brain sculpted? (iii) Do astrocytes or other glial cells contribute to Tau propagation? (iv) Does the accumulation of Tau in astrocytes contribute to neurodegeneration, and if so, what is the underlying mechanism?
To date, only a few published studies attempted to address these questions, which collectively paint an incomplete model. Tau accumulation in astrocytes was reported in some tauopathy mouse models [102,103]. More recently, using an in vivo reporter system, Anastasie et al. demonstrated bidirectional exchanges of Tau protein between neuron and astrocyte. They further showed that soluble Tau, but not Tau aggregates, is toxic to a subpopulation of hippocampal astrocytes [104]. This study hints at a role for astrocytes in tauopathy. A few studies investigated the disease relevance of astrocytic Tau in other experimental models. For example, expression of human Tau in glia in a Drosophila model led to neurotoxicity, suggesting that Tau, if propagated into glial cells, might have a pathogenic activity [105]. Likewise, in a transgenic mouse model, astrocyte-specific expression of human Tau leads to neurodegeneration [106]. A study by Richetin et al. also suggests astrocytic Tau as a causal factor for dementia. They detected Tau accumulation in astrocytes of the hilus, a portion of the hippocampus in AD patients; in mice, overexpression of the 3R Tau variant in hilar astrocytes of the dentate gyrus impaired mitochondrial function and thus ATP production [107]. Intriguingly, this work detected 3R Tau in astrocytes, unlike previous studies that attributed astrocytic Tau deposits predominantly to the 4R isoform [108].
Two recent papers further link Tau to the build-up of astrocytic senescent cells in the brain, which contribute to neurodegeneration. Musi et al. showed that destroying senescent cells in mice at early stages of tauopathy slows neurodegeneration and corrects aberrant brain blood flow [109], whereas Bussian et al. reported that specific elimination of senescent astrocytes is sufficient to prevent neurodegeneration and cognitive decline in a mouse model of tauopathy [110]. Although these studies both hinted at a critical role for astrocytic Tau in cell senescence, which in turn influences neurodegeneration, how the senescent state of microglia or astrocytes is aligned with other tauopathy-related features remains unclear. Altogether, the existing evidence suggests that in tauopathies, Tau proteopathy may exist beyond neurons, which warrants additional studies.
2.4. Tauopathies Are Associated with Widespread Reactive Astrogliosis
Under neurodegeneration conditions, astrocytes also undergo significant changes, which can fall into three morphologically defined categories: (i) atrophy/degeneration occurs as astrocytes lose their homeostatic function to support neuronal growth. (ii) Astroglial remodeling refers to morphologic alterations of astrocytes under disease or CNS injury conditions. (iii) Reactive astrogliosis refers to special responses of astrocytes to different insults in many CNS disorders, which result in astroglial hypertrophy (increased volume, thickened processes, and increased expression of GFAP etc. [111,112]).
Due to their sensitivity to the brain environment, astrocytes can enter a “reactive” or “activated” state now generally termed astrogliosis [113]. Many markers of reactive astrocytes [2,114,115] have been identified and used to characterize the neurodegenerative disease states. Under certain experimental conditions, reactive astrogliosis induced by lipopolysaccharide (LPS) increases the phagocytic activity of astrocytes, which may mitigate tauopathies if the activated astrocytes help to clear protein aggregates [116,117]. However, reactive astrogliosis under pathophysiological conditions can also be a major contributor of chronic neuroinflammation (Figure 1), which exacerbates neurodegeneration in several animal disease models [118,119]. Thus, it seems that upon activation, astrocytes might be transformed into multiple functional states, resulting in a heterogeneous population.
A recent transcription profiling study identified two gene expression signatures corresponding to two functional states of reactive astrogliosis termed as A1 and A2, respectively. A2 astrocytes have a neuron-supporting function and can restore neuronal activities after injury. By contrast, A1 astrocytes not only fail to promote synapse formation, but also release some neurotoxic factors. Complement C3 was later identified as an astrocyte-released factor that induces neuronal impairment, possibly through a C3 receptor (C3aR) because C3aR1 deficiency reverses plaque-proximal synapse loss in a Tau P301S mouse model [120]. Interestingly, astrocyte-released C3 appears to crosstalk to microglia as well, indicating a possible vicious cycle among neuron, astrocyte, and microglia in tauopathy [121].
The A2 to A1 switch of astrocytes, instigated by microglia, appears to convert astrocytes from a supporter of neuronal homeostasis to a cell death promoter in AD. Interleukin-1α (IL-1α), tumor necrosis factor α (TNFα), and complement component 1q (C1q) secreted from activated microglia were shown to collectively induce the A1 switch phenotype [122]. Hence, blocking the transformation of astrocytes to the A1 state by these factors may be a potential therapeutic strategy, as suggested by a recent study using a Parkinson’s disease (PD) mouse model [123]. However, this oversimplified model has recently been challenged. Concerns were raised regarding the potential overlook of astrocytic heterogeneity and the complexity of the factors implicated in shaping the astrocyte phenotypes during disease progression [124,125]. Recent advancements in single cell transcriptomics may help better define the various astrocytic states associated with different pathological conditions [126,127].
One of the common insults that change astrocyte state in neurodegenerative diseases is abnormal protein aggregates such as Aβ-, Tau-, and α-syn-containing fibrils. For instance, Aβ peptides, derived from abnormal processing of amyloid precursor protein (APP), can form distinct aggregated states, which activate different astrocytic receptors to induce a pro-inflammatory NFκB pathway [128,129]. Distinct Tau species also differentially activate integrin signaling in primary mouse astrocytes, which leads to NFκB activation and the release of pro-inflammatory cytokines and chemokines [39]. In rodent models of AD and Huntington’s disease (HD), NFκB activation in astrocytes was observed [119,130]. Thus, the NFκB pathway appears to be a critical link that connects extracellular proteotoxic insults to astrogliosis and neuroinflammation.
