

Synthesis and in Silico Investigation of Organoselenium-Clubbed Schiff Bases as Potential Mpro Inhibitors for the SARS-CoV-2 Replication


 ,
,  , ,
, ,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
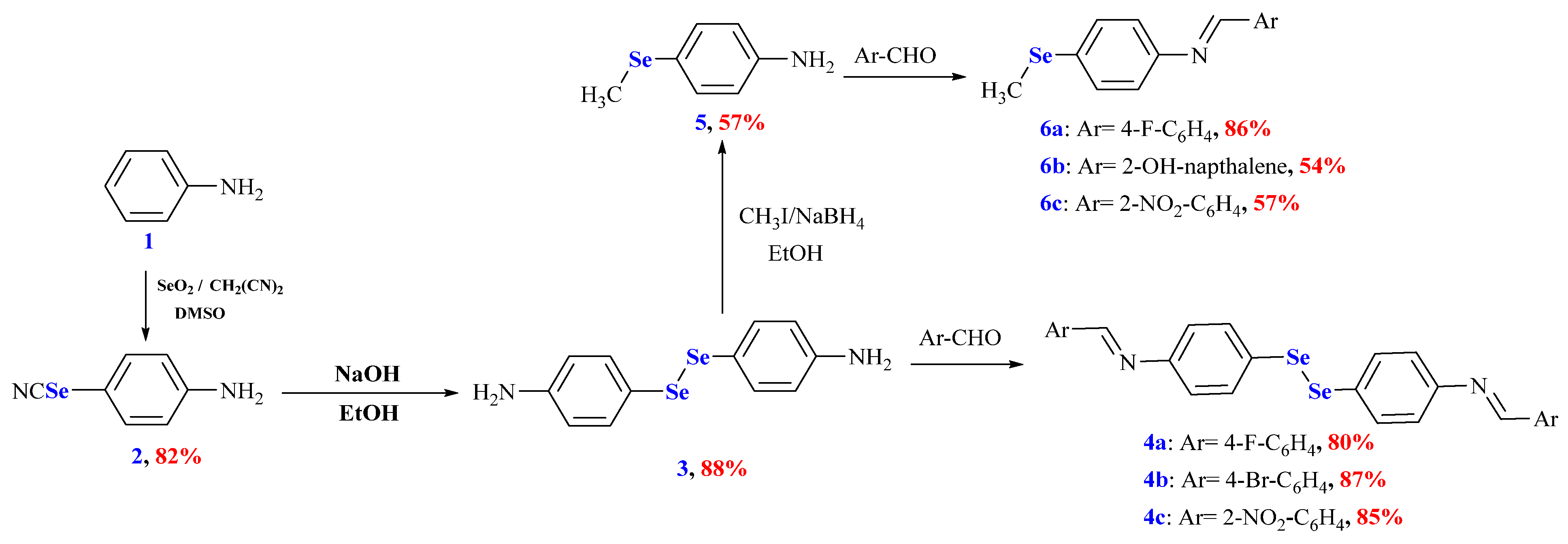
2.1. Synthesis and Characterization
2.2. Computational Calculations
3. Results
3.1. Design and Synthesis of the Organoselenium Compounds
3.2. DFT Calculations
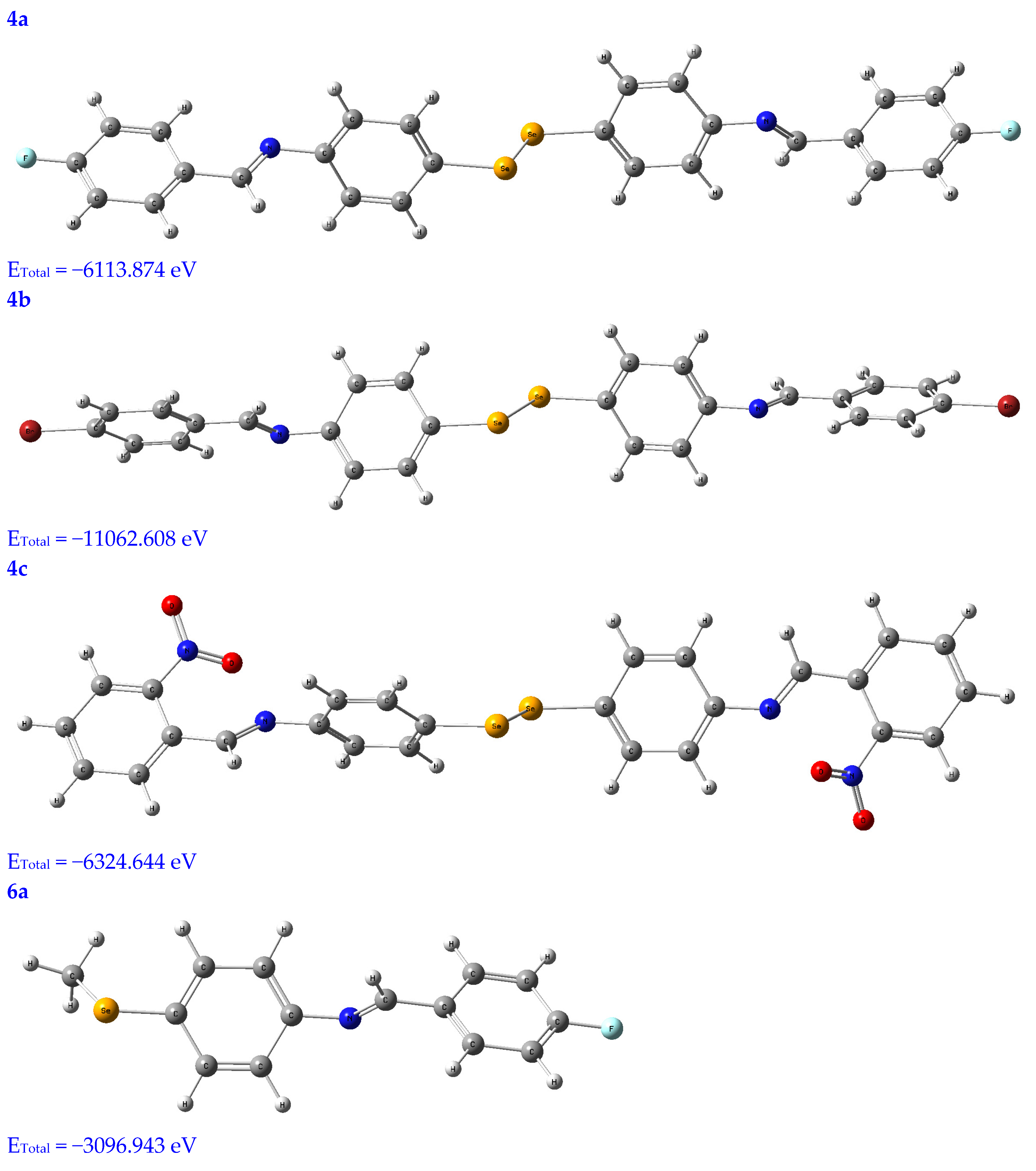
3.2.1. Geometry Optimization
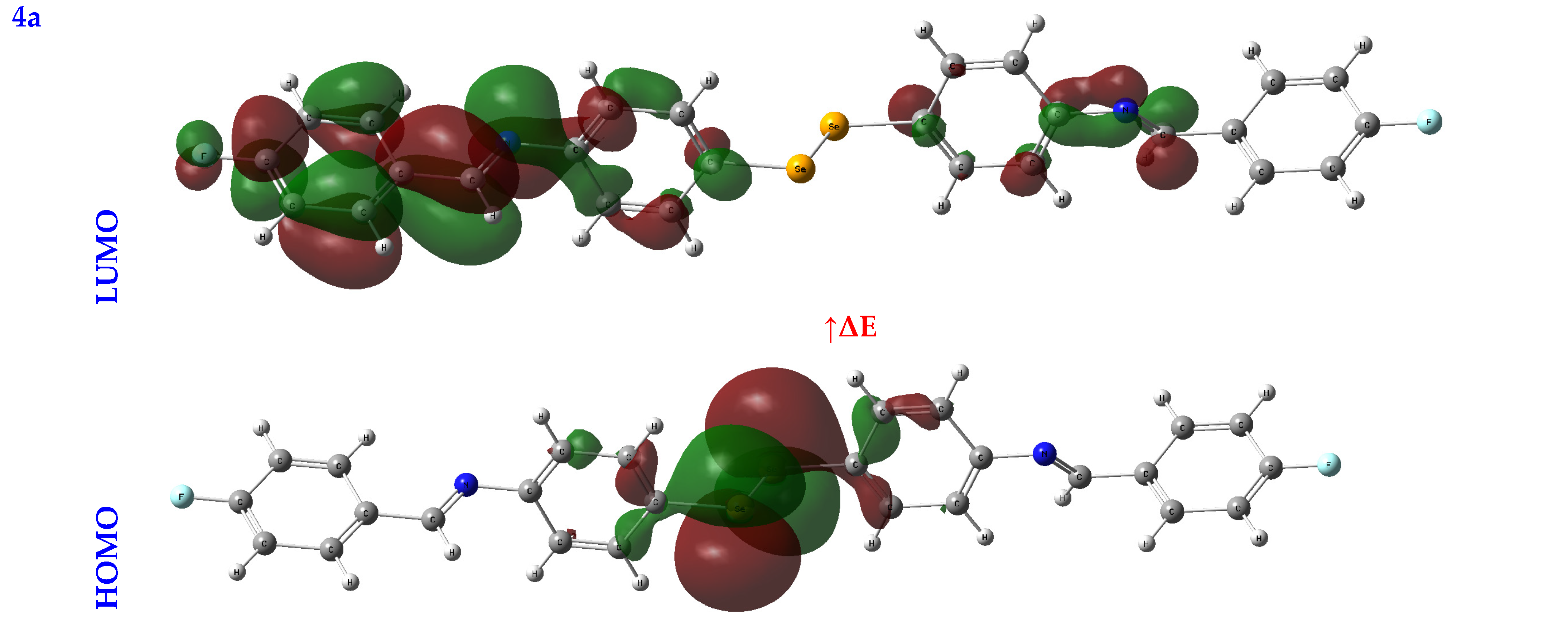
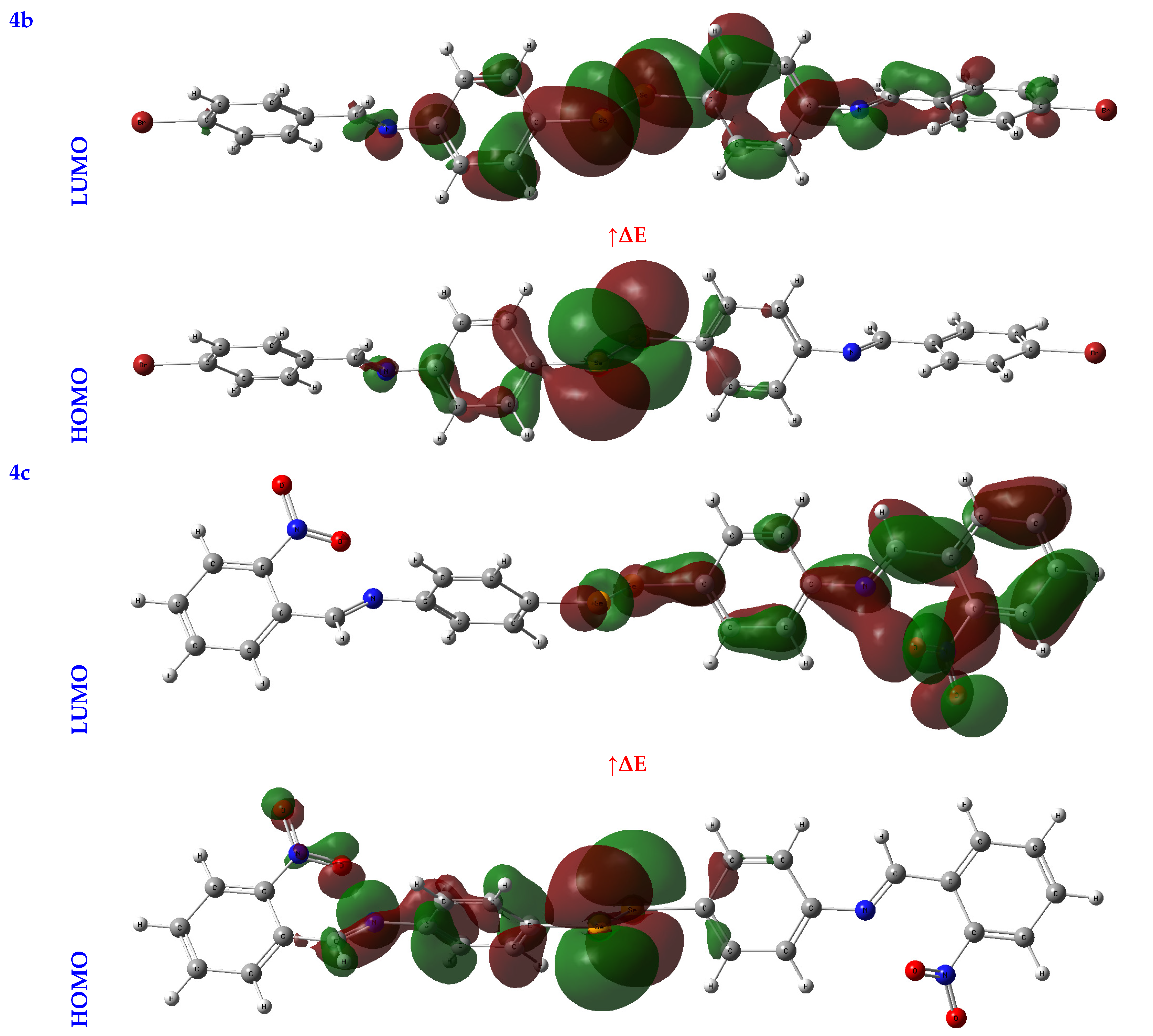
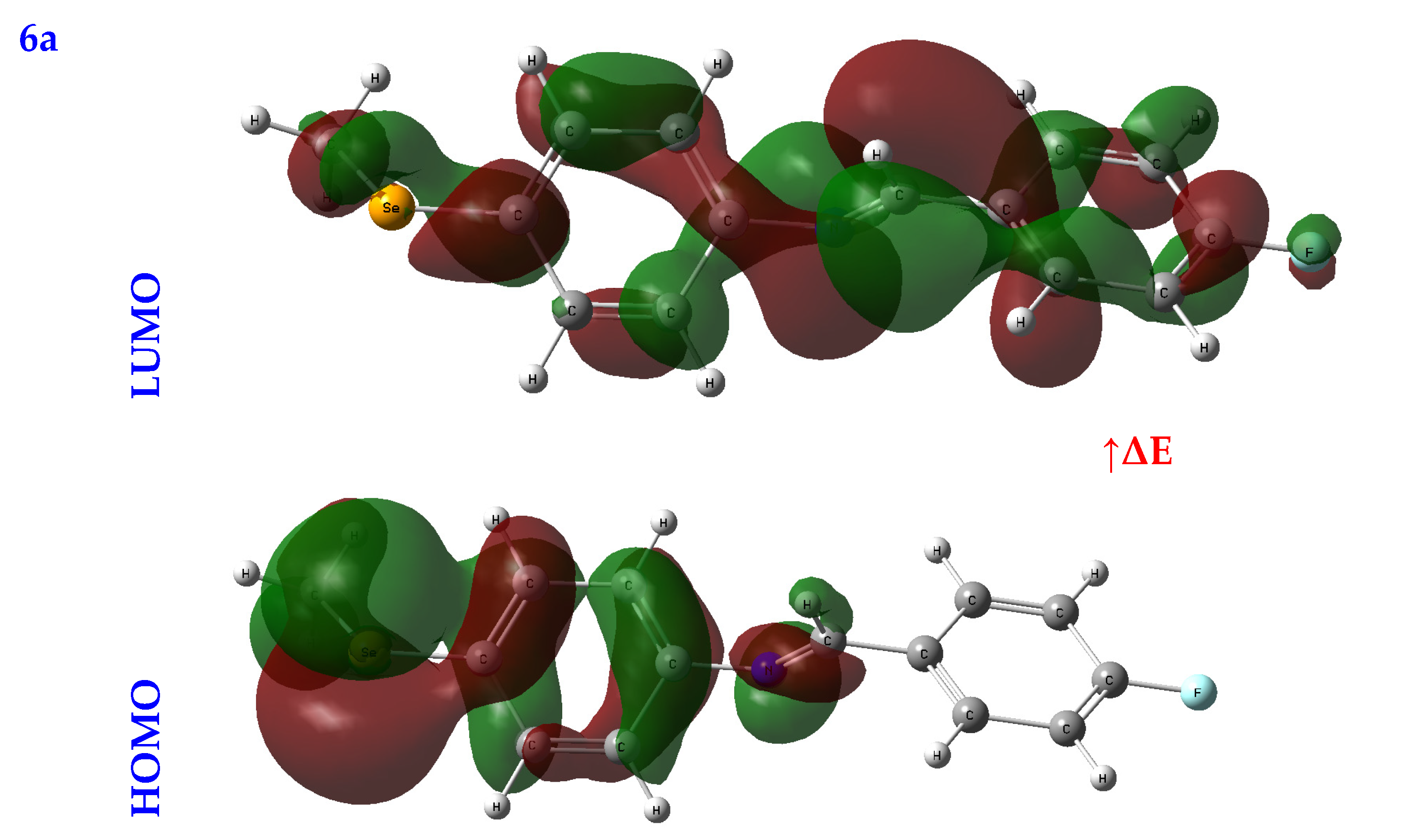
3.2.2. Frontier Molecular Orbital Analysis (FMO) Analysis
3.2.3. Global Reactive Indices
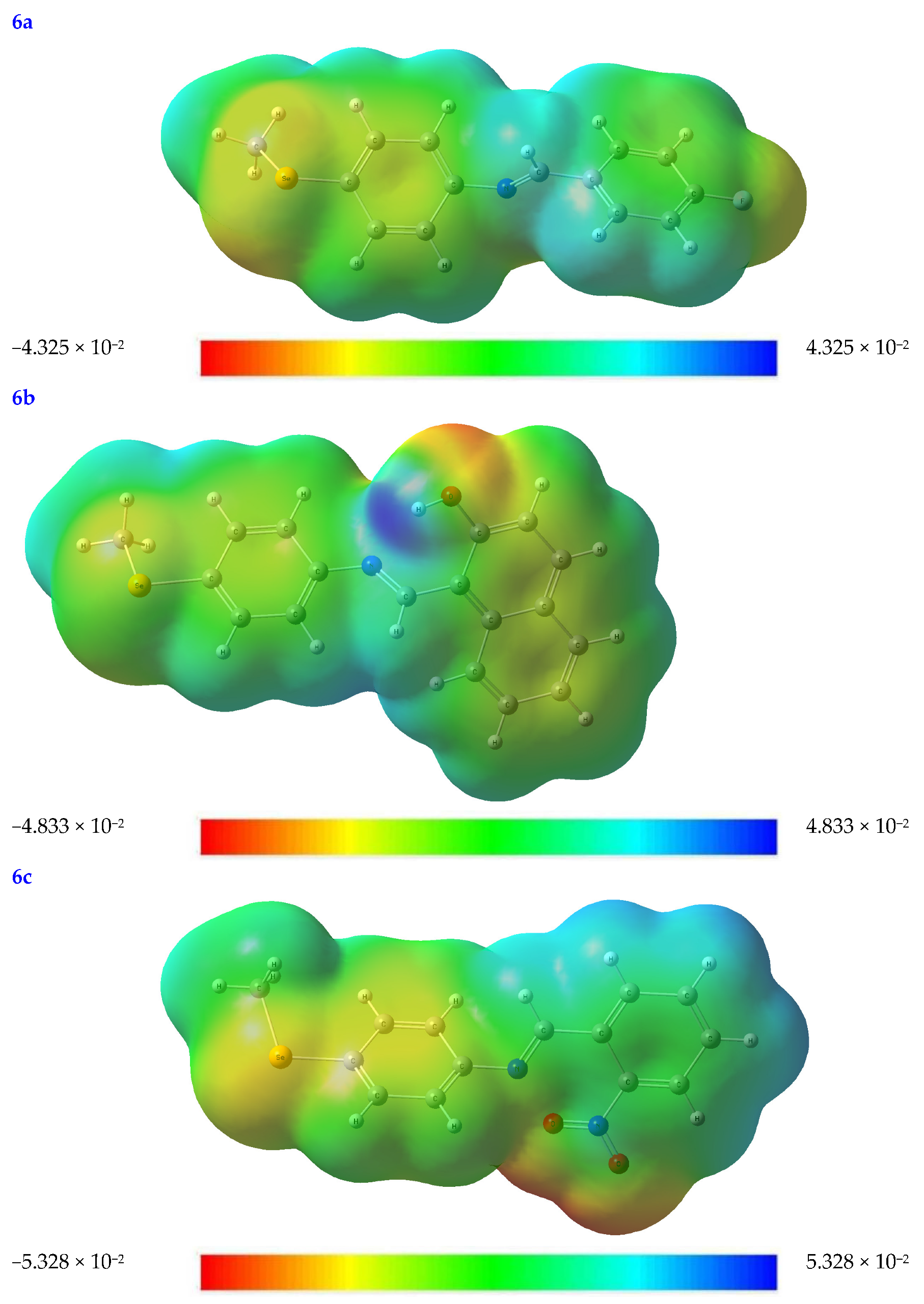
3.2.4. The Molecular Electrostatic Potential (MEP) Diagram
3.3. Drug-likeness Screening
3.4. Pharmacokinetic Properties Analysis
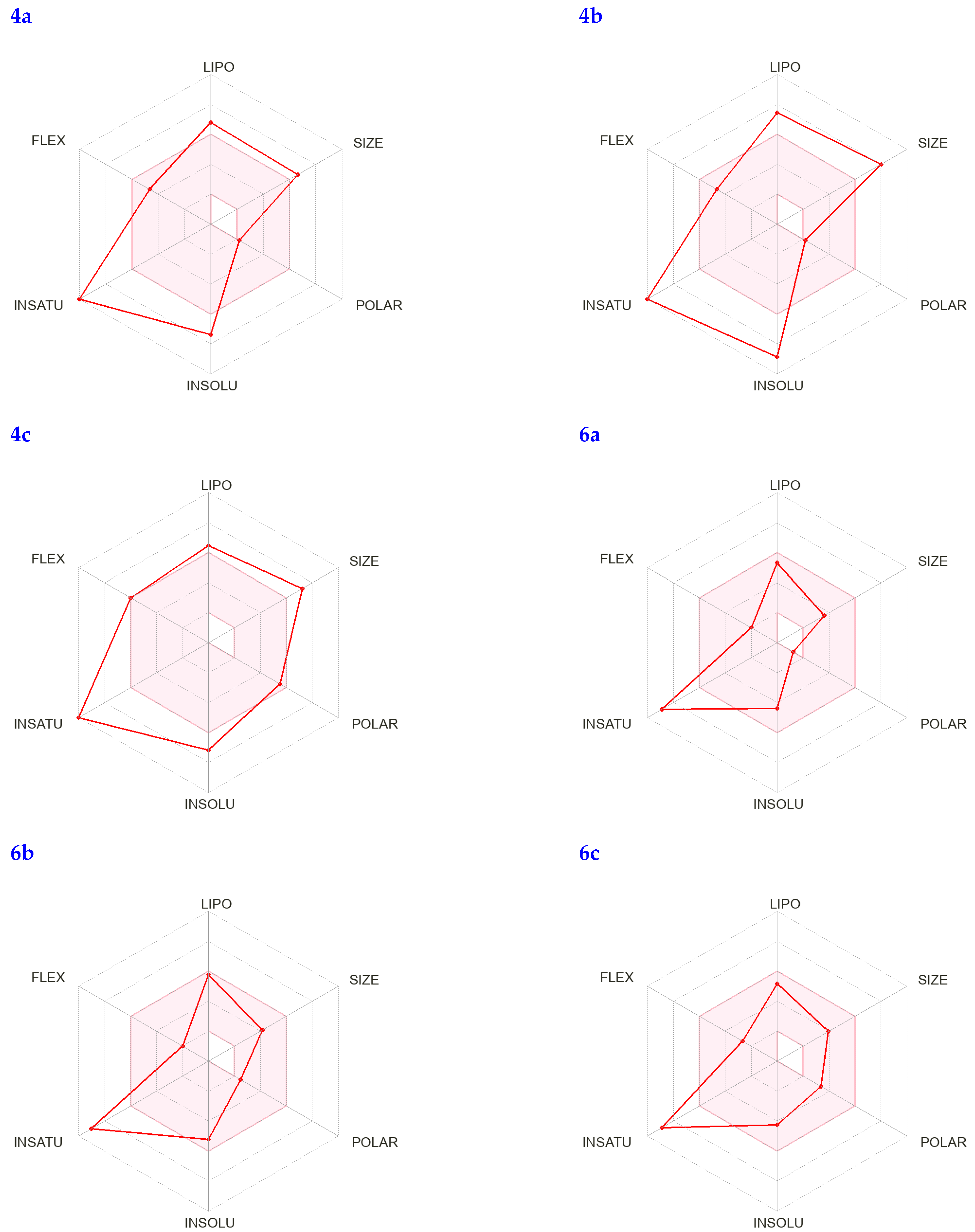
3.5. Pharmacophore Analysis
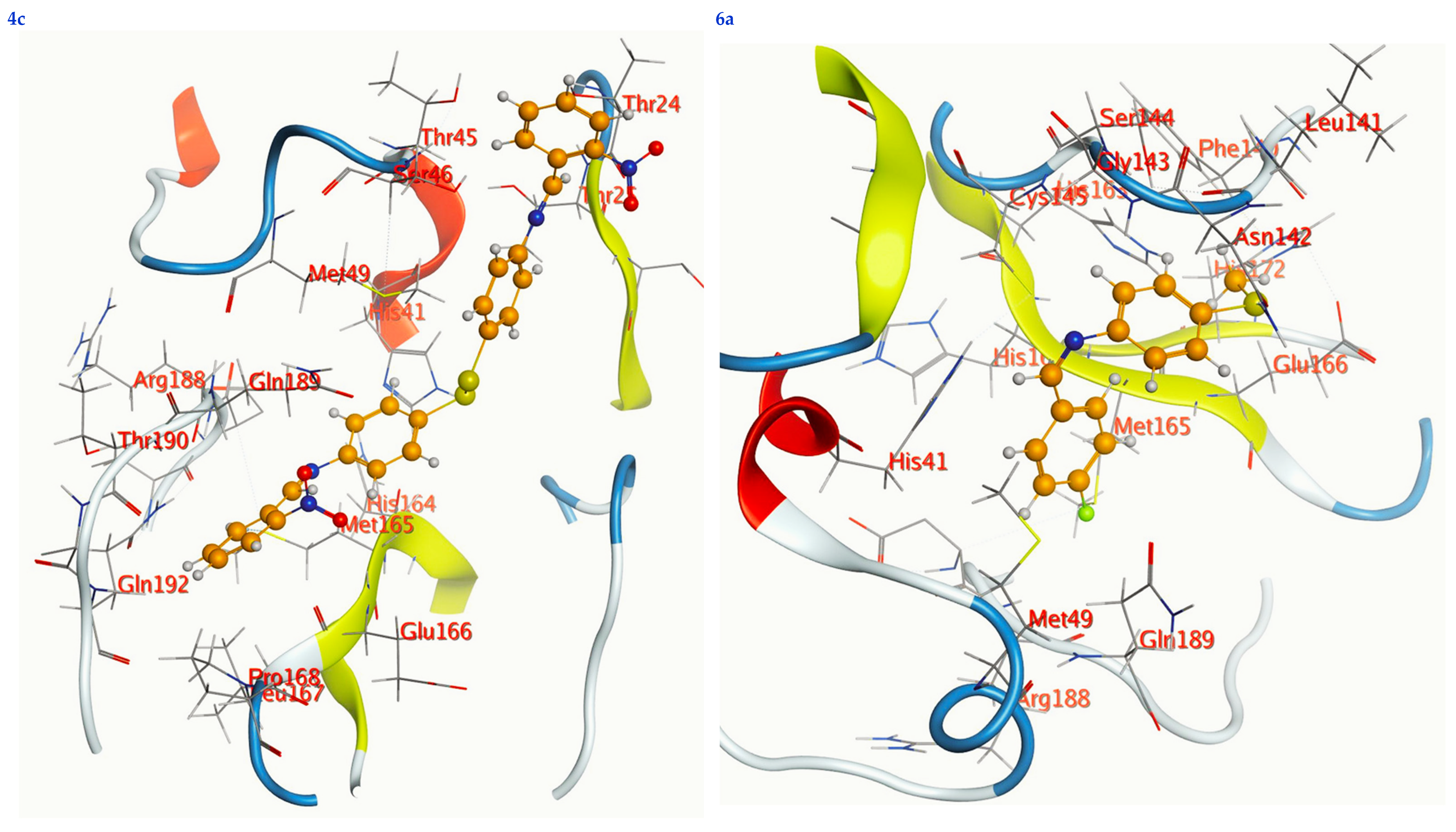
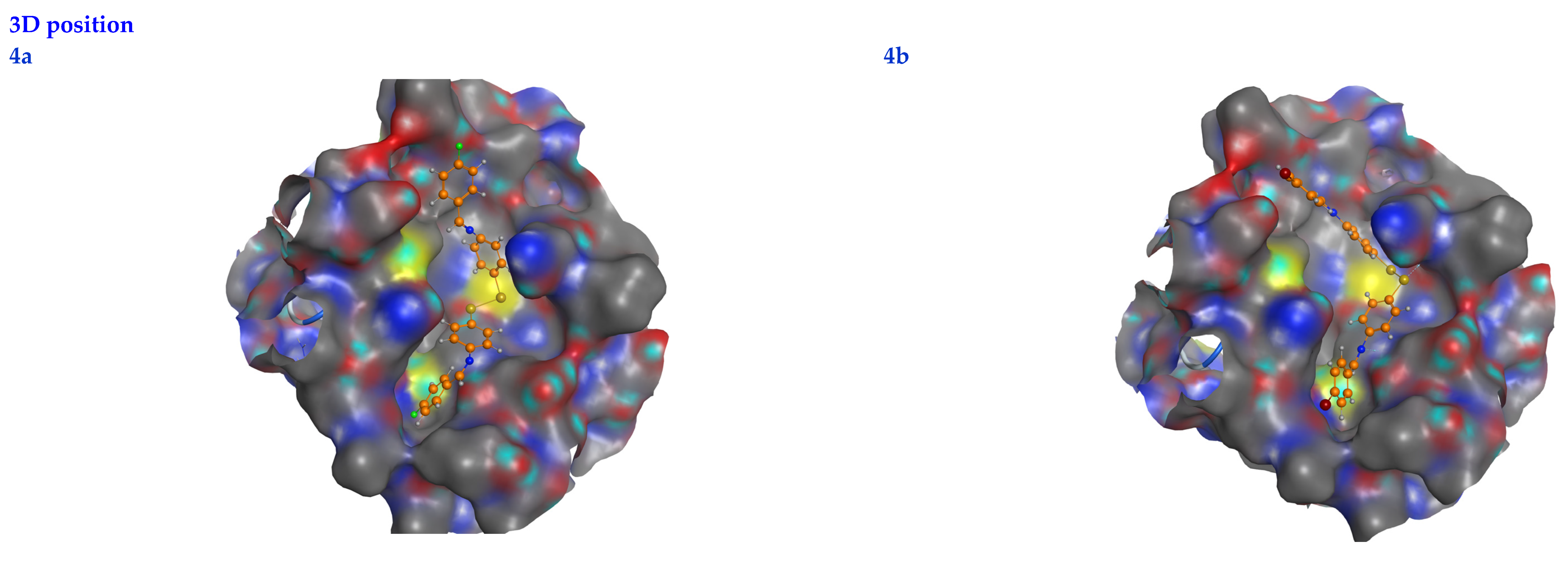
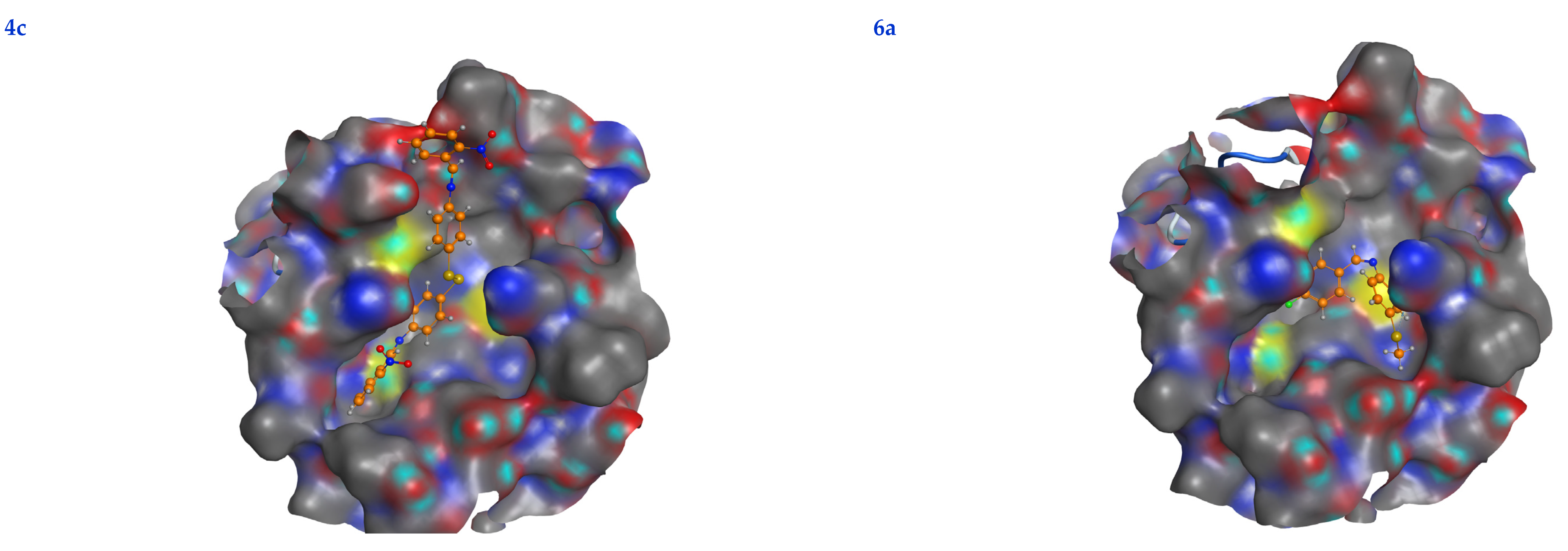
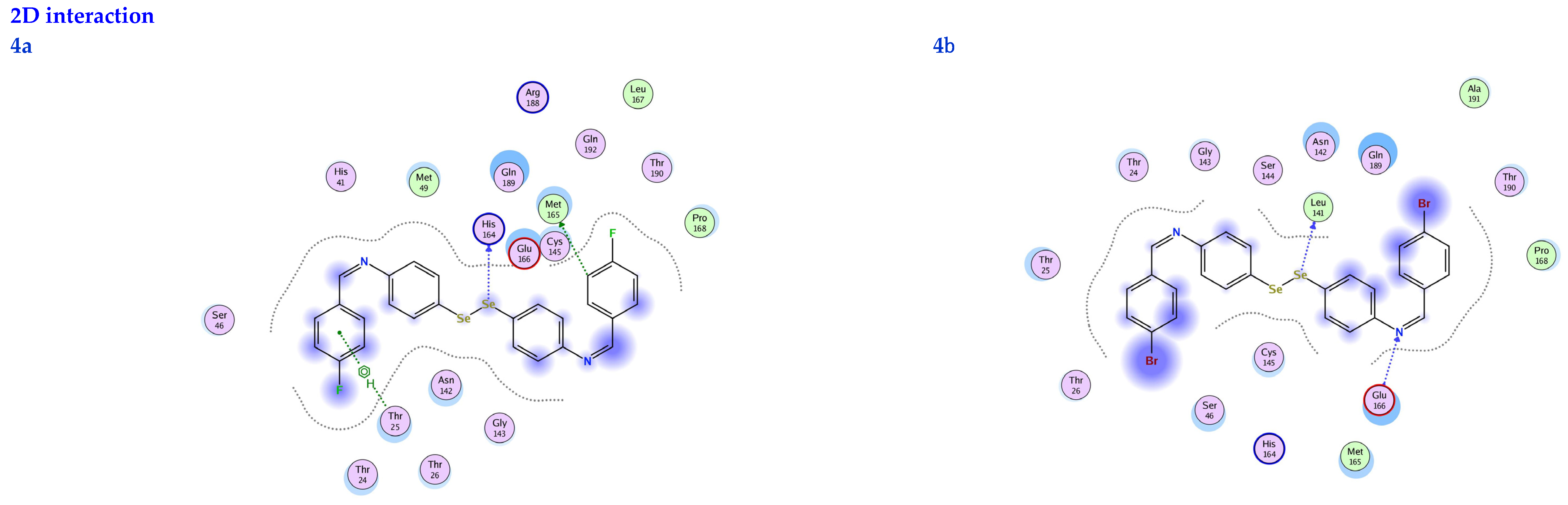
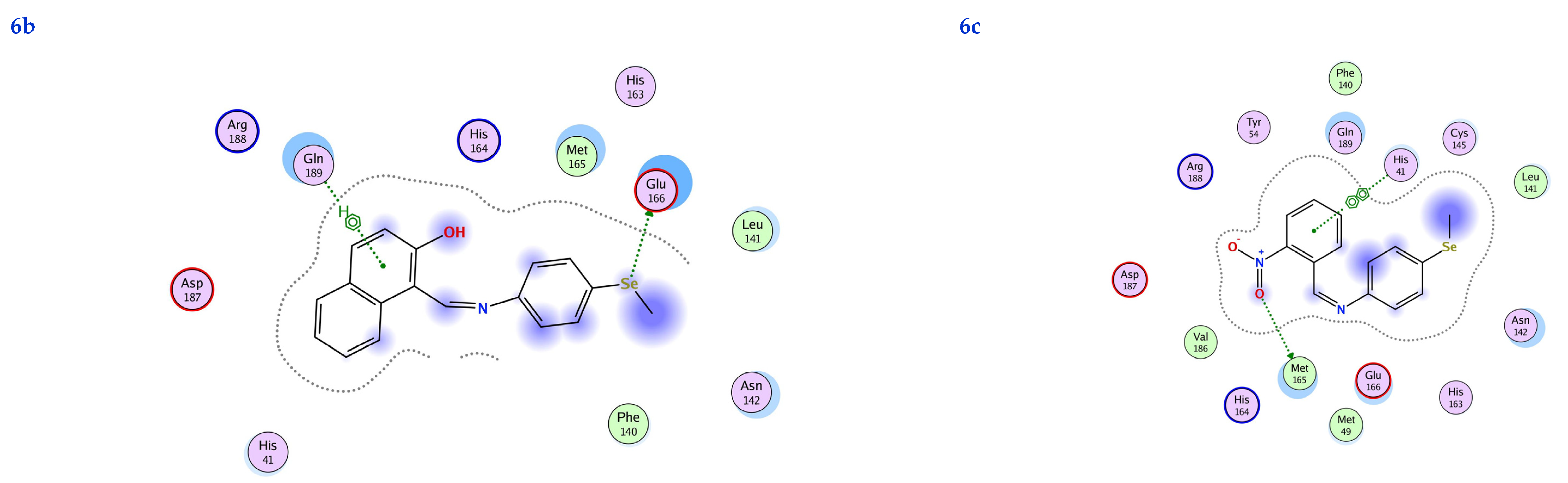
3.6. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shaaban, S.; El-Lateef, H.M.A.; Khalaf, M.M.; Gouda, M.; Youssef, I. One-Pot Multicomponent Polymerization, Metal-, and Non-Metal-Catalyzed Synthesis of Organoselenium Compounds. Polymers 2022, 14, 2208. [Google Scholar] [CrossRef] [PubMed]
- Chuai, H.; Zhang, S.-Q.; Bai, H.; Li, J.; Wang, Y.; Sun, J.; Wen, E.; Zhang, J.; Xin, M. Small molecule selenium-containing compounds: Recent development and therapeutic applications. Eur. J. Med. Chem. 2021, 223, 113621. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, K.; Keerthana, C.K.; Mohan, M.; Arivalagan, J.; Christyraj, J.R.S.S.; Firer, M.A.; Choudry, M.H.A.; Anto, R.J.; Lee, Y.J. The emerging role of selenium metabolic pathways in cancer: New therapeutic targets for cancer. J. Cell. Biochem. 