Coexistence of Growth Hormone Deficiency and Pituitary Microadenoma in a Child with Unique Mosaic Turner Syndrome: A Case Report and Literature Review


Abstract
:1. Introduction
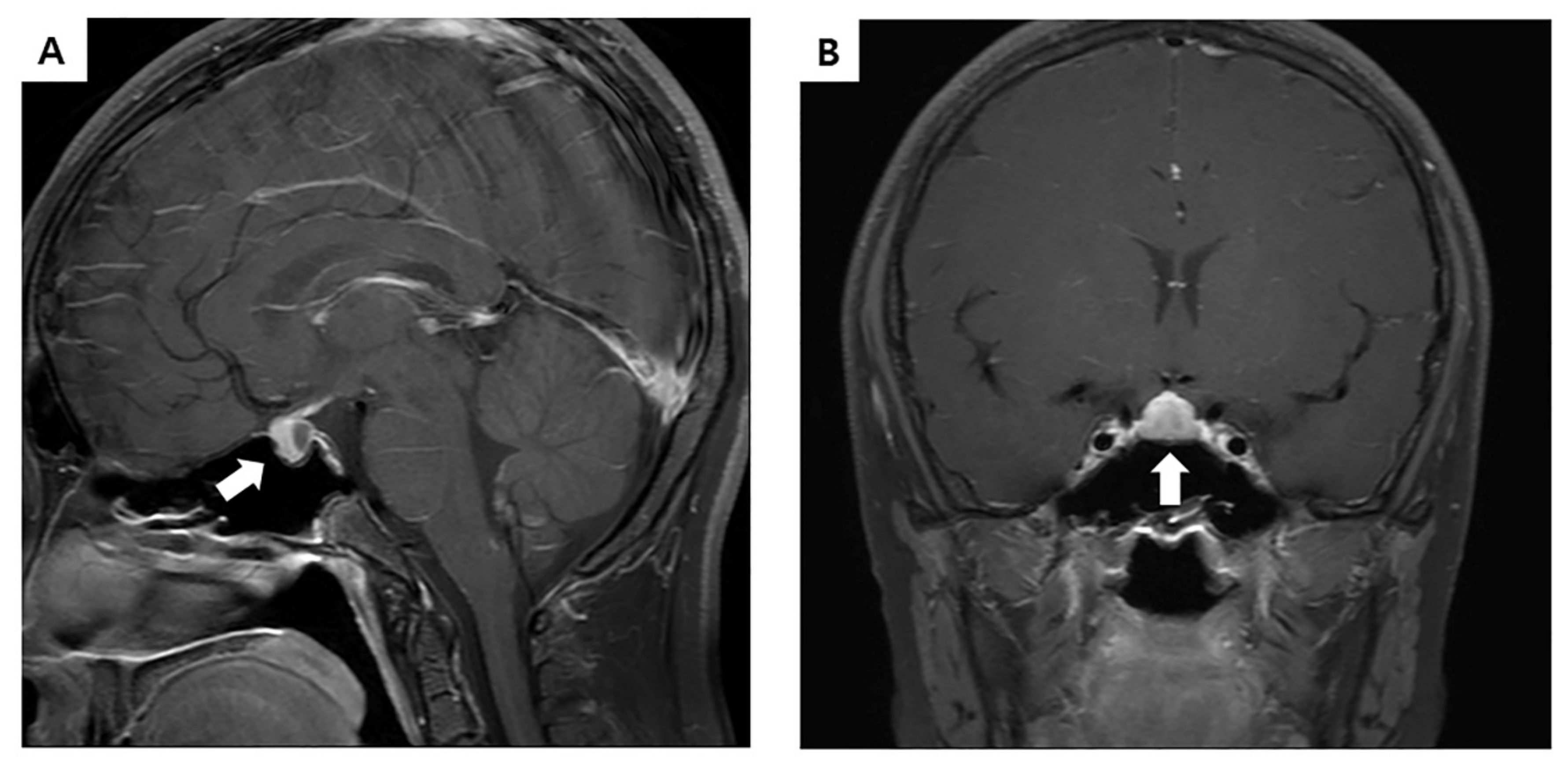
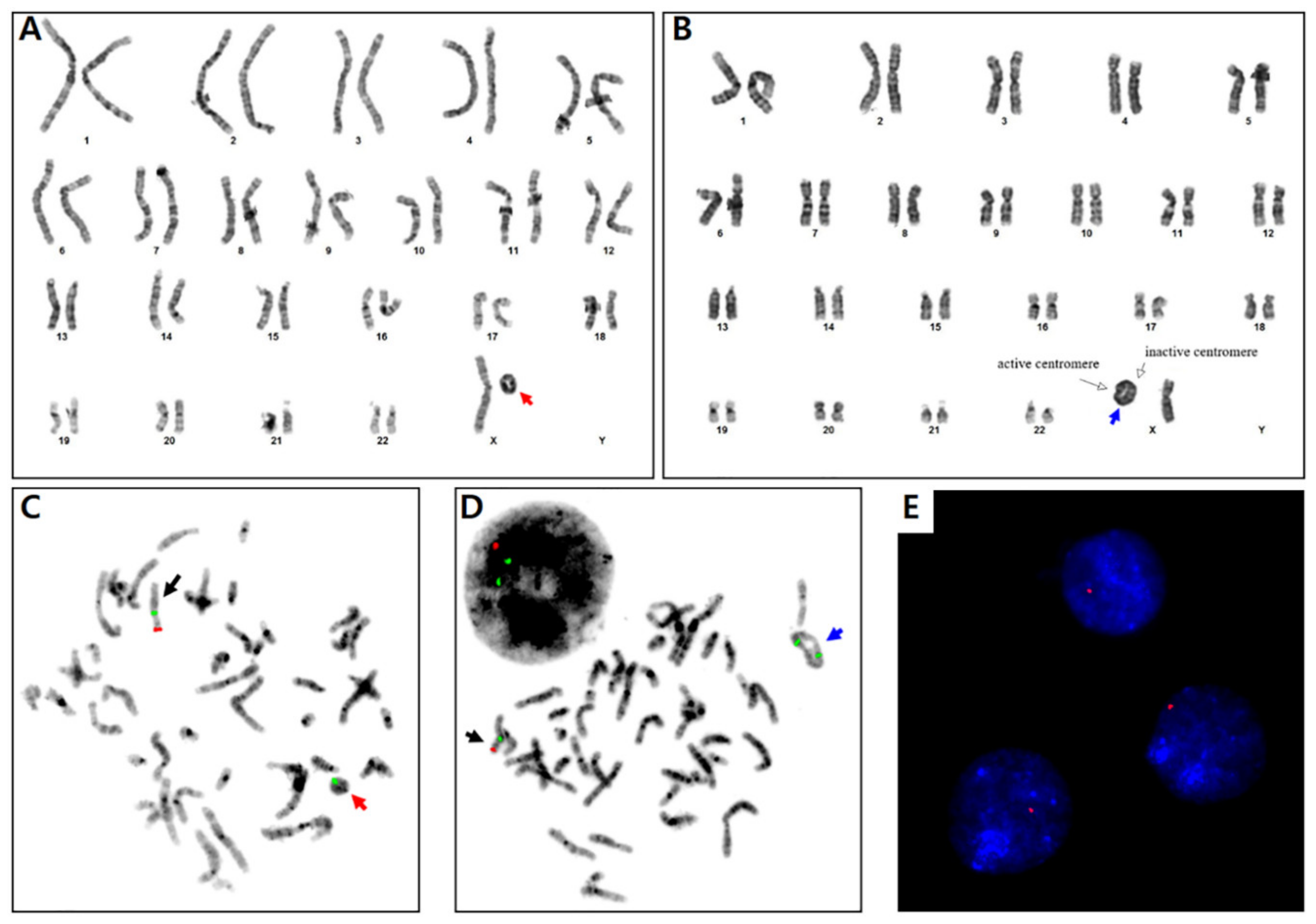
2. Case Presentation
3. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sybert, V.P.; McCauley, E. Turner’s syndrome. N. Engl. J. Med. 2004, 351, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H. Epidemiological, endocrine and metabolic features in Turner syndrome. Eur. J. Endocrinol. 2004, 151, 657–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondy, C.A.; Turner Syndrome Study Group. Care of girls and women with Turner syndrome: A guideline of the Turner Syndrome Study Group. J. Clin. Endocrinol. Metab. 2007, 92, 10–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; Dekkers, O.M.; Geffner, M.E.; Klein, K.O.; Lin, A.E.; Mauras, N.; Quigley, C.A.; Rubin, K.; et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: Proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur. J. Endocrinol. 2017, 177, G1–G70. [Google Scholar] [CrossRef]
- Milbrandt, T.; Thomas, E. Turner syndrome. Pediatr. Rev. 2013, 34, 420–421. [Google Scholar] [CrossRef]
- Jacobs, P.; Dalton, P.; James, R.; Mosse, K.; Power, M.; Robinson, D.; Skuse, D. Turner syndrome: A cytogenetic and molecular study. Ann. Hum. Genet. 1997, 61, 471–483. [Google Scholar] [CrossRef]
- De Groote, K.; Demulier, L.; De Backer, J.; De Wolf, D.; De Schepper, J.; T’Sjoen, G.; De Backer, T. Arterial hypertension in Turner syndrome: A review of the literature and a practical approach for diagnosis and treatment. J. Hypertens. 2015, 33, 1342–1351. [Google Scholar] [CrossRef]
- Yeh, T.; Soto, A.G.; Quintos, J.B.; Topor, L.S. Turner syndrome and pituitary adenomas: A case report and review of literature. J. Pediatr. Endocrinol. Metab. 2017, 30, 231–235. [Google Scholar] [CrossRef]
- Barstow, C.; Rerucha, C. Evaluation of Short and Tall Stature in Children. Am. Fam. Physician. 2015, 92, 43–50. [Google Scholar]
- Lindsay, R.; Feldkamp, M.; Harris, D.; Robertson, J.; Rallison, M. Utah Growth Study: Growth standards and the prevalence of growth hormone deficiency. J. Pediatr. 1994, 125, 29–35. [Google Scholar] [CrossRef]