Besides the NFκB pathway, the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway is also ubiquitously involved in cell proliferation, survival, and differentiation. STAT3 was recently suggested as a mediator of reactive astrogliosis under pathological conditions such as AD and HDs [131]. However, the contributions of STAT3-mediated reactive astrogliosis to these diseases are not entirely clear. For example, one study suggested that JAK/STAT3 activation is associated with a scar-forming astrocyte activity in a model of acute spinal cord injury [132], which limits inflammation spreading [133]. By contrast, in an APP/PS1 model of AD, STAT3 deficient animals showed reduced β-amyloid levels and plaque burden, decreased pro-inflammatory cytokines, and rescued memory decline. Similarly, in a Tau mouse AD model, inhibition of STAT3 also rescues Tau pathology, ameliorates neuroinflammation, and reverses synaptic deficits [120]. Thus, whether reactive astrogliosis is detrimental or beneficial for damaged neurons may depend on the cause of neurodegeneration.
3. Astrocytes and α-Synucleinopathies
3.1. α-Synuclein and Parkinson’s Disease
α-synuclein (α-syn) was originally identified as a protein recognized by an antiserum against purified cholinergic synaptic vesicles [134]. It was localized to a patch of nuclear membrane extended to the presynaptic nerve terminal, which gave rise to the name of synuclein (protein present in synapse and nucleus). In the CNS, α-syn is predominantly associated with a pool of synaptic vesicles [135,136] where it appears to regulate a SNARE complex [137,138] and hence the membrane fusion events in endocytosis and exocytosis [139]. Accordingly, it may regulate synaptic plasticity and neurotransmitter release [140,141,142]. Additionally, α-syn contains a lipid binding domain essential for membrane association, lipid packing, and membrane remodeling [143,144]. Lastly, a chaperone activity was demonstrated in vitro using high concentrations of recombinant α-syn, but the physiological relevance of this finding remains unclear [145].
Although the precise function of α-syn is unclear, numerous lines of evidence have linked its dysfunction to PD pathology. These include (1) the existence of familial PD cases caused by duplication or triplication of the α-syn gene [146], (2) the identification of mutations in α-syn gene that are causally linked to PD [147,148], (3) the α-syn-containing intraneuronal Lewy bodies (LB) or Lewy neurites (LN) are a major pathologic hallmark of PD and a heterogeneous group of disorders referred to as α-synucleinopathies [149], and (4) a close correlation of α syn oligomerization and fibril propagation with the PD progression [150,151].
The mechanism of Lewy body formation remains elusive. The cellular α-syn concentration is likely a key determinant because in animals, the fibrillization kinetics of α-syn are highly sensitive to the protein concentration [152,153,154,155]. For example, genetic alterations in other PD-associated genes such as leucine-rich repeat kinase 2 (LRRK2) and glucocerebrosidase (GBA) can influence lysosome-mediated α-syn turnover to promote its aggregation [156,157]. Likewise, GWAS has identified additional variations in genes associated with lysosomal degradation pathways such as CHMP2B, TMEM175, SCARB3 and BAG3, which also influence α-syn aggregation [158]. In addition to genetic factors, many aging-associated events such as mitochondrial dysfunction [159], oxidative and ER stress [160,161] can all impact endogenous α-syn levels to affect the build-up of α-syn aggregates. Besides α-syn concentration, PTMs likely also regulate its aggregation. These include phosphorylation, acetylation, and protease-mediated cleavage [162,163]. Most studies along this line were conducted either using recombinant proteins in vitro or in cell lines. However, a recent report showed that α-syn cleaved by matrix metalloproteinases is modified by a glutaminyl cyclase at the N-terminus. The resulting pyroglutamate79-α-syn, which was detected in patient Lewy bodies, is more prone to form toxic oligomers [164].
3.2. Intercellular Transmission of α-Syn in α-Synucleinopathies
Several lines of evidence suggested that α-syn can also propagate from neuron to neuron in a prion-like manner, which can be recapitulated by cell-based models and in animals inoculated with preformed α-syn fibrils. The accumulation of α-syn-containing inclusions has also been seen in non-neuronal cells across the spectrum of α-synucleinopathies. Clinical studies showed that the topographical distribution of astrocytic α-syn inclusions closely mirrors that of the cortical intraneuronal LN and LB in PD [165], suggesting that the build-up of astrocytic α-syn aggregates may contribute to the symptomatic progression of these diseases. However, subcortical astrocytes in multiple-system atrophy (MSA) and corticobasal degeneration (CBD) do not accumulate α-syn aggregates [166]. The heterogeneity of astrocytic α-synucleinopathies in various PD variants suggest the involvement of multiple factors besides distinct conformational strains of α-syn in α-synucleinopathies, which await further elucidation [167,168,169].
Since α-syn is predominantly expressed in neurons, the observed α-syn-containing inclusions in astrocytes suggest that astrocytes scavenge extracellular α-syn released from neurons via active endocytosis [170,171,172], which should reduce extracellular α-syn and thus, the inter-neuronal transmission of α-Syn (Figure 1). Additionally, α-syn is efficiently transferred from astrocyte to astrocyte, but less efficiently from astrocyte to neuron [173]. Thus, it is unlikely that astrocytes can serve as a mediator in α-syn propagation between neurons. Altogether, these findings suggest that astrocyte might promote the elimination of α-syn inclusions in early phases of α-synucleinopathies, which offers a beneficial effect. However, persistent accumulation of α-syn aggregates might overload astrocytes, inducing stress phenotypes to impair astrocyte functions.
3.3. Role of Astrocyte in α-Syn-Associated Neuroinflammation
Like tauopathies, a key aspect of PD pathology is neuroinflammation, which is associated with reactive astrogliosis and the loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) [174,175,176,177]. Inflammation-associated cytokines like interleukin-1 β (IL1-β), interleukin-6 (IL6), and tumor necrosis factor-α (TNFα) are all elevated in the cerebrospinal fluid (CSF) or serum of PD patients [178,179,180]. This neuroinflammation response used to be considered as an event downstream of dopaminergic neuron loss. However, emerging evidence suggests that disturbed astrocytes may play an active role in α-syn-associated neuroinflammation prior to the loss of dopaminergic neurons.