2021, 123, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gong, Y.; Sun, Y.; Cai, J.; Liu, Q.; Bao, J.; Yang, J.; Zhang, Z. Impact of Selenium Deficiency on Inflammation, Oxidative Stress, and Phagocytosis in Mouse Macrophages. Biol. Trace Elem. Res. 2020, 194, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.W.; Barbosa, N.V.; Rocha, J.B.T. Toxicology and pharmacology of synthetic organoselenium compounds: An update. Arch. Toxicol. 2021, 95, 1179–1226. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Rocha, J.B.T. Toxicology and pharmacology of selenium: Emphasis on synthetic organoselenium compounds. Arch. Toxicol. 2011, 85, 1313–1359. [Google Scholar] [CrossRef]
- Makhal, P.N.; Nandi, A.; Kaki, V.R. Insights into the recent synthetic advances of organoselenium compounds. ChemistrySelect 2021, 6, 663–679. [Google Scholar] [CrossRef]
- Lenardão, E.J.; Santi, C.; Sancineto, L. New Frontiers in Organoselenium Compounds; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Phadnis, P.P. Synthesis Strategies for Organoselenium Compounds and Their Potential Applications in Human Life. In Handbook on Synthesis Strategies for Advanced Materials; Springer: Berlin/Heidelberg, Germany, 2021; pp. 537–641. [Google Scholar]
- Zhao, X.; Liao, L. Modern Organoselenium Catalysis: Opportunities and Challenges. Synlett 2021, 32, 1262–1268. [Google Scholar] [CrossRef]
- Benelli, J.L.; Poester, V.R.; Munhoz, L.S.; Melo, A.M.; Trápaga, M.R.; A Stevens, D.; Xavier, M.O. Ebselen and diphenyl diselenide against fungal pathogens: A systematic review. Med. Mycol. 2021, 59, 409–421. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Mansour, M.A.; AboulMagd, A.M.; Abdel-Rahman, H.M. Quinazoline-Schiff base conjugates: In silico study and ADMET predictions as multi-target inhibitors of coronavirus (SARS-CoV-2) proteins. RSC Adv. 2020, 10, 34033–34045. [Google Scholar] [CrossRef]