- Yavas Abali, Z.; Darendeliler, F.; Neyzi, O. A Critical Appraisal of Growth Hormone Therapy in Growth Hormone Deficiency and Turner Syndrome Patients in Turkey. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 490–495. [Google Scholar] [CrossRef]
- Yu, J.; Shin, H.Y.; Lee, C.G.; Kim, J.H. Concomitant occurrence of Turner syndrome and growth hormone deficiency. Korean. J. Pediatr. 2016, 59, S121–S124. [Google Scholar] [CrossRef]
- Paul Wadwa, R.; Kappy, M.S. Short stature. In Berman’s Pediatric Decision Making, 5th ed.; Bajaj, L., Hambridge, S., Kerby, G., Nyquist, A.C., Eds.; Elsevier: Philadelphia, PA, USA, 2011. [Google Scholar]
- Brook, C.G. Growth hormone deficiency in Turner’s syndrome. N. Engl. J. Med. 1978, 298, 1203–1204. [Google Scholar] [PubMed]
- Efstathiadou, Z.; Tsatsoulis, A. Turner’s syndrome with concomitant hypopituitarism: Case report. Hum. Reprod. 2000, 15, 2388–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallicchio, C.T.; Alves, S.T.; Ramos, H.I.; Llerena, J.C.; Guimaraes, M.M. Association of Turner’s syndrome and hypopituitarism: A patient report. J. Pediatr. Endocrinol. Metab. 2003, 16, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Cheng, F.X.; Xiao, M.S.; Fan, Y.; Dong, W. Concurrent occurrence of chronic lymphocytic thyroiditis with hypothyroidism and growth hormone deficiency in a Turner’s syndrome patient. J. Pediatr. Endocrinol. Metab. 2011, 24, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Bolanowski, M.; Lomna-Bogdanov, E.; Kosmala, W.; Malczewska, J.; Slezak, R.; Zadrozna, B.; Podgorski, J.K. Turner’s syndrome followed by acromegaly in the third decade of life: An unusual coincidence of two rare conditions. Gynecol. Endocrinol. 2002, 16, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, L.; Julesz, J.; Kocsis, J.; Pasztor, E.; Laszlo, F. Mosaic Turner’s syndrome and pituitary microadenoma. Exp. Clin. Endocrinol. 1985, 86, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Mermilliod, J.A.; Gatchair-Rose, A.; Svec, F. Pituitary tumor and low gonadotropins in a patient with Turner’s syndrome. J. Louisiana. St. Med. Soc. 1995, 147, 540–543. [Google Scholar] [PubMed]
- Weibel, H.S.; Dahan, M.H. Pituitary mass and subsequent involution causing fluctuations of serum follicle-stimulating hormone levels in a Turner syndrome patient with premature ovarian failure: A case report. J. Reprod. Med. 2014, 59, 504–508. [Google Scholar] [PubMed]
