
The Case for Clinical Trials with Novel GABAergic Drugs in Diabetes Mellitus and Obesity

Centre of Discovery Brain Sciences, Deanery of Biomedical Sciences, University of Edinburgh, Edinburgh EH8 9XD, UK
Life 2022, 12(2), 322; https://0-doi-org.brum.beds.ac.uk/10.3390/life12020322
Submission received: 22 January 2022
/
Revised: 9 February 2022
/
Accepted: 16 February 2022
/
Published: 21 February 2022
(This article belongs to the Special Issue New Trends in Pharmaceutical Science)
{kind=link}
{kind=link}
Abstract
:Obesity and diabetes mellitus have become the surprising menaces of relative economic well-being worldwide. Gamma amino butyric acid (GABA) has a prominent role in the control of blood glucose, energy homeostasis as well as food intake at several levels of regulation. The effects of GABA in the body are exerted through ionotropic GABAA and metabotropic GABAB receptors. This treatise will focus on the pharmacologic targeting of GABAA receptors to reap beneficial therapeutic effects in diabetes mellitus and obesity. A new crop of drugs selectively targeting GABAA receptors has been under investigation for efficacy in stroke recovery and cognitive deficits associated with schizophrenia. Although these trials have produced mixed outcomes the compounds are safe to use in humans. Preclinical evidence is summarized here to support the rationale of testing some of these compounds in diabetic patients receiving insulin in order to achieve better control of blood glucose levels and to combat the decline of cognitive performance. Potential therapeutic benefits could be achieved (i) By resetting the hypoglycemic counter-regulatory response; (ii) Through trophic actions on pancreatic islets, (iii) By the mobilization of antioxidant defence mechanisms in the brain. Furthermore, preclinical proof-of-concept work, as well as clinical trials that apply the novel GABAA compounds in eating disorders, e.g., olanzapine-induced weight-gain, also appear warranted.
1. A Brief Primer of GABA and GABAA-RS
The main site of GABA production is the central nervous system (CNS) where it is an abundant neurotransmitter. The effects of GABA in the body are exerted through ionotropic (GABAA) and metabotropic GABAB receptors [1,2,3]. The biochemical synthetic, breakdown and uptake pathways for GABA are well known [4]. Reuptake into both neurons and glial cells has been well characterized. Importantly, extensive evidence shows that glial cells can synthesize/recycle as well as release GABA to influence neuronal activity [5,6,7,8]. The concentrations of GABA in the synaptic cleft are high, ranging from 10 to 1000 µM, usually saturating and desensitizing synaptic GABAA receptors (GABAA-R) [9]. Hence, the effects of synaptic GABA are generally thought of as “fast” and “phasic” [10]. Notably, a “slow” phasic GABAergic inhibition has been also reported [11]. Another significant category of GABA action is “tonic” inhibition, which is mediated by extra-synaptic GABAA-Rs [10] that are sensors for GABA in the interstitial space (“ambient GABA”) [12]. Tonic inhibition is fundamentally important in setting the firing threshold of cortical neurons [10,13]. The ambient GABA involved in this process is derived from overflow of the synaptic clefts, as well as through release from glial cells [6,7,8,14].
In addition to its prominent role in the CNS, potential sites of GABA action in a variety of peripheral tissues including the airways and the pancreas have been reported [15,16]. The physiological role of GABA in these systems remains to be fully explored. However, pharmacologic targeting of GABAA-Rs in these tissues appears to be effective in preclinical models [17,18,19,20] and offers new therapeutic opportunities.
GABAA-Rs are ligand-operated Cl- channels assembled from five integral membrane-protein subunits [1]. The repertory of GABAA-Rs subunits consists of several gene families (α, β, γ, δ, ε, θ, π, ρ) most of which have several isoforms. The most common GABAA-Rs configuration is 2α2βγ, a heteropentameric protein complex that spans the plasma membrane. The GABAA-R isoforms show distinct topographical distributions in the brain [2,21]. Moreover, molecular genetic dissection of the actions of diazepam, a non-selective positive allosteric modulator (PAM) of subunits, revealed that different GABAA α-subunits mediate its plethora of pharmacological effects. At a simplified level, α1 subunits were associated with sedation and protection from epilepsy, α2 with anxiety and α5 with effects on cognitive function [22]. The role of the α3 and α4 subunits remains moot, whilst α6 is primarily expressed in the cerebellum and is thus associated with motor coordination. Subsequent studies revealed that different GABAA receptor configurations underlie phasic and tonic effects of GABA [10,23,24]. Collectively, these findings provided a strong biological rationale for the development of compounds selective for GABAA-R isoforms [22,25,26].
2. Potential New GABA-ergic Drugs for Diabetes and Obesity
While GABAA receptors are targets for a wide range of drugs such as anxiolytics, hypnotics, anaesthetics and anticonvulsants [1,27], none of the compounds currently used in the clinic is selective for a particular GABAA-R isoform. The more recently characterized crop of isoform-selective compounds suitable for clinical trials is listed below.
2.1. Benzodiazepine-Site Modulators
Previously, compounds such as diazepam or alprazolam, that enhance the effects of GABA by acting at the benzodiazepine site were referred to as agonists, while those that reduce the effect of GABA e.g., Ro15-4513 or DMCM (methyl-6,7-dimethoxy-4-ethyl-beta-carboline-3-carboxylate) were called inverse agonists. At present, the terms positive and negative allosteric modulator are preferable instead of agonist and inverse agonist, respectively.
The main facets of the now-defunct GABA drug-development program at Merck, Sharpe and Dohme have been extensively reviewed [25,28,29]. Briefly, most of the compounds targeted the benzodiazepine allosteric modulatory site of GABAA-Rs. At the ligand-binding level, which is typically monitored as the displacement of 3H-flumazenil from high-affinity binding sites, these compounds had little if any isoform selectivity [28,29]. However, when tested in functional assays on recombinantly expressed receptors in vitro or in preclinical assays in vivo, they showed the characteristics of isoform-selective pharmacology. Thus the term “functional selectivity” was used for these molecules. Notable examples that have been tested in patients are MK-0777 (TPA-023; [30]), a positive allosteric modulator (PAM) preferring α2- and α3-GABAA-Rs [31] and α5IA [30,32] an α5-GABAA negative allosteric modulator (NAM). While deemed essentially safe in phase 1 clinical trials, the development of both compounds was eventually stopped as MK-0777 had unfavourable bioavailability and preclinical toxicity issues [28] while a metabolite of α5IA produced kidney damage in elderly patients [28,29]. Overall, a further problem was the lack of a good correlation between preclinical findings and the effects of the drugs in patients [26,28,29]. Nonetheless, both MK-0777 and α5IA are suitable for proof of concept pilot studies in humans e.g., see [31,32].
Basmisanil (RG1662, RO5186582, https://en.wikipedia.org/wiki/Basmisanil, (accessed on 15 February 2022) is a NAM selective for α5-GABAA-R developed by Roche. This compound shows close to 50-fold selectivity for α5-GABAA-Rs at the ligand-binding level when compared to other GABAA isoforms, thus is not just “functionally selective”. Basmisanil was safe in humans and showed clear CNS level efficacy [33]. It was tested in phase 2 clinical trials for Down’s syndrome to improve cognitive function in children (age 6–11), but the trial was terminated due to lack of efficacy https://www.downs-syndrome.org.uk/news/the-clematis-trial-conducted-by-roche/, (accessed on 15 February 2022), despite encouraging early EEG findings [34]. A phase 2 trial for improving cognitive deficits associated with schizophrenia in patients receiving non-specified anti-psychotics has also failed to meet the desired end-points https://clinicaltrials.gov/ct2/show/results/NCT02953639, (accessed on 15 February 2022).
2.2. Compounds Acting at the GABA-Site
Muscimol is the best-known non-selective GABA-site agonist. Isoform selective GABAA agonists have not been produced so far. A priori, given the high sequence homology of the α- and β-subunits that form the GABA-binding pocket [35,36,37], it would seem unlikely that this goal is achievable. However, following on from computer-modelling studies [38], recent data indicate that a segment (loop F) of the extracellular domain of the α-subunit, that shows significant sequence variation between isoforms, determines the potency of GABA on GABAA receptors [39]. In particular, these results raise the possibility of designing agonists selective for α2-GABAA receptors.
Gaboxadol (THIP, now OV101) is a potent agonist of GABA receptors that contain α4- or α6-subunits complemented with β- and δ-subunits, which have restricted anatomic distribution in the thalamus, hippocampus, and cerebellum and are mainly extra-synaptic in location [40,41]. This compound was also in clinical development and while it has been discontinued because of unwanted long-term side-effects, it is eminently suitable for shorter-term clinical work [42].
The variable segment of loop F of the α-subunit is also important in determining the isoform selectivity of novel GABAA competitive antagonists such as S44819 [43,44]. S44819 (Servier) appears to be selective for extra-synaptic α5-GABAA-Rs [43]. Moreover, in moderate doses, this compound is effective in modifying neuronal activity in the human motor cortex [45] and has been tested in phase 2 clinical trials for post-stroke recovery. No adverse effects were detected but ultimately the drug was discontinued due to lack of efficacy [46]. This trial has been criticized due to the selection of inadequate end-points [47,48]
Taken together, at least two different benzodiazepine site compounds and a GABA-site competitive antagonist are available for clinical trials to inhibit α5-GABAA-Rs. In addition, MK-777 is a PAM for the simultaneous enhancement of α2- and α3-GABAA-Rs that is also safe to use in humans in short-term trials [31]. Gaboxadol is a clinically tested agonist for α4 and α6 subunit-containing GABAA receptors.