- Alshammari, M.B.; Ramadan, M.; Aly, A.A.; El-Sheref, E.M.; Bakht, A.; Ibrahim, M.A.; Shawky, A.M. Synthesis of potentially new schiff bases of N-substituted-2-quinolonylacetohydrazides as anti-COVID-19 agents. J. Mol. Struct. 2020, 1230, 129649. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, S.; Adam, M.S.S.; El-Metwaly, N.M. Novel organoselenium-based N-mealanilic acid and its zinc (II) chelate: Catalytic, anticancer, antimicrobial, antioxidant, and computational assessments. J. Mol. Liq. 2022, 363, 119907. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Ma, Y.T.; Liu, M.C.; Zhou, Y.B.; Wu, H.Y. Synthesis of Organoselenium Compounds with Elemental Selenium. Adv. Synth. Catal. 2021, 363, 5386–5406. [Google Scholar] [CrossRef]
- Al-Gaber, M.A.I.; El-Lateef, H.M.A.; Khalaf, M.M.; Shaaban, S.; Shawky, M.; Mohamed, G.G.; Abdou, A.; Gouda, M.; Abu-Dief, A.M. Design, Synthesis, Spectroscopic Inspection, DFT and Molecular Docking Study of Metal Chelates Incorporating Azo Dye Ligand for Biological Evaluation. Materials 2023, 16, 897. [Google Scholar] [CrossRef]
- Shokr, E.K.; Kamel, M.S.; Abdel-Ghany, H.; Remaily, M.A.E.A.A.A.E.; Abdou, A. Synthesis, characterization, and DFT study of linear and non-linear optical properties of some novel thieno[2,3-b]thiophene azo dye derivatives. Mater. Chem. Phys. 2022, 290, 126646. [Google Scholar] [CrossRef]
- Elkanzi, N.A.A.; Ali, A.M.; Albqmi, M.; Abdou, A. New Benzimidazole-Based Fe (III) and Cr (III) Complexes: Characterization, Bioactivity Screening, and Theoretical Implementations Using DFT and Molecular Docking Analysis. Appl. Organomet. Chem. 2022, 36, e6868. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Shingu, H. A Molecular Orbital Theory of Reactivity in Aromatic Hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Nagata, C.; Shingu, H. Molecular Orbital Theory of Orientation in Aromatic, Heteroaromatic, and Other Conjugated Molecules. J. Chem. Phys. 1954, 22, 1433–1442. [Google Scholar] [CrossRef] [Green Version]