It turns out that extracellular α-syn aggregates can directly interact with astrocytes via a pattern recognition receptor such as Toll-like receptor (TLR) 4, inducing a TLR4-dependent inflammatory response [181,182]. Moreover, the induction of pro-inflammatory cytokines and chemokines correlates well with the level of astrocytic α-syn, suggesting that the neuron-to-astrocyte transmission of α-syn aggregates may be coupled to neuroinflammation [171]. α-syn also strongly upregulates IL6 and inflammatory mediator intercellular adhesion molecule-1 (ICAM-1) in human astrocytes and in a human U-373 MG astrocytoma cell line, which is further linked to the activation of the major mitogen-activated protein kinase (MAPK) pathway [183]. Cytokines released by activated astrocytes can induce neuronal death, but the underlying mechanisms remain unclear [184]. When PD-related A53T mutant α-syn was expressed in astrocytes in a mouse model, increased accumulation of α-syn aggregates in astrocytes was found in pre-symptomatic and asymptomatic mouse brains, correlating with the expansion of reactive astrogliosis. These mice also developed progressing paralysis before the onset of PD-like symptoms. This study argued for a critical involvement of astrocytic α-syn in neurodegeneration via a cell non-autonomous mechanism [185].
4. Conclusions and Perspectives
In summary, neurodegenerative diseases are not a disease of one cell type. Although neuronal cell death is the primary cause of disease symptoms, the underlying mechanisms are complex and can be influenced by both neuronal factors as well as non-autonomous factors from other cell types. Astrocytes, being the most abundant non-neuronal cells in human brains, can play a significant role in neurodegenerative diseases. More attention should be given to research along this direction. Many outstanding questions need to be addressed by both in vitro cell-based assays and animal models. For example, what controls the switch that changes astrocytes from a neuron supporter to a death promoter? Are misfolded Tau or α-syn by itself sufficient to activate astrocytes in vivo? What is the precise astrocyte-activating conformation of misfolded proteins? What are the membrane receptors that recognize these misfolded proteins? Are there functionally distinct populations of activated astrocytes, and if so, what is the population that causes neuronal cell death? What are the toxic factors released from astrocytes? Is there a way to maintain the astrocytes’ garage-cleaning function without stimulating them into an inflammatory state? Answers to these questions will fill an important gap in our understanding of these devastating diseases.
Author Contributions
P.W.; writing—original draft preparation, Y.Y.; writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The research in the Ye lab is supported by an intramural research program of the National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK) of NIH.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta. Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends. Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Gotz, J. Amyloid-beta and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahlson, M.A.; Colodner, K.J. Glial Tau Pathology in Tauopathies: Functional Consequences. J. Exp. Neurosci. 2015, 9, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyns, C.E.G.; Holtzman, D.M. Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef]
- Togo, T.; Dickson, D.W. Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol. 2002, 104, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Akiyama, H.; Kondo, H.; Haga, C.; Tanno, E.; Tokuda, T.; Ikeda, S. Thorn-shaped astrocytes: Possibly secondarily induced tau-positive glial fibrillary tangles. Acta Neuropathol. 1995, 90, 620–625. [Google Scholar] [CrossRef]
- Bullmann, T.; Holzer, M.; Mori, H.; Arendt, T. Pattern of tau isoforms expression during development in vivo. Int. J. Dev. Neurosci. 2009, 27, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2020, 11, 595532. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Li, C.; Gotz, J. Tau-based therapies in neurodegeneration: Opportunities and challenges. Nat. Rev. Drug Discov. 2017, 16, 863–883. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Kontaxi, C.; Piccardo, P.; Gill, A.C. Lysine-Directed Post-translational Modifications of Tau Protein in Alzheimer’s Disease and Related Tauopathies. Front Mol. Biosci. 2017, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Duong, D.M.; Kundinger, S.R.; Wang, K.; Williams, D.; DeTure, M.; Dickson, D.W.; Cook, C.N.; et al. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 2020, 180, 633–644 e612. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, L.; Dickson, D.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; De Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004, 13, 703–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, M.; Hyeon, S.J.; Das, T.; Suh, Y.S.; Kim, Y.K.; Lee, J.S.; Song, E.J.; Ryu, H.; Yu, K. UBE4B, a microRNA-9 target gene, promotes autophagy-mediated Tau degradation. Nat. Commun. 2021, 12, 3291. [Google Scholar] [CrossRef]
- Wang, P.; Joberty, G.; Buist, A.; Vanoosthuyse, A.; Stancu, I.C.; Vasconcelos, B.; Pierrot, N.; Faelth-Savitski, M.; Kienlen-Campard, P.; Octave, J.N.; et al. Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol. 2017, 133, 731–749. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Lopez-Gonzalez, I.; Carmona, M.; Arregui, L.; Dalfo, E.; Torrejon-Escribano, B.; Diehl, R.; Kovacs, G.G. Glial and neuronal tau pathology in tauopathies: Characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol. 2014, 73, 81–97. [Google Scholar] [CrossRef] [Green Version]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Fresard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; Van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Pitas, R.E.; Boyles, J.K.; Lee, S.H.; Foss, D.; Mahley, R.W. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta 1987, 917, 148–161. [Google Scholar] [CrossRef]
- Xu, Q.; Bernardo, A.; Walker, D.; Kanegawa, T.; Mahley, R.W.; Huang, Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 2006, 26, 4985–4994. [Google Scholar] [CrossRef]