- Dotsch, J.; Schoof, E.; Hensen, J.; Dorr, H.G. Prolactinoma causing secondary amenorrhea in a woman with Ullrich-Turner syndrome. Horm. Res. 1999, 51, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Willemse, C.H. A patient suffering from Turner’s syndrome and acromegaly. Acta. Endocrinol. 1962, 39, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Sato, A.; Nishio, S.; Takeda, T.; Miyamoto, T.; Katai, M.; Hashizume, K. Acromegaly accompanied by Turner syndrome with 47,XXX/45,X/46,XX mosaicism. Intern. Med. 2009, 48, 447–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelfand, R.A. Cushing’s disease associated with ovarian dysgenesis. Am. J. Med. 1984, 77, 1108–1110. [Google Scholar] [CrossRef]
- Artese, R.; D’Osvaldo, D.H.; Molocznik, I.; Benencia, H.; Oviedo, J.; Burdman, J.A.; Basso, A. Pituitary tumors in adolescent patients. Neurol. Res. 1998, 20, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Pack, S.D.; Qin, L.; Pak, E.; Wang, Y.; Ault, D.O.; Mannan, P.; Jaikumar, S.; Stratakis, C.A.; Oldfield, E.H.; Zhuang, Z.; et al. Common genetic changes in hereditary and sporadic pituitary adenomas detected by comparative genomic hybridization. Genes. Chromosomes. Cancer. 2005, 43, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Boikos, S.A.; Stratakis, C.A. Carney complex: The first 20 years. Curr. Opin. Oncol. 2007, 19, 24–29. [Google Scholar] [CrossRef]
- Thapar, K.; Kovacs, K.; Laws, E.R. The classification and molecular biology of pituitary adenomas. Adv. Tech. Stand. Neurosurg. 1995, 22, 3–53. [Google Scholar] [CrossRef]
- Chauhan, P.; Jaiswal, S.K.; Lakhotia, A.R.; Rai, A.K. Molecular cytogenetic characterization of two Turner syndrome patients with mosaic ring X chromosome. J. Assist. Reprod. Genet. 2016, 33, 1161–1168. [Google Scholar] [CrossRef] [Green Version]
- Leppig, K.A.; Disteche, C.M. Ring X and other structural X chromosome abnormalities: X inactivation and phenotype. Semin. Reprod. Med. 2001, 19, 147–157. [Google Scholar] [CrossRef]
- Mazzaschi, R.L.; Taylor, J.; Robertson, S.P.; Love, D.R.; George, A.M. A turner syndrome patient carrying a mosaic distal x chromosome marker. Case Rep. Genet. 2014, 2014, 597314. [Google Scholar] [CrossRef]
- De Moraes-Ruehsen, M.; Jones, G.S. Premature ovarian failure. Fertil. Steril. 1967, 18, 440–461. [Google Scholar] [CrossRef]