- Al-Wabli, R.I.; Resmi, K.; Mary, Y.S.; Panicker, C.Y.; Attia, M.I.; El-Emam, A.A.; Van Alsenoy, C. Vibrational spectroscopic studies, Fukui functions, HOMO-LUMO, NLO, NBO analysis and molecular docking study of (E)-1-(1,3-benzodioxol-5-yl)-4,4-dimethylpent-1-en-3-one, a potential precursor to bioactive agents. J. Mol. Struct. 2016, 1123, 375–383. [Google Scholar] [CrossRef]
- Missioui, M.; Said, M.A.; Demirtaş, G.; Mague, J.T.; Al-Sulami, A.; Al-Kaff, N.S.; Ramli, Y. A possible potential COVID-19 drug candidate: Diethyl 2-(2-(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetyl)hydrazono)malonate: Docking of disordered independent molecules of a novel crystal structure, HSA/DFT/XRD and cytotoxicity. Arab. J. Chem. 2021, 15, 103595. [Google Scholar] [CrossRef] [PubMed]
- Alghuwainem, Y.A.A.; El-Lateef, H.M.A.; Khalaf, M.M.; Amer, A.A.; Abdelhamid, A.A.; Alzharani, A.A.; Alfarsi, A.; Shaaban, S.; Gouda, M.; Abdou, A. Synthesis, DFT, Biological and Molecular Docking Analysis of Novel Manganese(II), Iron(III), Cobalt(II), Nickel(II), and Copper(II) Chelate Complexes Ligated by 1-(4-Nitrophenylazo)-2-naphthol. Int. J. Mol. Sci. 2022, 23, 15614. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.P.; Bharti, S.K.; Kumar, S.; Ved, K.; Padam, K. Study of molecular structure, chemical reactiv-ity and first hyperpolarizability of a newly synthesized N-(4-oxo-2-phenylquinazolin-3 (4H)-yl)-1H-indole-2-carboxamide using spectral analysis. J. Mol. Struct. 2017, 1148, 356–363. [Google Scholar] [CrossRef]
- Abdou, A.; Mostafa, H.M.; Abdel-Mawgoud, A.-M.M. Seven metal-based bi-dentate NO azocoumarine complexes: Synthesis, physicochemical properties, DFT calculations, drug-likeness, in vitro antimicrobial screening and molecular docking analysis. Inorg. Chim. Acta 2022, 539, 121043. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Elkanzi, N.A.; Hrichi, H.; Salah, H.; Albqmi, M.; Ali, A.M.; Abdou, A. Synthesis, physicochemical properties, biological, molecular docking and DFT investigation of Fe(III), Co(II), Ni(II), Cu(II) and Zn(II) complexes of the 4-[(5-oxo-4,5-dihydro-1,3-thiazol-2-yl)hydrazono]methyl}phenyl 4-methylbenzenesulfonate Schiff-base ligand. Polyhedron 2023, 230, 116219. [Google Scholar] [CrossRef]