- Ma, J.; Yee, A.; Brewer, H.B., Jr.; Das, S.; Potter, H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 1994, 372, 92–94. [Google Scholar] [CrossRef]
- Bales, K.R.; Verina, T.; Dodel, R.C.; Du, Y.; Altstiel, L.; Bender, M.; Hyslop, P.; Johnstone, E.M.; Little, S.P.; Cummins, D.J.; et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat. Genet. 1997, 17, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Bales, K.R.; Verina, T.; Cummins, D.J.; Du, Y.; Dodel, R.C.; Saura, J.; Fishman, C.E.; DeLong, C.A.; Piccardo, P.; Petegnief, V.; et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1999, 96, 15233–15238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [Green Version]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Ye, Y. Filamentous recombinant human Tau activates primary astrocytes via an integrin receptor complex. Nat. Commun. 2021, 12, 95. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Wojtas, A.M.; Carlomagno, Y.; Sens, J.P.; Kang, S.S.; Jensen, T.D.; Kurti, A.; Baker, K.E.; Berry, T.J.; Phillips, V.R.; Castanedes, M.C.; et al. Clusterin ameliorates tau pathology in vivo by inhibiting fibril formation. Acta Neuropathol. Commun. 2020, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Deming, Y.; Li, Z.; Kapoor, M.; Harari, O.; Del-Aguila, J.L.; Black, K.; Carrell, D.; Cai, Y.; Fernandez, M.V.; Budde, J.; et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017, 133, 839–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuis, J.; Flaig, A.; Grenier-Boley, B.; Eysert, F.; Pottiez, V.; Deloison, G.; Vandeputte, A.; Ayral, A.M.; Mendes, T.; Desai, S.; et al. Genome-wide, high-content siRNA screening identifies the Alzheimer’s genetic risk factor FERMT2 as a major modulator of APP metabolism. Acta Neuropathol. 2017, 133, 955–966. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, S.E.; Liao, M.; Smith, R.V.; White, C.; Lagomarsino, V.N.; Xu, J.; Taga, M.; Bennett, D.A.; De Jager, P.L.; Young-Pearse, T.L. Candidate-based screening via gene modulation in human neurons and astrocytes implicates FERMT2 in Abeta and TAU proteostasis. Hum. Mol. Genet. 2019, 28, 718–735. [Google Scholar] [CrossRef]
- Wang, H.Y.; Juo, L.I.; Lin, Y.T.; Hsiao, M.; Lin, J.T.; Tsai, C.H.; Tzeng, Y.H.; Chuang, Y.C.; Chang, N.S.; Yang, C.N.; et al. WW domain-containing oxidoreductase promotes neuronal differentiation via negative regulation of glycogen synthase kinase 3beta. Cell Death Differ. 2012, 19, 1049–1059. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.Y.; Chang, N.S. WWOX dysfunction induces sequential aggregation of TRAPPC6ADelta, TIAF1, tau and amyloid beta, and causes apoptosis. Cell Death Discov. 2015, 1, 15003. [Google Scholar] [CrossRef]
- Sze, C.I.; Su, M.; Pugazhenthi, S.; Jambal, P.; Hsu, L.J.; Heath, J.; Schultz, L.; Chang, N.S. Down-regulation of WW domain-containing oxidoreductase induces Tau phosphorylation in vitro. A potential role in Alzheimer’s disease. J. Biol. Chem. 2004, 279, 30498–30506. [Google Scholar] [CrossRef] [Green Version]
- Ramanan, V.K.; Risacher, S.L.; Nho, K.; Kim, S.; Shen, L.; McDonald, B.C.; Yoder, K.K.; Hutchins, G.D.; West, J.D.; Tallman, E.F.; et al. GWAS of longitudinal amyloid accumulation on 18F-florbetapir PET in Alzheimer’s disease implicates microglial activation gene IL1RAP. Brain 2015, 138, 3076–3088. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Shiroshima, T.; Lee, S.J.; Yasumura, M.; Uemura, T.; Chen, X.; Iwakura, Y.; Mishina, M. Interleukin-1 receptor accessory protein organizes neuronal synaptogenesis as a cell adhesion molecule. J. Neurosci. 2012, 32, 2588–2600. [Google Scholar] [CrossRef]
- Li, Y.Q.; Tan, M.S.; Wang, H.F.; Tan, C.C.; Zhang, W.; Zheng, Z.J.; Kong, L.L.; Wang, Z.X.; Tan, L.; Jiang, T.; et al. Common variant in PTK2B is associated with late-onset Alzheimer’s disease: A replication study and meta-analyses. Neurosci. Lett. 2016, 621, 83–87. [Google Scholar] [CrossRef]
- Dourlen, P.; Fernandez-Gomez, F.J.; Dupont, C.; Grenier-Boley, B.; Bellenguez, C.; Obriot, H.; Caillierez, R.; Sottejeau, Y.; Chapuis, J.; Bretteville, A.; et al. Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of Tau pathology. Mol. Psychiatry 2017, 22, 874–883. [Google Scholar] [CrossRef]
- Wang, Z.; Lei, H.; Zheng, M.; Li, Y.; Cui, Y.; Hao, F. Meta-analysis of the Association between Alzheimer Disease and Variants in GAB2, PICALM, and SORL1. Mol. Neurobiol. 2016, 53, 6501–6510. [Google Scholar] [CrossRef]
- Knupp, A.; Mishra, S.; Martinez, R.; Braggin, J.E.; Szabo, M.; Kinoshita, C.; Hailey, D.W.; Small, S.A.; Jayadev, S.; Young, J.E. Depletion of the AD Risk Gene SORL1 Selectively Impairs Neuronal Endosomal Traffic Independent of Amyloidogenic APP Processing. Cell Rep. 2020, 31, 107719. [Google Scholar] [CrossRef] [PubMed]
- Hinney, A.; Albayrak, O.; Antel, J.; Volckmar, A.L.; Sims, R.; Chapman, J.; Harold, D.; Gerrish, A.; Heid, I.M.; Winkler, T.W.; et al. Genetic variation at the CELF1 (CUGBP, elav-like family member 1 gene) locus is genome-wide associated with Alzheimer’s disease and obesity. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165B, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Liao, F.; Jiang, H.; Srivatsan, S.; Xiao, Q.; Lefton, K.B.; Yamada, K.; Mahan, T.E.; Lee, J.M.; Shaw, A.S.; Holtzman, D.M. Effects of CD2-associated protein deficiency on amyloid-beta in neuroblastoma cells and in an APP transgenic mouse model. Mol. Neurodegener. 2015, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Ubelmann, F.; Burrinha, T.; Salavessa, L.; Gomes, R.; Ferreira, C.; Moreno, N.; Guimas Almeida, C. Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 2017, 18, 102–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulman, J.M.; Imboywa, S.; Giagtzoglou, N.; Powers, M.P.; Hu, Y.; Devenport, D.; Chipendo, P.; Chibnik, L.B.; Diamond, A.; Perrimon, N.; et al. Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Hum. Mol. Genet. 2014, 23, 870–877. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xiong, M.; Gratuze, M.; Bao, X.; Shi, Y.; Andhey, P.S.; Manis, M.; Schroeder, C.; Yin, Z.; Madore, C.; et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 2021, 109, 1657–1674. [Google Scholar] [CrossRef] [PubMed]