3. Diabetes Mellitus: Replenishing the Pancreatic Beta Cell Pool
3.1. Unmet Medical Need
Diabetes mellitus is a serious metabolic disorder that is increasingly common globally. It is a dysfunction of the endocrine islets of the pancreas caused by variable etiologies. In Type 1 diabetes the primary lesion appears to be the autoimmune destruction of the pancreatic beta-cells that produce insulin, with secondary consequences to other cell types in the islets. The essence of Type 2 diabetes is functionally inadequate insulin secretion and/or marked insulin resistance of the target organs. Whilst numerous drugs, including various formulations of the beta-cell hormone insulin, are available to combat diabetes mellitus, adequate control of plasma glucose and the long-term cardiovascular and neuropsychiatric complications of the disorder remain a serious problem. The ideal therapy would be to restore the insulin secretory capacity and physiological regulation of pancreatic islet beta cells. This remains an unmet medical need.
3.2. Therapeutic Rationale
Pancreatic islets are intricate organs that show major species differences with respect to the microtopography and consequent functional orchestration of the constituent cells [49,50]. The arrangement of the cells in human pancreatic islets indicates a fundamental role for paracrine interactions between alpha- and beta-cells that secrete glucagon and insulin, respectively [49]. GABA is an important paracrine factor in pancreatic islets, its levels in the pancreas are comparable to those in the brain [51,52]. All the relevant components of GABA-ergic neurotransmission, including GABAA and GABAB receptors, have been demonstrated in pancreatic islets [49,52,53]. Beta-cells have a considerably higher GABA content than alpha-cells [53]. A further notable aspect of GABA action in islets is that KCC2, the K+-Cl− co-transporter protein that is required for the hyperpolarizing action of GABA in nerve cells [54] is expressed in alpha-cells but is absent from beta-cells [55]. Thus GABA depolarizes beta-cells, thereby facilitating insulin secretion and intracellular Ca2+ signals, while it hyperpolarizes alpha-cells and consequently inhibits the secretion of glucagon [52]. More significantly, it was shown by several groups that chronic administration of GABA in streptozotocin-diabetic mice converts islet alpha-cells into beta-cell-like insulin-producing cells and normalizes plasma glucose levels [19,56,57,58]. Treatment with GABAergic compounds also improves the survival and proliferation of beta cells of human islets grafted into mice [19,56,57,58]. Several of the GABAA-R subunits have been found in human pancreatic islets [52,59,60,61], and the localization of the α2-subunit in beta-cells appears particularly consistent and potentially significant [60,62].
Since the discovery that the GABA biosynthetic enzyme GAD65 is a target antigen in autoimmune diabetes [63,64], interest in the role of GABA in the immune system has intensified. Various immunomodulatory actions of GABAA receptors have been reported [20], which could be significant with respect to the autoimmune destruction of islet beta cells as well as the role of inflammation in insulin resistance in Type 2 diabetics. Evidence for the expression of several GABAA receptor subunits by immunocompetent cells has been provided at the mRNA level [20,65,66]. However, it is ultimately unclear which receptor configurations mediate the purported anti-inflammatory actions of GABA [20,67,68]. At this point, expression of the α1-subunit appears to be most frequently reported. Importantly, evidence for possible inflammatory effects of GABA has also been generated [69].
3.3. Potential Mode of Pharmacologic Intervention
The data outlined above indicate that activation of α2-GABAA receptors could prove beneficial in improving the deficiency of islet beta-cell function. Although GABA is available as a food supplement, it is unclear whether or not it is non-toxic at the dosage required for an islet-trophic effect. A recent report on the pharmacokinetics and tolerability of high doses of GABA has been published with no major adverse effects of 2 g of GABA three times daily for seven days. However, a recent preclinical study reported the appearance of nonalcoholic steatohepatitis (NASH) in mice fed a methionine-choline deficient diet and dosed with 2 mg/mL GABA in the drinking water for four weeks [69]. Moreover, inflammatory actions of GABA mediated by bicuculline-sensitive GABA receptors were also found in the same study. Further analysis is required to assess the significance of these findings with respect to the use of GABA as an islet-trophic agent. An option to reduce the dose of GABA required for islet-trophic effects would be to co-administer a GABAA PAM [19], preferably one that does not penetrate the CNS to avoid unwanted side effects. Most likely, GABAA PAMs that do not penetrate the blood-brain barrier already exist, except that until recently they were not thought to be of therapeutic value. However, there is a weakness to this approach as the unwanted side-effects of GABA could be also enhanced by a PAM that is not GABAA isoform-selective. The optimal choice appears to be the combination of an α2-GABAA-R selective PAM with a CNS-penetrating α5 inhibitor (NAM or GABA antagonist). This combination would enhance GABA action at the islets, potentially ameliorate the dysfunction of the hypoglycemia counter-regulatory response at the VMN and diminish the probability of cognitive impairment. The current crop of compounds, MK-0777 and α5IA or S44819 would be suitable for a clinical trial along this therapeutic principle. As a caveat, it should be mentioned that in vitro MK-0777 is substantially more potent on α3-GABAA receptors than on α2 containing ones [70,71]. Finally, experimental compounds that selectively potentiate α2-GABAA receptors and inhibit α5-GABAA containing ones are worth mentioning here [72].
4. Diabetes Mellitus: Prevention of Iatrogenic Hypoglycemia in Insulin-Dependent Diabetics
4.1. Unmet Medical Need
Recurrent iatrogenic hypoglycemia leading to hypoglycaemia associated autonomic failure is a serious complication in insulin-treated diabetes and is a limiting factor in maintaining proper glycemic control [73,74,75]. Repeated hypoglycaemia induced by insulin attenuates the endocrine counter-regulatory response to hypoglycemia, which normally involves a marked increase of plasma glucagon, norepinephrine and cortisol [73,74]. As a result, patients receiving chronic insulin therapy undergo episodes of poorly compensated hypoglycaemia and in many cases develop impaired perception of hypoglycemia. Hypoglycemia can lead to neuronal degeneration [76], and moreover diabetes facilitates brain small vessel disease, thereby potentially further aggravating the damage to neuronal function [77]. Currently, there are no adequate measures to prevent this complication of insulin therapy. It is of note, that the attenuation of the hypoglycemic counter-regulatory response can be also induced by repeated hypoglycemia in rodents as well as non-diabetic human subjects [78].
4.2. Therapeutic Rationale
The brain, and particularly the ventromedial nucleus of the hypothalamus (VMN), plays a crucial role in sensing hypoglycemia and initiating the physiological counter-regulatory responses that rapidly correct it [73,79,80,81]. There is a large body of preclinical evidence implicating GABAergic neurotransmission in the VMN in the etiopathology of recurrent iatrogenic hypoglycemia induced by insulin therapy [73,80]. Significantly, repeated hypoglycemia markedly increases the levels of the GABA synthetic enzyme GAD as well as GABA in the ventromedial hypothalamus of rats [82]. Moreover, in rats with an attenuated counter-regulatory response induced by repeated hypoglycemia, localized inhibition of ionotropic GABA (GABAA) receptors in the ventromedial hypothalamus restored the endocrine counter-regulatory response [82].
The anatomical topography of GABAergic neurotransmission in the VMN is remarkable in that the VMN contains virtually no cell bodies that express the key GABA-synthetic enzymes GAD 65 or 67 [83]. Thus, GABA in the VMN is largely derived from afferent nerve fibres originating outside of the nucleus. With respect to the potential sources of the GABAergic input to the VMN, the dorsomedial hypothalamic nucleus, the lateral hypothalamic area, the arcuate nucleus, the tuberomamillary nucleus and the amygdala are most notable [84]. In the case of the tuberomamillary nucleus, the GABAergic afferents also contain and release histamine [85]. With respect to the distribution of GABAA-R, the dorsomedial part of the VMN stands out as expressing high levels of α2 and α5 subunits, whereas the α3 subunit appears less abundant and tends to be localized in the ventrolateral aspect of the VMN [21,86,87,88,89]. By comparison, the expression of GABAA-R α-subunits in the arcuate nucleus, which is an important hypothalamic centre of nutrition and food intake, appeared to be much lower and consisted mainly of the α1 isoform [21,88].
4.3. Potential Mode of Pharmacologic Intervention
From a pharmacological point of view, it would be relevant to examine whether or not treatment with α5IA or S44819 would reverse the insulin-induced reprogramming of the counter-regulatory response to hypoglycemia or even prevent it from developing in the first place. Should these studies yield negative results further work with MK-0777 or the combination of MK-0777 and one of the α5-GABAA-R inhibitors appears warranted.