- Rajamanickam, R.; Mannangatty, R.; Sampathkumar, J.; Senthamaraikannan, K.; Diravidamani, B. Synthesis, crystal structure, DFT and molecular docking studies of N-acetyl-2,4-[diaryl-3-azabicyclo[3.3.1]nonan-9-yl]-9-spiro-4′-acetyl-2′-(acetylamino)-4′,9-dihydro-[1′,3′,4′]-thiadiazoles: A potential SARS-nCoV-2 Mpro (COVID-19) inhibitor. J. Mol. Struct. 2022, 1259, 132747. [Google Scholar] [CrossRef] [PubMed]
- Alghuwainem, Y.A.; El-Lateef, H.M.A.; Khalaf, M.M.; Abdelhamid, A.A.; Alfarsi, A.; Gouda, M.; Abdelbaset, M.; Abdou, A. Synthesis, structural, DFT, antibacterial, antifungal, anti-inflammatory, and molecular docking analysis of new VO(II), Fe(III), Mn(II), Zn(II), and Ag(I) complexes based on 4-((2-hydroxy-1-naphthyl)azo) benzenesulfonamide. J. Mol. Liq. 2023, 369, 120936. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Owens, J. Chris Lipinski discusses life and chemistry after the Rule of Five. Drug Discov. Today 2003, 8, 12–16. [Google Scholar] [CrossRef]
- Lin, J.H. Challenges in drug discovery: Lead optimization and prediction of human pharmacokinetics. In Pharmaceutical Profiling in Drug Discovery for Lead Selection; Borchardt, R., Kerns, E., Lipinski, C., Thakker, D., Wang, B., Eds.; Springer: New York, NY, USA, 2006; pp. 293–326. [Google Scholar]
- Martin, Y.C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Bento, A.P.; Gaulton, A.; Hersey, A.; Manners, E.J.; Radoux, C.J.; Leach, A.R. Target-Based Evaluation of “Drug-like” Properties and Ligand Efficiencies. J. Med. Chem. 2021, 64, 7210–7230. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Abdullahi, S.H.; Uzairu, A.; Shallangwa, G.A.; Uba, S.; Umar, A.B. Computational modeling, ligand-based drug design, drug-likeness and ADMET properties studies of series of chromen-2-ones analogues as anti-cancer agents. Bull. Natl. Res. Cent. 2022, 46, 177. [Google Scholar] [CrossRef]