- Zalocusky, K.A.; Najm, R.; Taubes, A.L.; Hao, Y.; Yoon, S.Y.; Koutsodendris, N.; Nelson, M.R.; Rao, A.; Bennett, D.A.; Bant, J.; et al. Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeg, M.; Stravalaci, M.; Romeo, M.; Carra, A.D.; Cagnotto, A.; Rossi, A.; Diomede, L.; Salmona, M.; Gobbi, M. Clusterin Binds to Abeta1-42 Oligomers with High Affinity and Interferes with Peptide Aggregation by Inhibiting Primary and Secondary Nucleation. J. Biol. Chem. 2016, 291, 6958–6966. [Google Scholar] [CrossRef] [Green Version]
- Narayan, P.; Orte, A.; Clarke, R.W.; Bolognesi, B.; Hook, S.; Ganzinger, K.A.; Meehan, S.; Wilson, M.R.; Dobson, C.M.; Klenerman, D. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-beta(1-40) peptide. Nat. Struct. Mol. Biol. 2011, 19, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Hammad, S.M.; Ranganathan, S.; Loukinova, E.; Twal, W.O.; Argraves, W.S. Interaction of apolipoprotein J-amyloid beta-peptide complex with low density lipoprotein receptor-related protein-2/megalin. A mechanism to prevent pathological accumulation of amyloid beta-peptide. J. Biol. Chem. 1997, 272, 18644–18649. [Google Scholar] [CrossRef] [Green Version]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons from Other Pathologies. Front Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Theodosiou, M.; Widmaier, M.; Bottcher, R.T.; Rognoni, E.; Veelders, M.; Bharadwaj, M.; Lambacher, A.; Austen, K.; Muller, D.J.; Zent, R.; et al. Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. eLife 2016, 5, e10130. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; McBride, J.D.; Guo, J.L.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 2015, 130, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.C.; Vasconcelos, B.; Ris, L.; Wang, P.; Villers, A.; Peeraer, E.; Buist, A.; Terwel, D.; Baatsen, P.; Oyelami, T.; et al. Templated misfolding of Tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in Tau transgenic mice. Acta Neuropathol. 2015, 129, 875–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Frost, B.; Diamond, M.I. Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 2010, 11, 155–159. [Google Scholar] [CrossRef]
- Lee, S.J.; Desplats, P.; Sigurdson, C.; Tsigelny, I.; Masliah, E. Cell-to-cell transmission of non-prion protein aggregates. Nat. Rev. Neurol. 2010, 6, 702–706. [Google Scholar] [CrossRef] [Green Version]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, B.; Stancu, I.C.; Buist, A.; Bird, M.; Wang, P.; Vanoosthuyse, A.; Van Kolen, K.; Verheyen, A.; Kienlen-Campard, P.; Octave, J.N.; et al. Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. 2016, 131, 549–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotz, J.; Chen, F.; Van Dorpe, J.; Nitsch, R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Gomes, L.A.; Hipp, S.A.; Rijal Upadhaya, A.; Balakrishnan, K.; Ospitalieri, S.; Koper, M.J.; Largo-Barrientos, P.; Uytterhoeven, V.; Reichwald, J.; Rabe, S.; et al. Abeta-induced acceleration of Alzheimer-related tau-pathology spreading and its association with prion protein. Acta Neuropathol. 2019, 138, 913–941. [Google Scholar] [CrossRef]
- Yamada, K.; Cirrito, J.R.; Stewart, F.R.; Jiang, H.; Finn, M.B.; Holmes, B.B.; Binder, L.I.; Mandelkow, E.M.; Diamond, M.I.; Lee, V.M.; et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J. Neurosci. 2011, 31, 13110–13117. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef]
- Karch, C.M.; Jeng, A.T.; Goate, A.M. Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies. J. Biol. Chem. 2012, 287, 42751–42762. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgrafe, K.; Mandelkow, E.M.; et al. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, X.; Dage, J.L.; Citron, M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 2012, 48, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Merezhko, M.; Brunello, C.A.; Yan, X.; Vihinen, H.; Jokitalo, E.; Uronen, R.L.; Huttunen, H.J. Secretion of Tau via an Unconventional Non-vesicular Mechanism. Cell Rep. 2018, 25, 2027–2035 e2024. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, S.N.; Zheng, D.; Sabbagh, J.J.; Martin, M.D.; Chaput, D.; Darling, A.; Trotter, J.H.; Stothert, A.R.; Nordhues, B.A.; Lussier, A.; et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549. [Google Scholar] [CrossRef]
- Xu, Y.; Cui, L.; Dibello, A.; Wang, L.; Lee, J.; Saidi, L.; Lee, J.G.; Ye, Y. DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell Discov. 2018, 4, 11. [Google Scholar] [CrossRef]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [Green Version]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef] [Green Version]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, L.D.; Wassmer, T.; Fraser, G.; Smith, J.; Perkinton, M.; Billinton, A.; Livesey, F.J. Extracellular Monomeric and Aggregated Tau Efficiently Enter Human Neurons through Overlapping but Distinct Pathways. Cell Rep. 2018, 22, 3612–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perea, J.R.; Lopez, E.; Diez-Ballesteros, J.C.; Avila, J.; Hernandez, F.; Bolos, M. Extracellular Monomeric Tau Is Internalized by Astrocytes. Front. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopschinski, B.E.; Holmes, B.B.; Miller, G.M.; Manon, V.A.; Vaquer-Alicea, J.; Prueitt, W.L.; Hsieh-Wilson, L.C.; Diamond, M.I. Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus alpha-synuclein and beta-amyloid aggregates. J. Biol. Chem. 2018, 293, 10826–10840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, J.N.; Chen, J.J.; Sorum, A.W.; Miller, G.M.; Sharf, T.; See, S.K.; Hsieh-Wilson, L.C.; Kampmann, M.; Kosik, K.S. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci. Rep. 2018, 8, 6382. [Google Scholar] [CrossRef] [Green Version]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Morozova, V.; Cohen, L.S.; Makki, A.E.; Shur, A.; Pilar, G.; El Idrissi, A.; Alonso, A.D. Normal and Pathological Tau Uptake Mediated by M1/M3 Muscarinic Receptors Promotes Opposite Neuronal Changes. Front. Cell. Neurosci. 2019, 13, 403. [Google Scholar] [CrossRef] [Green Version]