5. Diabetes Mellitus: Combating Cognitive Decline
5.1. Unmet Medical Need
An important long-term side-effect of insulin-dependent diabetes is the impairment of cognitive function, particularly attention and cognitive flexibility [90]. One causative factor in this process is likely to be recurrent hypoglycemia, especially if the subjects have impaired awareness of it [91]. In the case of type 2 diabetes, patients have been reported to have increased brain concentrations of GABA, which was correlated with the degree of the impairment of cognitive function observed in this condition [92]. In sum, both types of diabetes are associated with a slow decline in cognitive performance despite efforts to achieve glycemic control. Cognitive impairment is a serious impediment to self-sufficiency and thus an increasing social burden in the ageing human population. Taken together, adjuvant procognitive therapy to attenuate the decline of cognitive performance in diabetic patients is an unmet medical need.
5.2. Therapeutic Rationale
The mechanisms underlying the neurotoxic effect of hypoglycemic episodes in insulin-dependent diabetes are thought to be similar to those involved in stroke—hypoperfusion-reperfusion injury [76] a major element of which is oxidative damage [93]. Preclinical studies have demonstrated that inhibition of α-5 GABAARs ameliorates the impairment of cognitive performance induced by various etiologies [94,95,96,97,98] including stroke [99]. Significantly, these compounds also enhanced the post-stroke recovery of sensory-motor function in rodent models [99,100]. In a more recent study, S44819 was shown to prevent/reverse the impairment of working and recognition memory induced by mild stroke in rats or chronic cerebral hypoperfusion in mice [98]. The likely mechanisms underlying these effects of α-5 GABAA inhibition include facilitation of synaptic plasticity [29,43] and the enhancement of spontaneous neuronal network activity that favours antioxidant neuroprotective processes in the brain [98,101,102].
5.3. Potential Mode of Pharmacologic Intervention
Inhibition of α5-GABAARs (α5IA, basmisanil or S44819) as a therapy to improve cognitive function in diabetes appears to be worthwhile, because of the anticipated neuroprotective effects and the remediation of the dysfunction of synaptic plasticity. Moreover, the same agents may also ameliorate the defect in the counter-regulatory response to hypoglycemia as outlined in the previous section and thereby improve glycemic control. Last, but not least, α5-GABAAR inhibition could also prove beneficial with respect to comorbidities such as anxiety and depression [103] that are often associated with type 1 diabetes. With respect to Type 2 diabetes, the recent clinical findings indicating an increase of brain GABA levels and their potential impact on cognitive function also justify the therapeutic concept of α5-GABAA-R inhibition. A summary of the proposed adjunct GABAergic pharmacologic therapy of insulin-dependent diabetes mellitus is shown in Figure 1.
6. Obesity: The Case of Olanzapine Induced Weight Gain
6.1. Unmet Medical Need
Olanzapine (Zyprexa) is one of the most frequently prescribed antipsychotics. As with several other antipsychotics in this class, it is known to increase body weight, sometimes leading to metabolic syndrome and Type 2 diabetes [104]. As olanzapine and atypical antipsychotics are valuable for several therapeutic applications that require chronic administration, the control of olanzapine-induced body weight gain is an unmet medical need.
6.2. Principle of Therapeutic Intervention
Despite numerous studies addressing the underlying causes of olanzapine-induced weight gain, there is no consensus on the subject [105]. An important problem is the lack of good preclinical models: female rats readily gain weight on olanzapine, but males do not gain sufficient weight on a normal diet to achieve statistical power with realistic numbers of experimental animals [106]. Functional studies point to the role of the antagonism of histamine 1 (H1) receptors [107] and 5-HT2c receptors [108] by olanzapine. Hypothalamic AMP kinase activity is enhanced by H1 antagonists and provides a parsimonious explanation of weight gain [107]. However, there are studies that fail to confirm this [109]. A direct metabolic effect of olanzapine on cellular nutrient handling, independent of its antipsychotic receptor profile, has been also proposed [110].
Whilst food-intake studies in rodents have notoriously low translational potential, it is interesting to highlight here certain parallels between olanzapine-induced weight gain in humans and that induced by lesioning of the VMN in rats (see [111,112] for review). Both treatments produce a transient weight gain as if attaining a new set-point [113]. Both work most reliably in females [111,114] and disrupt the normal secretion of adrenal corticosteroids [115,116]. A recent study highlighted a potential sexually dimorphic mechanism by which metabolic GluR5 receptors interact with estrogen receptors to alter the function of steroidogenic factor 1 (SF1) positive VMN neurons [117], i.e., indicating the potential for female-specific regulation of metabolism at the level of the VMN. The SF1 neurons are known to regulate the sensitivity to insulin in peripheral tissues [118] as well as to suppress food intake through projections to the paraventricular thalamic nuclei [119]. In a yet further aspect, histamine H1 receptors, implicated in the weight-gaining effects of olanzapine in humans, are prominent in the VMN and histaminergic activation of the VMN in rats reduces food intake [120,121,122]. On the basis of these findings, it is attractive to speculate that a major action of olanzapine is to impair the anorexic functionality of the VMN. As the VMN prominently expresses α2- and α5-GABAA receptors, and in view of the reported interaction between the actions of histamine and GABA in the control of the excitability of VMN neurons [123] further work exploring the role of these receptors in olanzapine-induced weight gain appears justified.
As the first human genetic approach to the problem, a recent genome-wide association study was targeted at weight gain induced by the atypical antipsychotics clozapine or olanzapine [124]. A highly significant association of antipsychotic-induced weight gain with polymorphisms in the GABRA2 gene that encodes the GABAA-R α2-subunit was found. Previous genome-wide association studies have implicated GABRA2 in the obesity of normal subjects [125].
6.3. Potential Mode of Pharmacologic Intervention
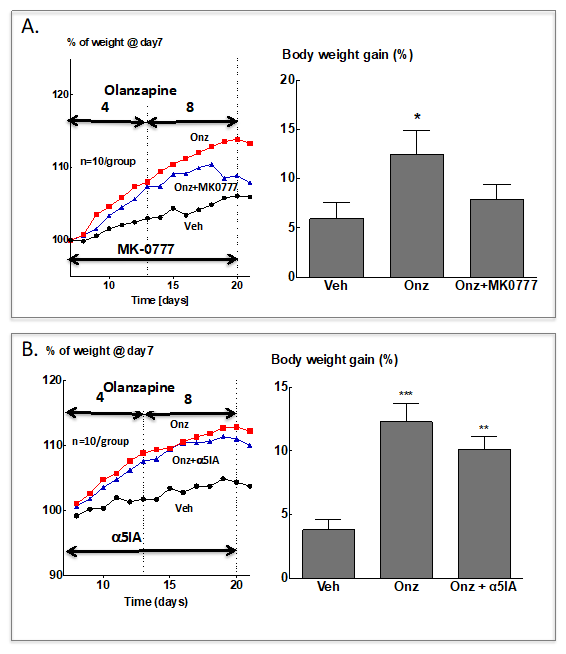
The findings outlined above prompted us to examine the effect of the α2/α3-GABAA PAM, MK-0777 on olanzapine-induced weight gain in female Sprague–Dawley rats. Interestingly, MK-0777 (10 mg/kg twice daily) failed to prevent the weight gain induced by olanzapine administration. However, once the typical plateau of olanzapine-induced weight gain was reached, a significant degree of weight loss was observed so that body weight gain after two weeks of simultaneous treatment with olanzapine and MK-0777 was not different from that in the vehicle-treated group (Figure 2A). Under identical conditions the α5-GABAA NAM α5IA had no significant effect on olanzapine induced weight gain (Figure 2B).
Thus, further exploration of the effects of MK-0777 or further α2/α3-GABAA PAMs on olanzapine-induced weight gain appears warranted. An adjuvant therapy achieving α2-GABA enhancement as well as α5-GABA inhibition could be also advantageous, as olanzapine-induced weight gain, and disturbances of plasma glucose regulation, as well as cognitive deficits associated with schizophrenia [126] may be all improved.
7. Future Perspectives
A major advance for GABAergic therapeutics would be a more detailed structural understanding of the way in which drugs interact with the heteropentameric complex [129]. Cryo-electronmicroscopy should help in this respect [130], but will have to be aided by special pharmacological agents that allow exploration of the role played by, for instance, loop F of the extracellular domain in ligand recognition [39,131] and various other sites of drug-receptor interaction [132]. This approach may well lead to selective α2-GABAA agonists that could significantly improve the therapy of diabetes mellitus. Moreover, such compounds could also have favourable properties for the control of stress, anxiety and overeating [133]. In addition to the supply of novel drugs, studies of human genetic polymorphisms and epigenetics are likely to yield important new applications of existing GABAergic compounds.
Footnote: + Note that it in some publications, ventromedial hypothalamus and ventromedial nucleus (VMN) are used interchangeably. However, in addition to the VMN, the ventromedial hypothalamus also contains the arcuate nucleus and the median eminence with their respective afferent inputs.
Funding
The cited study was supported by the Egis PLC (Budapest)—Les Laboratoires Servier (Suresnes) research cooperation agreement.
Institutional Review Board Statement
All experimental protocols were approved by the Animal Care and Use Ethical Committee of Egis Pharmaceuticals PLC and complied with the Hungarian Law of Animal Care and Use (1998. XVIII) approved 15 March 2015.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
I would like to thank István Gacsályi and Gábor Gigler for fruitful discussions and permission to publish our unpublished data.