- Atkins, L.; Sceery, R.T.; Keenan, M.E. An unstable ring chromosome in a female infant with hypotonia, seizures, and retarded development. J. Med. Genet. 1966, 3, 134–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, E.; Weiss, B.; Fukami, M.; Rump, A.; Niesler, B.; Mertz, A.; Muroya, K.; Binder, G.; Kirsch, S.; Winkelmann, M.; et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet. 1997, 16, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T. SHOX: Pseudoautosomal homeobox containing gene for short stature and dyschondrosteosis. Growth. Horm. IGF. Res. 1999, 9, 53–57. [Google Scholar] [CrossRef]
- Clement-Jones, M.; Schiller, S.; Rao, E.; Blaschke, R.J.; Zuniga, A.; Zeller, R.; Robson, S.C.; Binder, G.; Glass, I.; Strachan, T.; et al. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum. Mol. Genet. 2000, 9, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Rogol, A.D.; Hayden, G.F. Etiologies and early diagnosis of short stature and growth failure in children and adolescents. J. Pediatr. 2014, 164, S1–S14.e16. [Google Scholar] [CrossRef]
- Haymond, M.; Kappelgaard, A.M.; Czernichow, P.; Biller, B.M.; Takano, K.; Kiess, W.; The Participants in the Global Advisory Panel Meeting on the Effects of Growth Hormone. Early recognition of growth abnormalities permitting early intervention. Acta. Paediatr. 2013, 102, 787–796. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Case in This Study | Yu et al. [12] | Yu et al. [12] | Brook et al. [13] | Efstathiadou et al. [14] | Gallicchino et al. [15] | Jin et al. [16] | |
|---|---|---|---|---|---|---|---|
| Age at diagnosis (y) | 12.3 | 8.9 | 12.3 | 9.1 | 30 | 11 | 11 |
| Turner syndrome | 12.3 | 7.5 | 12.3 | 9.6 | 17 | 12 | 11 |
| GH deficiency | |||||||
| Height (SDS) at diagnosis | −3.4 | −1.89 | −1.72 † | −3.6 | −2.35 | −4.2 | −3.69 |
| Turner syndrome | −3.4 | −2.30 | −1.72 † | NA | −6.0 | −4.9 | −3.69 |
| GH deficiency | |||||||
| Karyotype | 46,X,r(X)/45,X/46,X,psu dic r(X;X) | 45,X/45,X+mar | 45,X/46,XX | 45,X | 45,X | 45,X/46,XX | 45,X |
| Peak GH on GH provocation test (ng/mL) | 3.63 | 6.17 | 7.38 | 6.1 | 4.65 | 0.14 | <5 |
| Other pituitary hormone deficiencies | ACTH | None | None | None | TSH, gonadotropin | TSH, gonadotropin | None |
| Associated conditions | Subclinical hypothyroidism, pituitary microadenoma | Partial empty sella, horseshoe kidney | None | None | None | Empty sella | Chronic lymphocytic thyroiditis |