- Silva, I.; Silva, J.; Ferreira, R.; Trigo, D. Glymphatic system, AQP4, and their implications in Alzheimer’s disease. Neurol. Res. Pract. 2021, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Shoji, M.; Kawarai, T.; Kawarabayashi, T.; Matsubara, E.; Murakami, T.; Sasaki, A.; Tomidokoro, Y.; Ikarashi, Y.; Kuribara, H.; et al. Accumulation of filamentous tau in the cerebral cortex of human tau R406W transgenic mice. Am. J. Pathol. 2005, 166, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Dawson, H.N.; Cantillana, V.; Chen, L.; Vitek, M.P. The tau N279K exon 10 splicing mutation recapitulates frontotemporal dementia and parkinsonism linked to chromosome 17 tauopathy in a mouse model. J. Neurosci. 2007, 27, 9155–9168. [Google Scholar] [CrossRef]
- Mate de Gerando, A.; D’Orange, M.; Augustin, E.; Josephine, C.; Auregan, G.; Gaudin-Guerif, M.; Guillermier, M.; Herard, A.S.; Stimmer, L.; Petit, F.; et al. Neuronal tau species transfer to astrocytes and induce their loss according to tau aggregation state. Brain 2021, 144, 1167–1182. [Google Scholar] [CrossRef] [PubMed]
- Colodner, K.J.; Feany, M.B. Glial fibrillary tangles and JAK/STAT-mediated glial and neuronal cell death in a Drosophila model of glial tauopathy. J. Neurosci. 2010, 30, 16102–16113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, M.S.; Lal, D.; Zhang, B.; Dabir, D.V.; Swanson, E.; Lee, V.M.; Trojanowski, J.Q. Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J. Neurosci. 2005, 25, 3539–3550. [Google Scholar] [CrossRef]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Begard, S.; Pythoud, C.; Rey, M.; et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef]
- Schoch, K.M.; DeVos, S.L.; Miller, R.L.; Chun, S.J.; Norrbom, M.; Wozniak, D.F.; Dawson, H.N.; Bennett, C.F.; Rigo, F.; Miller, T.M. Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model. Neuron. 2016, 90, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Ferrer, I. Diversity of astroglial responses across human neurodegenerative disorders and brain aging. Brain Pathol. 2017, 27, 645–674. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhauser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Burchett, J.M.; Schuler, D.R.; Cirrito, J.R.; et al. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014, 34, 9607–9620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, H.Y.; Chen, Y.C.; Chen, H.M.; Tu, P.H.; Chern, Y. A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington’s disease. Hum. Mol. Genet. 2013, 22, 1826–1842. [Google Scholar] [CrossRef] [Green Version]
- Litvinchuk, A.; Wan, Y.W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337–1353 e1335. [Google Scholar] [CrossRef]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123 e2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Dunne, A.; Lopez-Rodriguez, A.B. Astrocytes: Heterogeneous and Dynamic Phenotypes in Neurodegeneration and Innate Immunity. Neuroscientist 2019, 25, 455–474. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; De Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [Green Version]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, e167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akama, K.T.; Van Eldik, L.J. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNFalpha)-dependent, and involves a TNFalpha receptor-associated factor- and NFkappaB-inducing kinase-dependent signaling mechanism. J. Biol. Chem. 2000, 275, 7918–7924. [Google Scholar] [CrossRef] [Green Version]
- Carrero, I.; Gonzalo, M.R.; Martin, B.; Sanz-Anquela, J.M.; Arevalo-Serrano, J.; Gonzalo-Ruiz, A. Oligomers of beta-amyloid protein (Abeta1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp. Neurol. 2012, 236, 215–227. [Google Scholar] [CrossRef]
- Ben Haim, L.; Ceyzeriat, K.; Carrillo-de Sauvage, M.A.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E.; et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef]
- Herrmann, J.E.; Imura, T.; Song, B.; Qi, J.; Ao, Y.; Nguyen, T.K.; Korsak, R.A.; Takeda, K.; Akira, S.; Sofroniew, M.V. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci. 2008, 28, 7231–7243. [Google Scholar] [CrossRef]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; De Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef]
- Sharma, M.; Burre, J.; Sudhof, T.C. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 2011, 13, 30–39. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Muller, V.; Jacobsen, H.; Schindzielorz, A.; Okochi, M.; Leimer, U.; Van Der Putten, H.; Probst, A.; et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosc.i 2000, 20, 6365–6373. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, C.; Zhu, Y.; Cai, Q.; Chan, P.; Ueda, K.; Yu, S.; Yang, H. Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: An immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 2008, 1244, 40–52. [Google Scholar] [CrossRef]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Ouberai, M.M.; Wang, J.; Swann, M.J.; Galvagnion, C.; Guilliams, T.; Dobson, C.M.; Welland, M.E. Alpha-Synuclein senses lipid packing defects and induces lateral expansion of lipids leading to membrane remodeling. J. Biol. Chem. 2013, 288, 20883–20895. [Google Scholar] [CrossRef] [Green Version]
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. Alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rekas, A.; Ahn, K.J.; Kim, J.; Carver, J.A. The chaperone activity of alpha-synuclein: Utilizing deletion mutants to map its interaction with target proteins. Proteins 2012, 80, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann. Neurol. 2008, 63, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Stefanis, L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, R.; Vieira-Saecker, A.M.; Kuhn, W.; Berg, D.; Muller, T.; Kuhnl, N.; Fuchs, G.A.; Storch, A.; Hungs, M.; Woitalla, D.; et al. Increased susceptibility to sporadic Parkinson’s disease by a certain combined alpha-synuclein/apolipoprotein E genotype. Ann. Neurol. 1999, 45, 611–617. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinsons Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Kalia, S.K.; McLean, P.J.; Lozano, A.M.; Lang, A.E. Alpha-Synuclein oligomers and clinical implications for Parkinson disease. Ann. Neurol. 2013, 73, 155–169. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.W.; Hague, S.M.; Clarimon, J.; Baptista, M.; Gwinn-Hardy, K.; Cookson, M.R.; Singleton, A.B. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 2004, 62, 1835–1838. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrer, M.; Kachergus, J.; Forno, L.; Lincoln, S.; Wang, D.S.; Hulihan, M.; Maraganore, D.; Gwinn-Hardy, K.; Wszolek, Z.; Dickson, D.; et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 2004, 55, 174–179. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 Expression Augments alpha-Synuclein Sequestration into Inclusions in Neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Heilbron, K.; Vallerga, C.L.; Bandres-Ciga, S.; von Coelln, R.; Pihlstrom, L.; Simon-Sanchez, J.; Schulte, C.; Sharma, M.; Krohn, L.; et al. Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov. Disord. 2019, 34, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Coppola-Segovia, V.; Cavarsan, C.; Maia, F.G.; Ferraz, A.C.; Nakao, L.S.; Lima, M.M.; Zanata, S.M. ER Stress Induced by Tunicamycin Triggers alpha-Synuclein Oligomerization, Dopaminergic Neurons Death and Locomotor Impairment: A New Model of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 5798–5806. [Google Scholar] [CrossRef]
- Barrett, P.J.; Timothy Greenamyre, J. Post-translational modification of alpha-synuclein in Parkinson’s disease. Brain Res 2015, 1628, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Li, J.D. The Roles of Post-translational Modifications on alpha-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [Green Version]
- Hartlage-Rubsamen, M.; Bluhm, A.; Moceri, S.; Machner, L.; Koppen, J.; Schenk, M.; Hilbrich, I.; Holzer, M.; Weidenfeller, M.; Richter, F.; et al. A glutaminyl cyclase-catalyzed alpha-synuclein modification identified in human synucleinopathies. Acta Neuropathol. 2021, 142, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.J.; Halliday, G.M.; Holton, J.L.; Lashley, T.; O’Sullivan, S.S.; McCann, H.; Lees, A.J.; Ozawa, T.; Williams, D.R.; Lockhart, P.J.; et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J. Neuropathol. Exp. Neurol. 2009, 68, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Nussbaum, R.L. Genetics of Synucleinopathies. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Alegre-Abarrategui, J.; Brimblecombe, K.R.; Roberts, R.F.; Velentza-Almpani, E.; Tilley, B.S.; Bengoa-Vergniory, N.; Proukakis, C. Selective vulnerability in alpha-synucleinopathies. Acta Neuropathol. 2019, 138, 681–704. [Google Scholar] [CrossRef] [Green Version]
- Rostami, J.; Holmqvist, S.; Lindstrom, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergstrom, J.; Roybon, L.; Erlandsson, A. Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J. Neurosci. 2017, 37, 11835–11853. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Xu, Y.; Lee, J.; Jarnik, M.; Wu, X.; Bonifacino, J.S.; Shen, J.; Ye, Y. A myosin-7B-dependent endocytosis pathway mediates cellular entry of alpha-synuclein fibrils and polycation-bearing cargos. Proc. Natl. Acad. Sci. USA 2020, 117, 10865–10875. [Google Scholar] [CrossRef]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. Alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.W.; Yung, K.L.; Chan, Y.S. Reactive astrocytes as potential manipulation targets in novel cell replacement therapy of Parkinson’s disease. Curr. Drug Targets 2005, 6, 821–833. [Google Scholar] [CrossRef]
- Blum-Degen, D.; Muller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Stypula, G.; Kunert-Radek, J.; Stepien, H.; Zylinska, K.; Pawlikowski, M. Evaluation of interleukins, ACTH, cortisol and prolactin concentrations in the blood of patients with parkinson’s disease. Neuroimmunomodulation 1996, 3, 131–134. [Google Scholar] [CrossRef]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC. Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klegeris, A.; Giasson, B.I.; Zhang, H.; Maguire, J.; Pelech, S.; McGeer, P.L. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 2006, 20, 2000–2008. [Google Scholar] [CrossRef]
- Chavarria, C.; Rodriguez-Bottero, S.; Quijano, C.; Cassina, P.; Souza, J.M. Impact of monomeric, oligomeric and fibrillar alpha-synuclein on astrocyte reactivity and toxicity to neurons. Biochem. J. 2018, 475, 3153–3169. [Google Scholar] [CrossRef]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
A dual role of microglia and astrocytes in neuronal growth and neurodegenerative diseases. Accumulating evidence suggests that the activation of microglia and astrocytes may be a double-edged sword. Under healthy conditions, microglia and astrocytes engulf neuron-derived misfolded proteins such as Tau and α-syn to promote protein homeostasis in the brain micro-environment. Astrocytes can also provide other supportive functions including axonal guidance and synaptic support. However, when these cells are overactivated by toxic factors (e.g., LPS or excess amount of extracellular Tau or α-syn), they release pro-inflammatory cytokines and chemokines to disrupt neuronal integrity. Reactive microglia can also cross-activate astrocytes by releasing cytokines such as TNFα, IL1-α and C1q. Conversely, astrocytes release complement C3, which can act on both microglia and neurons to further enhance neuroinflammation. Image created in BioRender.com.
Figure 1.
A dual role of microglia and astrocytes in neuronal growth and neurodegenerative diseases. Accumulating evidence suggests that the activation of microglia and astrocytes may be a double-edged sword. Under healthy conditions, microglia and astrocytes engulf neuron-derived misfolded proteins such as Tau and α-syn to promote protein homeostasis in the brain micro-environment. Astrocytes can also provide other supportive functions including axonal guidance and synaptic support. However, when these cells are overactivated by toxic factors (e.g., LPS or excess amount of extracellular Tau or α-syn), they release pro-inflammatory cytokines and chemokines to disrupt neuronal integrity. Reactive microglia can also cross-activate astrocytes by releasing cytokines such as TNFα, IL1-α and C1q. Conversely, astrocytes release complement C3, which can act on both microglia and neurons to further enhance neuroinflammation. Image created in BioRender.com.