Conflicts of Interest
The author confirms that the contents of this chapter have no conflict of interest.
References
- Olsen, R.W.; Sieghart, W. International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: Classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 2008, 60, 243–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, U.; Möhler, H. GABAA receptor subtypes: Therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 483–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belelli, D.; Hales, T.G.; Lambert, J.J.; Luscher, B.; Olsen, R.; Peters, J.A.; Rudolph, U.; Sieghart, W. GABAA receptors in GtoPdb v.2021.3. IUPHAR/BPS Guide Pharmacol CITE 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Roth, F.C.; Draguhn, A. GABA metabolism and transport: Effects on synaptic efficacy. Neural Plast. 2012, 2012, 805830. [Google Scholar] [CrossRef] [Green Version]
- Héja, L.; Nyitrai, G.; Kékesi, O.; Dobolyi, A.; Szabó, P.; Fiath, R.; Ulbert, I.; Pál-Szenthe, B.; Palkovits, M.; Kardos, J. Astrocytes convert network excitation to tonic inhibition of neurons. BMC Biol. 2012, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Karayannis, T.; Elfant, D.; Huerta-Ocampo, I.; Teki, S.; Scott, R.S.; Rusakov, D.A.; Jones, M.V.; Capogna, M. Slow GABA transient and receptor desensitization shape synaptic responses evoked by hippocampal neurogliaform cells. J. Neurosci. 2010, 30, 9898–9909. [Google Scholar] [CrossRef]
- Yoon, B.E.; Woo, J.; Chun, Y.E.; Chun, H.; Jo, S.; Bae, J.Y.; An, H.; Min, J.O.; Oh, S.J.; Han, K.S.; et al. Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. 2014, 592, 4951–4968. [Google Scholar] [CrossRef]
- Lee, S.; Yoon, B.E.; Berglund, K.; Oh, S.J.; Park, H.; Shin, H.S.; Augustine, G.J.; Lee, C.J. Channel-mediated tonic GABA release from glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef]
- Jones, M.V.; Westbrook, G.L. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron 1995, 15, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Brickley, S.G.; Módy, I. Extrasynaptic GABA(A) receptors: Their function in the CNS and implications for disease. Neuron 2012, 73, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Capogna, M.; Pearce, R.A. GABAA,slow: Causes and consequences. Trends Neurosci. 2011, 34, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.Y.; Canning, K.J.; Zhu, G.; Pennefather, P.; Macdonald, J.F.; Orser, B.A. Tonically activated GABAA receptors in hippocampal neurons are high-affinity, low-conductance sensors for extracellular GABA. Mol. Pharmacol. 2003, 63, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonin, R.P.; Martin, L.J.; Macdonald, J.F.; Orser, B.A. Alpha5GABAA receptors regulate the intrinsic excitability of mouse hippocampal pyramidal neurons. J. Neurophysiol. 2007, 98, 2244–2254. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.C.; Kullmann, D.M. Tonic GABAA Receptor-Mediated Signaling in Epilepsy; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK98181/ (accessed on 15 February 2022).
- Ong, J.; Kerr, D.I. GABA-receptors in peripheral tissues. Life Sci. 1990, 46, 1489–1501. [Google Scholar] [CrossRef]
- Erdő, S.L.; Wolff, J.R. Gamma-Aminobutyric acid outside the mammalian brain. J. Neurochem. 1990, 54, 363–372. [Google Scholar] [CrossRef]
- Lee, W.S.; Limmroth, V.; Ayata, C.; Cutrer, F.M.; Waeber, C.; Yu, X.; Moskowitz, M.A. Peripheral GABAA receptor-mediated effects of sodium valproate on dural plasma protein extravasation to substance P and trigeminal stimulation. Br. J. Pharmacol. 1995, 116, 1661–1667. [Google Scholar] [CrossRef] [Green Version]
- Mizuta, K.; Xu, D.; Pan, Y.; Comas, G.; Sonett, J.R.; Zhang, Y.; Panettieri, R.A., Jr.; Yang, J.; Emala, C.W., Sr. GABAA receptors are expressed and facilitate relaxation in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1206–L1216. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Dang, H.; Middleton, B.; Kaufman, D.L. Clinically applicable GABA receptor positive allosteric modulators promote ss-cell replication. Sci. Rep. 2017, 7, 374. [Google Scholar] [CrossRef] [Green Version]
- Barragan, A.; Weidner, J.M.; Jin, Z.; Korpi, E.R.; Birnir, B. GABAergic signalling in the immune system. Acta Physiol. 2015, 213, 819–827. [Google Scholar] [CrossRef]
- Pirker, S.; Schwarzer, C.; Wieselthaler, A.; Sieghart, W.; Sperk, G. GABA(A) receptors: Immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 2000, 101, 815–850. [Google Scholar] [CrossRef]
- Möhler, H.; Crestani, F.; Rudolph, U. GABA(A)-receptor subtypes: A new pharmacology. Curr. Opin. Pharmacol. 2001, 1, 22–25. [Google Scholar] [CrossRef]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: Phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 2005, 6, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Ferando, I.; Mody, I. Interneuronal GABAA receptors inside and outside of synapses. Curr. Opin. Neurobiol. 2014, 26, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, G.R.; Collinson, N.; Atack, J.R. Development of subtype selective GABAA modulators. CNS Spectr. 2005, 10, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Skolnick, P. Anxioselective anxiolytics: On a quest for the Holy Grail. Trends Pharmacol. Sci. 2012, 33, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.J.; Gharpure, A.; Teng, J.; Zhuang, Y.; Howard, R.J.; Zhu, S.; Noviello, C.M.; Walsh, R.M., Jr.; Lindahl, E.; Hibbs, R.E. Shared structural mechanisms of general anaesthetics and benzodiazepines. Nature 2020, 585, 303–308. [Google Scholar] [CrossRef]
- Atack, J.R. GABAA receptor subtype-selective modulators. I. alpha2/alpha3-selective agonists as non-sedating anxiolytics. Curr. Top. Med. Chem. 2011, 11, 1176–1202. [Google Scholar] [CrossRef]
- Atack, J.R. GABAA receptor subtype-selective modulators. II. alpha5-selective inverse agonists for cognition enhancement. Curr. Top. Med. Chem. 2011, 11, 1203–1214. [Google Scholar] [CrossRef]
- Atack, J.R. GABA(A) receptor subtype-selective efficacy: TPA023, an alpha2/alpha3 selective non-sedating anxiolytic and alpha5IA, an alpha5 selective cognition enhancer. CNS Neurosci. Ther. 2008, 14, 25–35. [Google Scholar] [CrossRef]
- Buchanan, R.W.; Keefe, R.S.; Lieberman, J.A.; Barch, D.M.; Csernansky, J.G.; Goff, D.C.; Gold, J.M.; Green, M.F.; Jarskog, L.F.; Javitt, D.C.; et al. A randomized clinical trial of MK-0777 for the treatment of cognitive impairments in people with schizophrenia. Biol. Psychiatry 2011, 69, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Nutt, D.J.; Besson, M.; Wilson, S.J.; Dawson, G.R.; Lingford-Hughes, A.R. Blockade of alcohol’s amnestic activity in humans by an alpha5 subtype benzodiazepine receptor inverse agonist. Neuropharmacology 2007, 53, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Hipp, J.F.; Knoflach, F.; Comley, R.; Ballard, T.M.; Honer, M.; Trube, G.; Gasser, R.; Prinssen, E.; Wallace, T.L.; Rothfuss, A.; et al. Basmisanil, a highly selective GABA(A)-α5 negative allosteric modulator: Preclinical pharmacology and demonstration of functional target engagement in man. Sci. Rep. 2021, 11, 7700. [Google Scholar] [CrossRef] [PubMed]
- Goeldner, C.; Kishnani, P.S.; Skotko, B.G.; Casero, J.L.; Hipp, J.F.; Derks, M.; Hernandez, M.C.; Khwaja, O.; Lennon-Chrimes, S.; Noeldeke, J.; et al. A randomized, double-blind, placebo-controlled phase II trial to explore the effects of a GABAA-alpha5 NAM (basmisanil) on intellectual disability associated with Down syndrome. J. Neurodev. Disord. 2022, 14, 10. [Google Scholar] [CrossRef]