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1399–1404. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Han, L.; Pang, K.; Fu, T.; Phillips, C.J.C.; Gao, T. Nano-selenium Supplementation Increases Selenoprotein (Sel) Gene Expression Profiles and Milk Selenium Concentration in Lactating Dairy Cows. Biol. Trace Elem. Res. 2020, 199, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Balaramnavar, V.M.; Ahmad, K.; Saeed, M.; Ahmad, I.; Kamal, M.; Jawed, T. Pharmacophore-based approaches in the rational repurposing technique for FDA approved drugs targeting SARS-CoV-2 Mpro. RSC Adv. 2020, 10, 40264–40275. [Google Scholar] [CrossRef]
- Maji, S.; Pattanayak, S.K.; Sen, A.; Badavath, V.N. Chapter 6—Pharmacophore modeling in drug design. In Drug Discovery Update, Computer Aided Drug Design (CADD): From Ligand-Based Methods to Structure-Based Approaches; Rudrapal, M., Egbuna, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 157–179. [Google Scholar] [CrossRef]
- Sahu, S.N.; Satpathy, S.S.; Pattnaik, S.; Mohanty, C.; Pattanayak, S.K. Boerhavia diffusa plant extract can be a new potent therapeutics against mutant nephrin protein responsible for type1 nephrotic syndrome: Insight into hydrate-ligand docking interactions and molecular dynamics simulation study. J. Indian Chem. Soc. 2022, 99, 100669. [Google Scholar] [CrossRef]
- Mengist, H.M.; Fan, X.; Jin, T. Designing of improved drugs for COVID-19: Crystal structure of SARS-CoV-2 main protease Mpro. Signal Transduct. Target. Ther. 2020, 5, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Moharana, M.; Pattanayak, S.K. Molecular recognition of bio-active triterpenoids from Swertia chirayita towards hepatitis Delta antigen: A mechanism through docking, dynamics simulation, Gibbs free energy landscape. J. Biomol. Struct. Dyn. 2023, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Al-Khafaji, K.; Tok, T.T. Molecular dynamics simulation, free energy landscape and binding free energy computations in exploration the anti-invasive activity of amygdalin against metastasis. Comput. Methods Programs Biomed. 