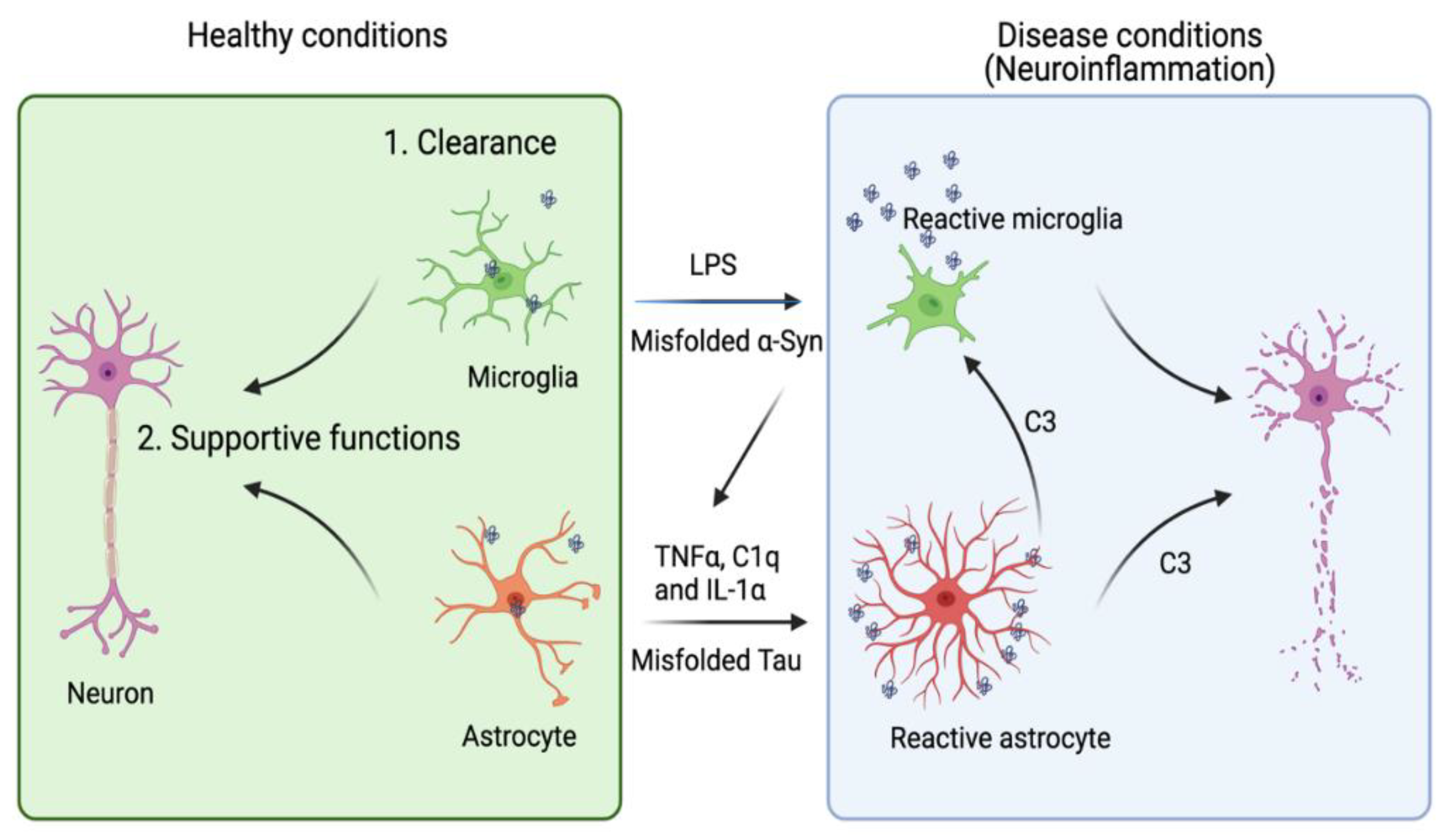{kind=link}
Table 1.
A list of astrocyte- or microglia-specific AD and tauopathy modulators.
| Gene | Glia Cell Type | Pathway | Effect on Aβ | Effect on Tau |
|---|---|---|---|---|
| APOE [30] | Astrocyte | Lipid metabolism, immune response | Aβ clearance [34] | Tau aggregation and toxicity [38,39] |
| CLU(APOJ) [40,41] | Astrocyte | Lipid metabolism, immune response | Amyloid formation [42] | Unknown |
| FERMT2 [43] | Astrocyte | Integrin signaling, and cell adhesion, angiogenesis | Aβ production [44] | Tau proteostasis [45] |
| WWOX [29] | Astrocyte | Putative oxidoreductase, neuronal differentiation [46] | Aβ aggregation [47] | Tau phosphorylation, NFT formation [47,48] |
| IL1RAP [49] | Astrocyte, oligodendrocyte | Neuronal synaptogenesis [50] | Unknown | Unknown |
| PTK2B [51] | Microglia, astrocyte | Immune response, endocytosis, synaptic transmission | Unknown | Tau toxicity [52] |
| SORL1 [53] | Microglia, astrocyte | Endosomal traffic | APP trafficking [54] | Unknown |
| CELF1 [55] | Astrocyte, oligodendrocyte, microglia | Unknown | Unknown | Unknown |
| EPHA1 [56,57] | Astrocyte, oligodendrocyte, microglia | Cell migration and proliferation, immune response | Unknown | Tau toxicity [52] |
| CD2AP [56,57] | Astrocyte, oligodendrocyte, microglia | Neurite structure modulation and blood-brain barrier integrity | Aβ production [58,59] | Tau toxicity [60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, P.; Ye, Y. Astrocytes in Neurodegenerative Diseases: A Perspective from Tauopathy and α-Synucleinopathy. Life 2021, 11, 938. https://0-doi-org.brum.beds.ac.uk/10.3390/life11090938
AMA Style
Wang P, Ye Y. Astrocytes in Neurodegenerative Diseases: A Perspective from Tauopathy and α-Synucleinopathy. Life. 2021; 11(9):938. https://0-doi-org.brum.beds.ac.uk/10.3390/life11090938
Chicago/Turabian StyleWang, Peng, and Yihong Ye. 2021. "Astrocytes in Neurodegenerative Diseases: A Perspective from Tauopathy and α-Synucleinopathy" Life 11, no. 9: 938. https://0-doi-org.brum.beds.ac.uk/10.3390/life11090938
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.