- Ernst, M.; Bruckner, S.; Boresch, S.; Sieghart, W. Comparative models of GABAA receptor extracellular and transmembrane domains: Important insights in pharmacology and function. Mol. Pharmacol. 2005, 68, 1291–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurny, R.; Ramerstorfer, J.; Price, K.; Brams, M.; Ernst, M.; Nury, H.; Verheij, M.; Legrand, P.; Bertrand, D.; Bertrand, S.; et al. Pentameric ligand-gated ion channel ELIC is activated by GABA and modulated by benzodiazepines. Proc. Natl. Acad. Sci. USA 2012, 109, E3028–E3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, R.; Kongsbak, K.; Sorensen, P.L.; Sander, T.; Balle, T. A unified model of the GABA(A) receptor comprising agonist and benzodiazepine binding sites. PLoS ONE 2013, 8, e52323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, T.S.; Lau, E.Y.; Lightstone, F.C. A role for loop F in modulating GABA binding affinity in the GABA(A) receptor. J. Mol. Biol. 2012, 422, 310–323. [Google Scholar] [CrossRef]
- Pálvölgyi, A.; Móricz, K.; Pataki, A.; Mihalik, B.; Gigler, G.; Megyeri, K.; Udvari, S.; Gacsályi, I.; Antoni, F.A. Loop F of the GABAA receptor alpha subunit governs GABA potency. Neuropharmacology 2018, 128, 408–415. [Google Scholar] [CrossRef]
- Krogsgaard-Larsen, P.; Frølund, B.; Liljefors, T.; Ebert, B. GABA(A) agonists and partial agonists: THIP (Gaboxadol) as a non-opioid analgesic and a novel type of hypnotic. Biochem. Pharmacol. 2004, 68, 1573–1580. [Google Scholar] [CrossRef]
- Harrison, N.L. Mechanisms of sleep induction by GABA(A) receptor agonists. J. Clin. Psychiatry 2007, 68 (Suppl. S5), 6–12. [Google Scholar]
- Lankford, D.A.; Corser, B.C.; Zheng, Y.-P.; Li, Z.; Snavely, D.B.; Lines, C.R.; Deacon, S. Effect of gaboxadol on sleep in adult and elderly patients with primary insomnia: Results from two randomized, placebo-controlled, 30-night polysomnography studies. Sleep 2008, 31, 1359–1370. [Google Scholar] [PubMed]
- Etherington, L.A.; Pálvölgyi, A.; Mihalik, B.; Ling, I.; Pallagi, K.; Kertész, S.; Gunn, B.G.; Brown, A.R.; Livesey, M.R.; Belelli, D.; et al. Selective targeting of extra-synaptic α5-GABAA receptors receptors by a new therapeutic agent S44819 (Egis-13529). Neuropharmacology 2017, 125, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihalik, B.; Pálvölgyi, A.; Bogár, F.; Megyeri, K.; Ling, I.; Barkóczy, J.; Bartha, F.; Martinek, T.; Gacsályi, I.; Antoni, F.A. Loop-F of the α-subunit determines the pharmacologic profile of novel competitive inhibitors of GABAA receptors. Eur. J. Pharmacol. 2017, 798, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Darmani, G.; Zipser, C.M.; Bohmer, G.M.; Deschet, K.; Muller-Dahlhaus, F.; Belardinelli, P.; Schwab, M.; Ziemann, U. Effects of the selective alpha5-GABAAR Antagonist S44819 on excitability in the human brain: A TMS-EMG and TMS-EEG Phase I study. J. Neurosci. 2016, 36, 12312–12320. [Google Scholar] [CrossRef] [Green Version]
- Chabriat, H.; Bassetti, C.L.; Marx, U.; Audoli-Inthavong, M.L.; Sors, A.; Lambert, E.; Wattez, M.; Hermann, D.M. Safety and efficacy of GABA(A) α5 antagonist S44819 in patients with ischaemic stroke: A multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2020, 19, 226–233. [Google Scholar] [CrossRef]
- Cramer, S.C. Issues important to the design of stroke recovery trials. Lancet Neurol. 2020, 19, 197–198. [Google Scholar] [CrossRef]
- Hermann, D.M.; Bassetti, C.L.; Marx, U.; Audoli-Inthavong, M.-L.; Chabriat, H. Refining endpoints for stroke recovery trials. Lancet Neurol. 2020, 19, 381–382. [Google Scholar] [CrossRef]
- Caicedo, A. Paracrine and autocrine interactions in the human islet: More than meets the eye. Semin. Cell Dev. Biol. 2013, 24, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A.; Hodson, D.J. Minireview: Intraislet regulation of insulin secretion in humans. Mol. Endocrinol. 2013, 27, 1984–1995. [Google Scholar] [CrossRef] [Green Version]
- Michalik, M.; Erecinska, M. GABA in pancreatic islets: Metabolism and function. Biochem. Pharmacol. 1992, 44, 1–9. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, Q.; Prud’homme, G.J. GABAergic system in the endocrine pancreas: A new target for diabetes treatment. Diabetes Metab. Syndr. Obes. 2015, 8, 79–87. [Google Scholar] [PubMed] [Green Version]
- Jenstad, M.; Chaudhry, F.A. The amino acid transporters of the glutamate/GABA-glutamine cycle and their impact on insulin and glucagon secretion. Front. Endocrinol. 2013, 4, 199. [Google Scholar] [CrossRef] [Green Version]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, S.L.; Roussa, E.; Le Rouzic, P.; Thevenod, F.; Alper, S.L.; Best, L.; Brown, P.D. Expression of K+-Cl- cotransporters in the alpha-cells of rat endocrine pancreas. Biochim. Biophys. Acta 2004, 1667, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Purwana, I.; Zheng, J.; Li, X.; Deurloo, M.; Son, D.O.; Zhang, Z.; Liang, C.; Shen, E.; Tadkase, A.; Feng, Z.P.; et al. GABA promotes human beta-cell proliferation and modulates glucose homeostasis. Diabetes 2014, 63, 4197–4205. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, T.; Avolio, F.; Vieira, A.; Ben-Othman, N.; Courtney, M.; Gjernes, E.; Hadzic, B.; Druelle, N.; Navarro Sanz, S.; Silvano, S.; et al. GABA signaling stimulates alpha-cell-mediated beta-like cell neogenesis. Commun. Integr. Biol. 2017, 10, e1300215. [Google Scholar] [CrossRef]
- Ben-Othman, N.; Vieira, A.; Courtney, M.; Record, F.; Gjernes, E.; Avolio, F.; Hadzic, B.; Druelle, N.; Napolitano, T.; Navarro-Sanz, S.; et al. Long-term GABA administration induces alpha cell-mediated beta-like cell neogenesis. Cell 2017, 168, 73–85.e11. [Google Scholar] [CrossRef] [Green Version]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Clark, A.; Walker, J.N.; Johnson, P.R.; Rorsman, P. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 2010, 59, 1694–1701. [Google Scholar] [CrossRef] [Green Version]
- Taneera, J.; Jin, Z.; Jin, Y.; Muhammed, S.J.; Zhang, E.; Lang, S.; Salehi, A.; Korsgren, O.; Renstrom, E.; Groop, L.; et al. Gamma-Aminobutyric acid (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia 2012, 55, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honore, C.; Courtney, M.; Huber, K.V.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins target GABAA receptor signaling and impair alpha cell identity. Cell 2017, 168, 86–100.e115. [Google Scholar] [CrossRef] [Green Version]
- Korol, S.V.; Jin, Z.; Jin, Y.; Bhandage, A.K.; Tengholm, A.; Gandasi, N.R.; Barg, S.; Espes, D.; Carlsson, P.O.; Laver, D.; et al. Functional characterization of native, high-affinity GABAa receptors in human pancreatic beta cells. EBioMedicine 2018, 30, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, D.L.; Clare-Salzler, M.; Tian, J.; Forsthuber, T.; Ting, G.S.; Robinson, P.; Atkinson, M.A.; Sercarz, E.E.; Tobin, A.J.; Lehmann, P.V. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature 1993, 366, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.L.; Erlander, M.G.; Clare-Salzler, M.; Atkinson, M.A.; Maclaren, N.K.; Tobin, A.J. Autoimmunity to two forms of glutamate decarboxylase in insulin-dependent diabetes mellitus. J. Clin. Investig. 1992, 89, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Lu, Y.; Zhang, H.; Chau, C.H.; Dang, H.N.; Kaufman, D.L. Gamma-aminobutyric acid inhibits T cell autoimmunity and the development of inflammatory responses in a mouse type 1 diabetes model. J. Immunol. 2004, 173, 5298–5304. [Google Scholar] [CrossRef] [PubMed]