2020, 195, 105660–105673. [Google Scholar] [CrossRef] [PubMed]
- Al-Abdallah, B.; Al-Faiyz, Y.S.; Shaaban, S. Organoselenocyanates Tethered Methyl Anthranilate Hybrids with Promising Anticancer, Antimicrobial, and Antioxidant Activities. Inorganics 2022, 10, 246. [Google Scholar] [CrossRef]
- Al-Abdallah, B.; Al-Faiyz, Y.S.; Shaaban, S. Anticancer, Antimicrobial, and Antioxidant Activities of Or-ganodiselenide-Tethered Methyl Anthranilates. Biomolecules 2022, 12, 1765. [Google Scholar] [CrossRef] [PubMed]
- Abu-Dief, A.M.; Alotaibi, N.H.; Al-Farraj, E.S.; Qasem, H.A.; Alzahrani, S.; Mahfouz, M.K.; Abdou, A. Fabrication, structural elucidation, theoretical, TD-DFT, vibrational calculation and molecular docking studies of some novel adenine imine chelates for biomedical applications. J. Mol. Liq. 2022, 365, 119961. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EHOMO (eV) | ELUMO (eV) | ∆E (eV) | IP (eV) | EA (eV) | χ (eV) | CP (eV) | η (eV) | σ (eV−1) | ω (eV) | Nu (eV−1) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a | −5.18 | −2.86 | 2.32 | 5.18 | 2.86 | 4.02 | −4.02 | 1.16 | 0.43 | 6.97 | 0.14 |
| 4b | −4.33 | −1.90 | 2.43 | 4.33 | 1.90 | 3.12 | −3.12 | 1.22 | 0.41 | 3.99 | 0.25 |
| 4c | −4.23 | −2.55 | 1.68 | 4.23 | 2.55 | 3.39 | −3.39 | 0.84 | 0.59 | 6.84 | 0.15 |
| 6a | −5.69 | −2.10 | 3.59 | 5.69 | 2.10 | 3.89 | −3.89 | 1.79 | 0.28 | 4.23 | 0.24 |
| 6b | −5.50 | −1.46 | 4.04 | 5.50 | 1.46 | 3.48 | −3.48 | 2.02 | 0.25 | 3.00 | 0.33 |
| 6c | −5.18 | −2.09 | 3.09 | 5.18 | 2.09 | 3.64 | −3.64 | 1.55 | 0.32 | 4.28 | 0.23 |
| Ligand | Receptor | Interaction | Distance | E (kcal/mol) | S (kcal/mol) | RMSD | Ki (μM) | |