| Case in This Study | Yeh et al. [8] | Bolanowski et al. [18] | Gaspar et al. [19] | Mermilliod et al. [20] | Weibel et al. [21] | Dotsch et al. [22] | Willemse et al. [23] | Yamazaki et al. [24] | Gelfand et al. [25] | |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at TS diagnosis (yr) | 13 | 16 | 10 | 16 | 16 | 43 | 12 | 19 | 33 | 26 |
| Age at pituitary disease diagnosis (yr) | 13 | 16 | 33 | 25 | 18 | 43 | 19 | 26 | 33 | 29 |
| Karyotype | 46,X,r(X)/45,X/46,X,psu dic r(X;X) | 45,X | 45,X/46,X,i(X) (q10) | 45,X/46,XX | 45,X | 45,X/46,XX/47,XXX | 45,X/46,XX | 45,X | 47,XXX/45,X/46,XX | 45,X/47,XXX |
| Symptoms or labs related to pituitary disease | Short stature | Headache, vomiting, cranial nerve IV palsy | Facial changes, increased hand/foot size | Secondary amenorrhea, galactorrhea | Hypogonado-tropic hypogonadism | Unexpected normalization of FSH level | Secondary amenorrhea, hyperprolactinemia | Change in appearance, enlarged feet | Dysphagia due to soft palate edema, enlarged hands/feet | Weight gain, ankle edema, acne, hirsutism |
| Pituitary hormone abnormalities | Deficiency in GH, ACTH | Deficiency in GnRH | GH excess | Prolactin excess | Deficiency in GnRH | Deficiency in GnRH | Prolactin excess | GH excess | GH excess | Cortisol excess |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, E.G.; Kim, E.-J.; Kim, E.-J.; Kim, H.-Y.; Kim, S.-H.; Yang, A. Coexistence of Growth Hormone Deficiency and Pituitary Microadenoma in a Child with Unique Mosaic Turner Syndrome: A Case Report and Literature Review. Diagnostics 2020, 10, 783. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10100783
Park EG, Kim E-J, Kim E-J, Kim H-Y, Kim S-H, Yang A. Coexistence of Growth Hormone Deficiency and Pituitary Microadenoma in a Child with Unique Mosaic Turner Syndrome: A Case Report and Literature Review. Diagnostics. 2020; 10(10):783. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10100783
Chicago/Turabian StylePark, Eu Gene, Eun-Jung Kim, Eun-Jee Kim, Hyun-Young Kim, Sun-Hee Kim, and Aram Yang. 2020. "Coexistence of Growth Hormone Deficiency and Pituitary Microadenoma in a Child with Unique Mosaic Turner Syndrome: A Case Report and Literature Review" Diagnostics 10, no. 10: 783. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10100783