- Bergeret, M.; Khrestchatisky, M.; Tremblay, E.; Bernard, A.; Gregoire, A.; Chany, C. GABA modulates cytotoxicity of immunocompetent cells expressing GABAA receptor subunits. Biomed. Pharmacother. 1998, 52, 214–219. [Google Scholar] [CrossRef]
- Dionisio, L.; Arias, V.; Bouzat, C.; Esandi Mdel, C. GABAA receptor plasticity in Jurkat T cells. Biochimie 2013, 95, 2376–2384. [Google Scholar] [CrossRef]
- Tian, J.; Dang, H.N.; Yong, J.; Chui, W.S.; Dizon, M.P.; Yaw, C.K.; Kaufman, D.L. Oral treatment with gamma-aminobutyric acid improves glucose tolerance and insulin sensitivity by inhibiting inflammation in high fat diet-fed mice. PLoS ONE 2011, 6, e25338. [Google Scholar] [CrossRef] [Green Version]
- Seok Roh, Y.; Cho, A.; Zhou, Z.; Jeong, H.; Park, J.-E.; Cha, Y.-S.; Oh, S.-H.; Lim, C.-W.; Kim, B. Γ-Aminobutyric acid promotes methionine-choline deficient diet-induced nonalcoholic steatohepatitis. J. Biomed. Res. 2017, 31, 65–73. [Google Scholar]
- Atack, J.R.; Wafford, K.A.; Tye, S.J.; Cook, S.M.; Sohal, B.; Pike, A.; Sur, C.; Melillo, D.; Bristow, L.; Bromidge, F.; et al. TPA023 [7-(1,1-dimethylethyl)-6-(2-ethyl-2H-1,2,4-triazol-3-ylmethoxy)-3-(2-fluorophenyl )-1,2,4-triazolo[4,3-b]pyridazine], an agonist selective for alpha2- and alpha3-containing GABAA receptors, is a nonsedating anxiolytic in rodents and primates. J. Pharmacol. Exp. Ther. 2006, 316, 410–422. [Google Scholar] [CrossRef]
- De Lucas, A.G.; Ahring, P.K.; Larsen, J.S.; Rivera-Arconada, I.; Lopez-Garcia, J.A.; Mirza, N.R.; Munro, G. GABAA alpha5 subunit-containing receptors do not contribute to reversal of inflammatory-induced spinal sensitization as indicated by the unique selectivity profile of the GABAA receptor allosteric modulator NS16085. Biochem. Pharmacol. 2015, 93, 370–379. [Google Scholar] [CrossRef]
- Kenéz, Á.; Bertha, F.; Barkóczy, J.; Antoni, F.A.; Gacsályi, I.; Mihalik, B.; Gigler, G.; Móricz, K.; Németh, G.; Angyalne Pataki, Á.; et al. Dihydro-Oxazinobenzodiazepine Compounds, a Process for Their Preparation and Pharmaceutical Compositions Containing Them. U.S. Patent WO2015110848A1, 30 July 2015. [Google Scholar]
- McCrimmon, R.J.; Sherwin, R.S. Hypoglycemia in type 1 diabetes. Diabetes 2010, 59, 2333–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, D.L.; Cryer, P.E. Mechanisms of hypoglycemia-associated autonomic failure in diabetes. N. Engl. J. Med. 2013, 369, 362–372. [Google Scholar]
- Halimi, S. Severe hypoglycaemia the “tip of the iceberg”: An underestimated risk in both type 1 and type 2 diabetic patients. Diabetes Metab. 2015, 41, 105–106. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, G.M.I.; Wang, P.; Ciardi, A.; Mamillapalli, R.; Johnson, J.; Zhu, W.; Eid, T.; Behar, K.; Chan, O. Impaired Glutamatergic Neurotransmission in the Ventromedial Hypothalamus May Contribute to Defective Counterregulation in Recurrently Hypoglycemic Rats. Diabetes 2017, 66, 1979–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrington, M.S.; Farmerie, S.; Ertl, A.C.; Wang, Z.; Tate, D.B.; Davis, S.N. Effects of antecedent GABAA activation with alprazolam on counterregulatory responses to hypoglycemia in healthy humans. Diabetes 2010, 59, 1074–1081. [Google Scholar] [CrossRef] [Green Version]
- Mccrimmon, R. Glucose sensing during hypoglycemia: Lessons from the lab. Diabetes Care 2009, 32, 1357–1363. [Google Scholar] [CrossRef] [Green Version]
- Chan, O.; Sherwin, R. Influence of VMH fuel sensing on hypoglycemic responses. Trends Endocrinol. Metab. 2013, 24, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Meek, T.H.; Nelson, J.T.; Matsen, M.E.; Dorfman, M.D.; Guyenet, S.J.; Damian, V.; Allison, M.B.; Scarlett, J.M.; Nguyen, H.T.; Thaler, J.P.; et al. Functional identification of a neurocircuit regulating blood glucose. Proc. Natl. Acad. Sci. USA 2016, 113, E2073–E2082. [Google Scholar] [CrossRef] [Green Version]
- Chan, O.; Cheng, H.; Herzog, R.; Czyzyk, D.; Zhu, W.; Wang, A.; Mccrimmon, R.J.; Seashore, M.R.; Sherwin, R.S. Increased GABAergic tone in the ventromedial hypothalamus contributes to suppression of counterregulatory responses after antecedent hypoglycemia. Diabetes 2008, 57, 1363–1370. [Google Scholar] [CrossRef] [Green Version]
- Sunkin, S.M.; Ng, L.; Lau, C.; Dolbeare, T.; Gilbert, T.L.; Thompson, C.L.; Hawrylycz, M.; Dang, C. Allen Brain Atlas: An integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 2013, 41, D996–D1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamitakahara, A.; Xu, B.; Simerly, R. Ventromedial hypothalamic expression of BDNF is required to establish normal patterns of afferent GABAergic connectivity and responses to hypoglycemia. Mol. Metab. 2016, 5, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ye, Z.; Houston, C.M.; Zecharia, A.Y.; Ma, Y.; Zhang, Z.; Uygun, D.S.; Parker, S.; Vyssotski, A.L.; Yustos, R.; et al. Wakefulness is governed by GABA and histamine cotransmission. Neuron 2015, 87, 164–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sur, C.; Fresu, L.; Howell, O.; Mckernan, R.M.; Atack, J.R. Autoradiographic localization of alpha5 subunit-containing GABAA receptors in rat brain. Brain Res. 1999, 822, 265–270. [Google Scholar] [CrossRef]
- Li, M.; Szabo, A.; Rosenberg, H.C. Evaluation of native GABA(A) receptors containing an alpha 5 subunit. Eur. J. Pharmacol. 2001, 413, 63–72. [Google Scholar] [CrossRef]
- Bäckberg, M.; Ultenius, C.; Fritschy, J.M.; Meister, B. Cellular localization of GABA receptor alpha subunit immunoreactivity in the rat hypothalamus: Relationship with neurones containing orexigenic or anorexigenic peptides. J. Neuroendocrinol. 2004, 16, 589–604. [Google Scholar] [CrossRef]
- Atack, J.R.; Alder, L.; Cook, S.M.; Smith, A.J.; Mckernan, R.M. In vivo labelling of alpha5 subunit-containing GABA(A) receptors using the selective radioligand [(3)H]L-655,708. Neuropharmacology 2005, 49, 220–229. [Google Scholar] [CrossRef]
- Kodl, C.T.; Seaquist, E.R. Cognitive dysfunction and diabetes mellitus. Endocr. Rev. 2008, 29, 494–511. [Google Scholar] [CrossRef]
- Hansen, T.I.; Olsen, S.E.; Haferstrom, E.C.D.; Sand, T.; Frier, B.M.; Haberg, A.K.; Bjorgaas, M.R. Cognitive deficits associated with impaired awareness of hypoglycaemia in type 1 diabetes. Diabetologia 2017, 60, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Van Bussel, F.C.; Backes, W.H.; Hofman, P.A.; Puts, N.A.; Edden, R.A.; Van Boxtel, M.P.; Schram, M.T.; Stehouwer, C.D.; Wildberger, J.E.; Jansen, J.F. Increased GABA concentrations in type 2 diabetes mellitus are related to lower cognitive functioning. Medicine 2016, 95, e4803. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Ballard, T.M.; Knoflach, F.; Prinssen, E.; Borroni, E.; Vivian, J.A.; Basile, J.; Gasser, R.; Moreau, J.L.; Wettstein, J.G.; Buettelmann, B.; et al. RO4938581, a novel cognitive enhancer acting at GABAA alpha5 subunit-containing receptors. Psychopharmacology 2009, 202, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Dawson, G.R.; Maubach, K.A.; Collinson, N.; Cobain, M.; Everitt, B.J.; Macleod, A.M.; Choudhury, H.I.; Mcdonald, L.M.; Pillai, G.; Rycroft, W.; et al. An inverse agonist selective for alpha5 subunit-containing GABAA receptors enhances cognition. J. Pharmacol. Exp. Ther. 2006, 316, 1335–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redrobe, J.P.; Elster, L.; Frederiksen, K.; Bundgaard, C.; De Jong, I.E.; Smith, G.P.; Bruun, A.T.; Larsen, P.H.; Didriksen, M. Negative modulation of GABAA alpha5 receptors by RO4938581 attenuates discrete sub-chronic and early postnatal phencyclidine (PCP)-induced cognitive deficits in rats. Psychopharmacology 2012, 221, 451–468. [Google Scholar] [CrossRef] [PubMed]