|---|---|---|---|---|---|---|---|---|
| 4a | Se 7 | HIS 164 | H-donor | 2.82 | −1.04 | −7.33 | 1.37 | 4.33 |
| C 29 | MET 165 | H-donor | 2.86 | −0.80 | ||||
| 6-ring | THR 25 | pi-H | 3.38 | −1.00 | ||||
| 4b | Se 7 | LEU 141 | H-donor | 2.78 | −1.00 | −7.21 | 1.46 | 5.29 |
| N 24 | GLU 166 | H-acceptor | 2.83 | −0.80 | ||||
| 4c | N 15 | MET 165 | H-donor | 2.96 | −0.10 | −8.19 | 1.35 | 1.01 |
| C 19 | MET 165 | H-donor | 3.17 | −0.80 | ||||
| 6-ring | GLN 189 | pi-H | 3.51 | −0.60 | ||||
| 6a | Se 1 | ASN 142 | H-donor | 2.72 | −1.50 | −6.20 | 1.85 | 28.90 |
| 6-ring | ASN 142 | pi-H | 2.90 | −1.20 | ||||
| 6-ring | GLY 143 | pi-H | 3.29 | −0.70 | ||||
| 6b | Se 1 | GLU 166 | H-donor | 3.26 | −4.10 | −6.10 | 1.73 | 34.26 |
| 6-ring | GLN 189 | pi-H | 3.69 | −0.70 | ||||
| 6c | O 19 | MET 165 | H-donor | 2.59 | −0.20 | −6.58 | 1.55 | 15.12 |
| 6-ring | HIS 41 | pi-pi | 2.99 | −0.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaaban, S.; Abdou, A.; Alhamzani, A.G.; Abou-Krisha, M.M.; Al-Qudah, M.A.; Alaasar, M.; Youssef, I.; Yousef, T.A. Synthesis and in Silico Investigation of Organoselenium-Clubbed Schiff Bases as Potential Mpro Inhibitors for the SARS-CoV-2 Replication. Life 2023, 13, 912. https://0-doi-org.brum.beds.ac.uk/10.3390/life13040912
Shaaban S, Abdou A, Alhamzani AG, Abou-Krisha MM, Al-Qudah MA, Alaasar M, Youssef I, Yousef TA. Synthesis and in Silico Investigation of Organoselenium-Clubbed Schiff Bases as Potential Mpro Inhibitors for the SARS-CoV-2 Replication. Life. 2023; 13(4):912. https://0-doi-org.brum.beds.ac.uk/10.3390/life13040912
Chicago/Turabian StyleShaaban, Saad, Aly Abdou, Abdulrahman G. Alhamzani, Mortaga M. Abou-Krisha, Mahmoud A. Al-Qudah, Mohamed Alaasar, Ibrahim Youssef, and Tarek A. Yousef. 2023. "Synthesis and in Silico Investigation of Organoselenium-Clubbed Schiff Bases as Potential Mpro Inhibitors for the SARS-CoV-2 Replication" Life 13, no. 4: 912. https://0-doi-org.brum.beds.ac.uk/10.3390/life13040912