- Milic, M.; Timic, T.; Joksimovic, S.; Biawat, P.; Rallapalli, S.; Divljakovic, J.; Radulovic, T.; Cook, J.M.; Savic, M.M. PWZ-029, an inverse agonist selective for alpha(5) GABAAreceptors, improves object recognition, but not water-maze memory in normal and scopolamine-treated rats. Behav. Brain Res. 2013, 241, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gacsályi, I.; Móricz, K.; Gigler, G.; Megyeri, K.; Machado, P.; Antoni, F.A. Persistent therapeutic effect of a novel alpha5-GABAA receptor antagonist in rodent preclinical models of vascular cognitive impairment. Eur. J. Pharmacol. 2018, 834, 118–125. [Google Scholar] [CrossRef]
- Wang, Y.C.; Dzyubenko, E.; Sanchez-Mendoza, E.H.; Sardari, M.; Silva De Carvalho, T.; Doeppner, T.R.; Kaltwasser, B.; Machado, P.; Kleinschnitz, C.; Bassetti, C.L.; et al. Postacute delivery of GABAA alpha5 antagonist promotes postischemic neurological recovery and peri-infarct brain remodeling. Stroke 2018, 49, 2495–2503. [Google Scholar] [CrossRef]
- Clarkson, A.N.; Huang, B.S.; Macisaac, S.E.; Mody, I.; Carmichael, S.T. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 2010, 468, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Samson, A.J.; Robertson, G.; Zagnoni, M.; Connolly, C.N. Neuronal networks provide rapid neuroprotection against spreading toxicity. Sci. Rep. 2016, 6, 33746. [Google Scholar] [CrossRef] [Green Version]
- Zanos, P.; Nelson, M.E.; Highland, J.N.; Krimmel, S.R.; Georgiou, P.; Gould, T.D.; Thompson, S.M. A negative allosteric modulator for alpha5 subunit-containing GABA receptors exerts a rapid and persistent antidepressant-like action without the side effects of the NMDA receptor antagonist ketamine in mice. eNeuro 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Rojo, L.E.; Gaspar, P.A.; Silva, H.; Risco, L.; Arena, P.; Cubillos-Robles, K.; Jara, B. Metabolic syndrome and obesity among users of second generation antipsychotics: A global challenge for modern psychopharmacology. Pharmacol. Res. 2015, 101, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.P.; Kirk, S.L. Metabolic side effects of antipsychotic drug treatment—pharmacological mechanisms. Pharmacol. Ther. 2010, 125, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zwaal, E.M.; Janhunen, S.K.; La Fleur, S.E.; Adan, R.A. Modelling olanzapine-induced weight gain in rats. Int. J. Neuropsychopharmacol. 2014, 17, 169–186. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Deng, C.; Huang, X.F. The role of hypothalamic H1 receptor antagonism in antipsychotic-induced weight gain. CNS Drugs 2013, 27, 423–434. [Google Scholar] [CrossRef]
- Lord, C.C.; Wyler, S.C.; Wan, R.; Castorena, C.M.; Ahmed, N.; Mathew, D.; Lee, S.; Liu, C.; Elmquist, J.K. The atypical antipsychotic olanzapine causes weight gain by targeting serotonin receptor 2C. J. Clin. Investig. 2017, 127, 3402–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferno, J.; Varela, L.; Skrede, S.; Vazquez, M.J.; Nogueiras, R.; Dieguez, C.; Vidal-Puig, A.; Steen, V.M.; Lopez, M. Olanzapine-induced hyperphagia and weight gain associate with orexigenic hypothalamic neuropeptide signaling without concomitant AMPK phosphorylation. PLoS ONE 2011, 6, e20571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, R.H.; Jokinen, J.D.; Massey, V.L.; Falkner, K.C.; Shi, X.; Yin, X.; Zhang, X.; Beier, J.I.; Arteel, G.E. Olanzapine activates hepatic mammalian target of rapamycin: New mechanistic insight into metabolic dysregulation with atypical antipsychotic drugs. J. Pharmacol. Exp. Ther. 2013, 347, 126–135. [Google Scholar] [CrossRef] [Green Version]
- King, B.M. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol. Behav. 2006, 87, 221–244. [Google Scholar] [CrossRef]
- Khodai, T.; Luckman, S.M. Ventromedial Nucleus of the Hypothalamus Neurons Under the Magnifying Glass. Endocrinology 2021, 162, bqab141. [Google Scholar] [CrossRef]
- Sindelar, D.K.; Carson, M.W.; Morin, M.; Shaw, J.; Barr, R.J.; Need, A.; Alexander-Chacko, J.; Coghlan, M.; Gehlert, D.R. LLY-2707, a novel nonsteroidal glucocorticoid antagonist that reduces atypical antipsychotic-associated weight gain in rats. J. Pharmacol. Exp. Ther. 2014, 348, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhardt, S.; Haberhausen, M.; Heinzel-Gutenbrunner, M.; Gebhardt, N.; Remschmidt, H.; Krieg, J.C.; Hebebrand, J.; Theisen, F.M. Antipsychotic-induced body weight gain: Predictors and a systematic categorization of the long-term weight course. J. Psychiatr. Res. 2009, 43, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Horsley, C.; Aguila, S.; Dallman, M.F. The hypothalamic ventromedial nuclei couple activity in the hypothalamo-pituitary-adrenal axis to the morning fed or fasted state. J. Neurosci. 1996, 16, 8170–8180. [Google Scholar] [CrossRef] [PubMed]
- Assie, M.B.; Carilla-Durand, E.; Bardin, L.; Maraval, M.; Aliaga, M.; Malfetes, N.; Barbara, M.; Newman-Tancredi, A. The antipsychotics clozapine and olanzapine increase plasma glucose and corticosterone levels in rats: Comparison with aripiprazole, ziprasidone, bifeprunox and F15063. Eur. J. Pharmacol. 2008, 592, 160–166. [Google Scholar] [CrossRef]
- Fagan, M.P.; Ameroso, D.; Meng, A.; Rock, A.; Maguire, J.; Rios, M. Essential and sex-specific effects of mGluR5 in ventromedial hypothalamus regulating estrogen signaling and glucose balance. Proc. Natl. Acad. Sci. USA 2020, 117, 19566–19577. [Google Scholar] [CrossRef]
- Coutinho, E.A.; Okamoto, S.; Ishikawa, A.W.; Yokota, S.; Wada, N.; Hirabayashi, T.; Saito, K.; Sato, T.; Takagi, K.; Wang, C.-C.; et al. Activation of SF1 Neurons in the Ventromedial Hypothalamus by DREADD Technology Increases Insulin Sensitivity in Peripheral Tissues. Diabetes 2017, 66, 2372–2386. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, D.; Sweeney, P.; Yang, Y. An excitatory ventromedial hypothalamus to paraventricular thalamus circuit that suppresses food intake. Nat. Commun. 2020, 11, 6326. [Google Scholar] [CrossRef] [PubMed]
- Panula, P.; Nuutinen, S. The histaminergic network in the brain: Basic organization and role in disease. Nat. Rev. Neurosci. 2013, 14, 472–487. [Google Scholar] [CrossRef]
- Provensi, G.; Blandina, P.; Passani, M.B. The histaminergic system as a target for the prevention of obesity and metabolic syndrome. Neuropharmacology 2016, 106, 3–12. [Google Scholar] [CrossRef]
- Clapp, R.H.; Luckman, S.M. Proxyfan acts as a neutral antagonist of histamine H3 receptors in the feeding-related hypothalamic ventromedial nucleus. Br. J. Pharmacol. 2012, 167, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Jang, I.S.; Rhee, J.S.; Watanabe, T.; Akaike, N.; Akaike, N. Histaminergic modulation of GABAergic transmission in rat ventromedial hypothalamic neurones. J. Physiol. 2001, 534, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Zai, C.C.; Tiwari, A.K.; Chowdhury, N.I.; Brandl, E.J.; Shaikh, S.A.; Freeman, N.; Lieberman, J.A.; Meltzer, H.Y.; Muller, D.J.; Kennedy, J.L. Association study of GABAA alpha2 receptor subunit gene variants in antipsychotic-associated weight gain. J. Clin. Psychopharmacol. 2015, 35, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Speliotes, E.K.; Loos, R.J.; Li, S.; Lindgren, C.M.; Heid, I.M.; Berndt, S.I.; Elliott, A.L.; Jackson, A.U.; Lamina, C.; et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 2009, 41, 25–34. [Google Scholar] [PubMed]
- Fioravanti, M.; Bianchi, V.; Cinti, M.E. Cognitive deficits in schizophrenia: An updated metanalysis of the scientific evidence. BMC Psychiatry 2012, 12, 64. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Young, A.J.; Ehli, E.A.; Nowotny, D.; Davies, P.S.; Droke, E.A.; Soundy, T.J.; Davies, G.E. Metformin and berberine prevent olanzapine-induced weight gain in rats. PLoS ONE 2014, 9, e93310. [Google Scholar] [CrossRef]
- Atack, J.R. Preclinical and clinical pharmacology of the GABAA receptor alpha5 subtype-selective inverse agonist alpha5IA. Pharmacol. Ther. 2010, 125, 11–26. [Google Scholar] [CrossRef]
- Puthenkalam, R.; Hieckel, M.; Simeone, X.; Suwattanasophon, C.; Feldbauer, R.V.; Ecker, G.F.; Ernst, M. Structural studies of GABAA receptor binding sites: Which experimental structure tells us what? Front. Mol. Neurosci. 2016, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Laverty, D.; Desai, R.; Uchanski, T.; Masiulis, S.; Stec, W.J.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Steyaert, J.; Miller, K.W.; et al. Cryo-EM structure of the human alpha1beta3gamma2 GABAA receptor in a lipid bilayer. Nature 2019, 565, 516–520. [Google Scholar] [CrossRef]
- Krall, J.; Balle, T.; Krogsgaard-Larsen, N.; Sorensen, T.E.; Krogsgaard-Larsen, P.; Kristiansen, U.; Frølund, B. GABAA receptor partial agonists and antagonists: Structure, binding mode, and pharmacology. Adv. Pharmacol. 2015, 72, 201–227. [Google Scholar]
- Masiulis, S.; Desai, R.; Uchański, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABA(A) receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459. [Google Scholar] [CrossRef]
- Dallman, M.F.; Pecoraro, N.; Akana, S.F.; La Fleur, S.E.; Gomez, F.; Houshyar, H.; Bell, M.E.; Bhatnagar, S.; Laugero, K.D.; Manalo, S. Chronic stress and obesity: A new view of “comfort food”. Proc. Natl. Acad. Sci. USA 2003, 100, 11696–11701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
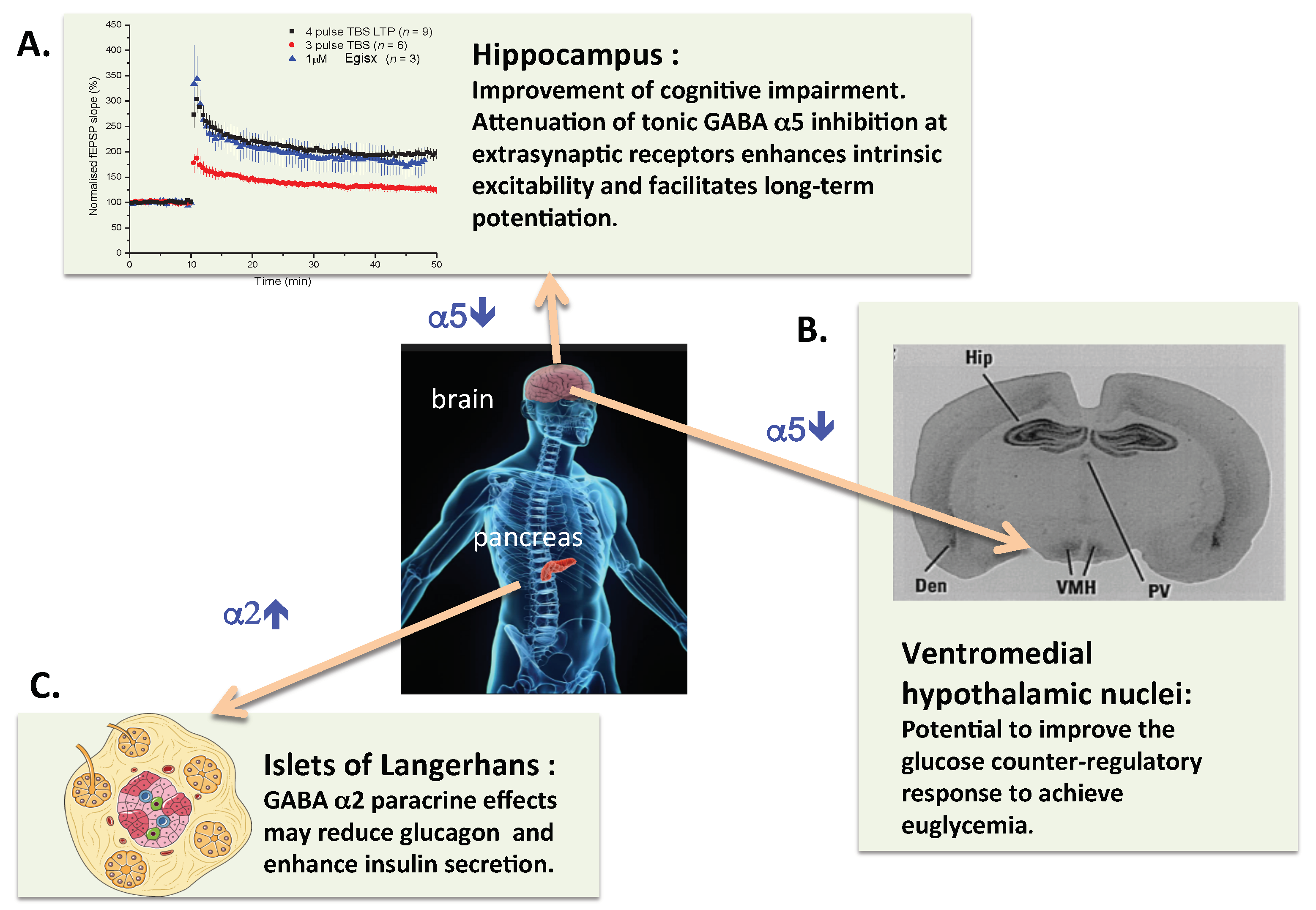
Figure 1.
Schematic representation of the potential therapeutic benefits of treatment of insulin-dependent diabetic patients with a combination of an α2-GABAA enhancer and an α5-GABAA inhibitor compound. (A) It is well established in preclinical models that inhibitors of α5-GABAA-R facilitate hippocampal synaptic plasticity and can improve impairment of learning and memory. (B) In addition to the hippocampus, the hypothalamic ventromedial nuclei stand out with high levels of α5-GABAA binding sites in rat brain [86]. This has been confirmed by mRNA in situ hybridization in human brain. Thus it is justified to hypothesize that inhibition of α5-GABAA-R could reduce the GABAergic impairment of the glucose counterregulatory response in insulin-dependent diabetics. (C) The insulinotrophic effects of GABA appear to be mediated by α2-GABAA-R, thus an α2-GABAA PAM could improve pancreatic islet beta cell function. Finally, it is of note that compounds with dual, α2-GABAA enhancer and α5-GABAA inhibitor activity have been produced [72].
Figure 1.
Schematic representation of the potential therapeutic benefits of treatment of insulin-dependent diabetic patients with a combination of an α2-GABAA enhancer and an α5-GABAA inhibitor compound. (A) It is well established in preclinical models that inhibitors of α5-GABAA-R facilitate hippocampal synaptic plasticity and can improve impairment of learning and memory. (B) In addition to the hippocampus, the hypothalamic ventromedial nuclei stand out with high levels of α5-GABAA binding sites in rat brain [86]. This has been confirmed by mRNA in situ hybridization in human brain. Thus it is justified to hypothesize that inhibition of α5-GABAA-R could reduce the GABAergic impairment of the glucose counterregulatory response in insulin-dependent diabetics. (C) The insulinotrophic effects of GABA appear to be mediated by α2-GABAA-R, thus an α2-GABAA PAM could improve pancreatic islet beta cell function. Finally, it is of note that compounds with dual, α2-GABAA enhancer and α5-GABAA inhibitor activity have been produced [72].

Figure 2.
Effect of treatment with GABAA-R modulators on olanzapine-induced increases of body weight in female Sprague–Dawley rats (G. Gigler, I. Gacsályi, F. A. Antoni, unpublished data). Female rats were allowed to accommodate in individual cages and were weighed daily for 7 days. Vehicle (0.9% saline w/v) and olanzapine treatment were started on day 8, 4 mg/kg p.o. per day for 7 days and subsequently 8 mg/kg for the next 7 days [127]. (A) MK-0777 was suspended in 1% Tween 80 and given at 10 mg/kg i.p. at the same time (2 h before lights off) as olanzapine; vehicle and olanzapine only treated animals received 1% Tween 80 (2 mL/kg) i.p. Left panel: Means, error bars are not shown for the sake of clarity. Right panel: Mean body weights on day 21 after the last administration of drugs. Means ± S.E.M. are shown, N = 10/group. Data were analysed by one-way ANOVA followed by Dunnett’s test for multiple comparisons. * p < 0.05 vs. the vehicle group. (B) As in A. except that α5IA [128] was the GABAA-R modulator used. ** p < 0.01, *** p < 0.001 vs. the vehicle group.
Figure 2.
Effect of treatment with GABAA-R modulators on olanzapine-induced increases of body weight in female Sprague–Dawley rats (G. Gigler, I. Gacsályi, F. A. Antoni, unpublished data). Female rats were allowed to accommodate in individual cages and were weighed daily for 7 days. Vehicle (0.9% saline w/v) and olanzapine treatment were started on day 8, 4 mg/kg p.o. per day for 7 days and subsequently 8 mg/kg for the next 7 days [127]. (A) MK-0777 was suspended in 1% Tween 80 and given at 10 mg/kg i.p. at the same time (2 h before lights off) as olanzapine; vehicle and olanzapine only treated animals received 1% Tween 80 (2 mL/kg) i.p. Left panel: Means, error bars are not shown for the sake of clarity. Right panel: Mean body weights on day 21 after the last administration of drugs. Means ± S.E.M. are shown, N = 10/group. Data were analysed by one-way ANOVA followed by Dunnett’s test for multiple comparisons. * p < 0.05 vs. the vehicle group. (B) As in A. except that α5IA [128] was the GABAA-R modulator used. ** p < 0.01, *** p < 0.001 vs. the vehicle group.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Antoni, F.A. The Case for Clinical Trials with Novel GABAergic Drugs in Diabetes Mellitus and Obesity. Life 2022, 12, 322. https://0-doi-org.brum.beds.ac.uk/10.3390/life12020322
AMA Style
Antoni FA. The Case for Clinical Trials with Novel GABAergic Drugs in Diabetes Mellitus and Obesity. Life. 2022; 12(2):322. https://0-doi-org.brum.beds.ac.uk/10.3390/life12020322
Chicago/Turabian StyleAntoni, Ferenc A. 2022. "The Case for Clinical Trials with Novel GABAergic Drugs in Diabetes Mellitus and Obesity" Life 12, no. 2: 322. https://0-doi-org.brum.beds.ac.uk/10.3390/life12020322
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.