Complications in Idiopathic Pulmonary Fibrosis: Focus on Their Clinical and Radiological Features


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Study Design, Setting and Participants
2.2. Statistical and Data Analysis
3. Results
4. Discussion
- Lung cancer;
- Acute exacerbation;
- Pulmonary hypertension;
- Pneumothorax;
- Pulmonary infection.
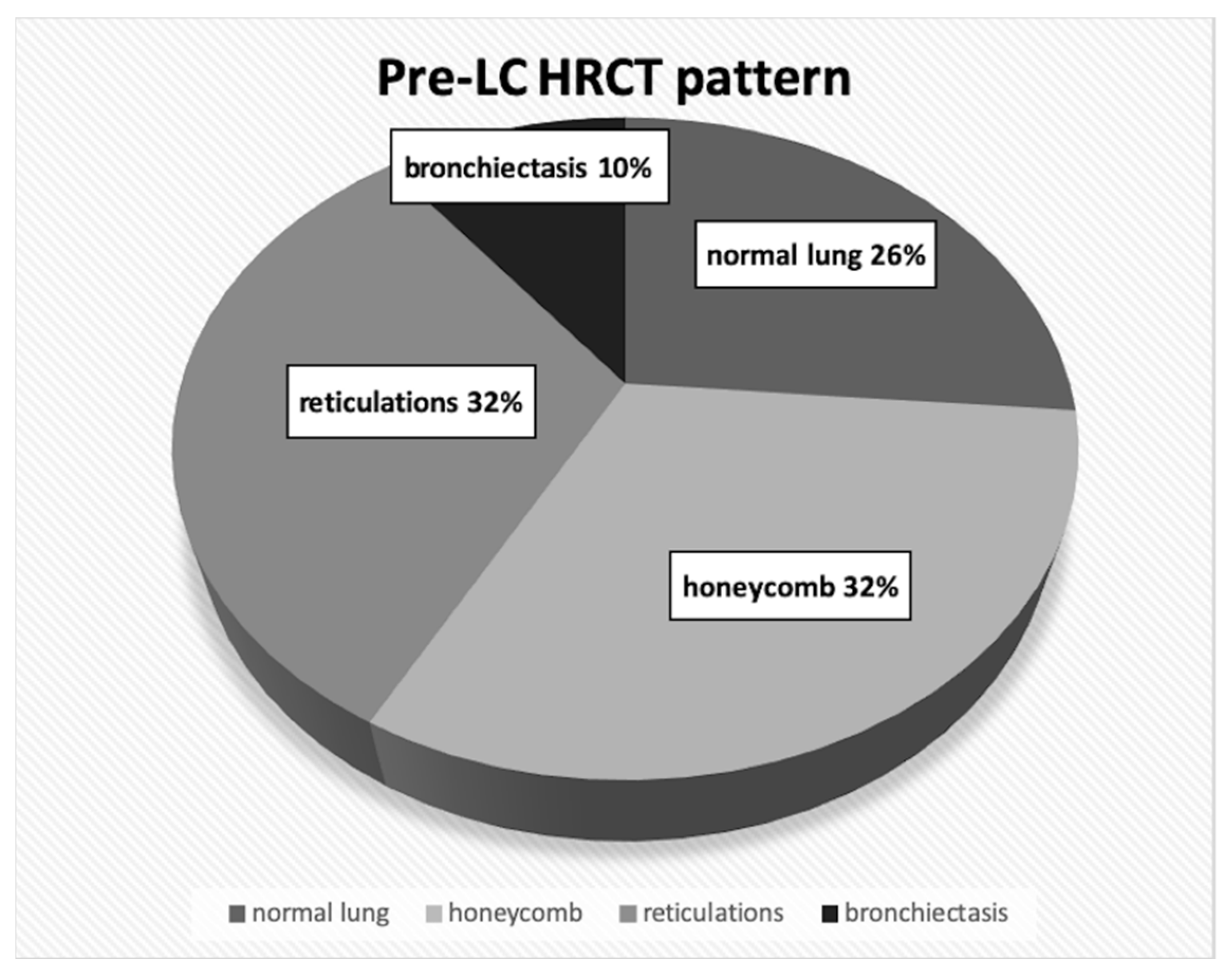
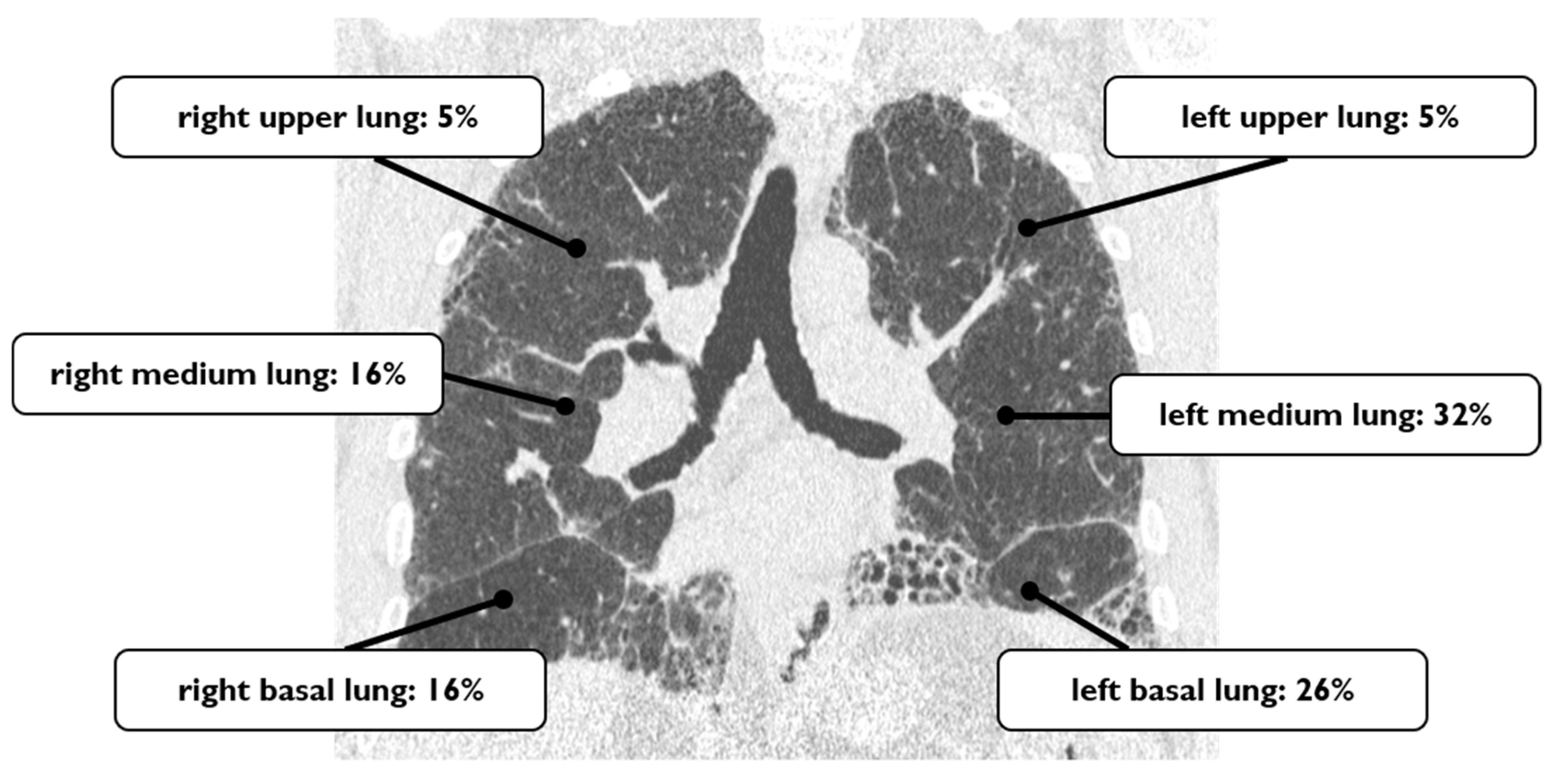
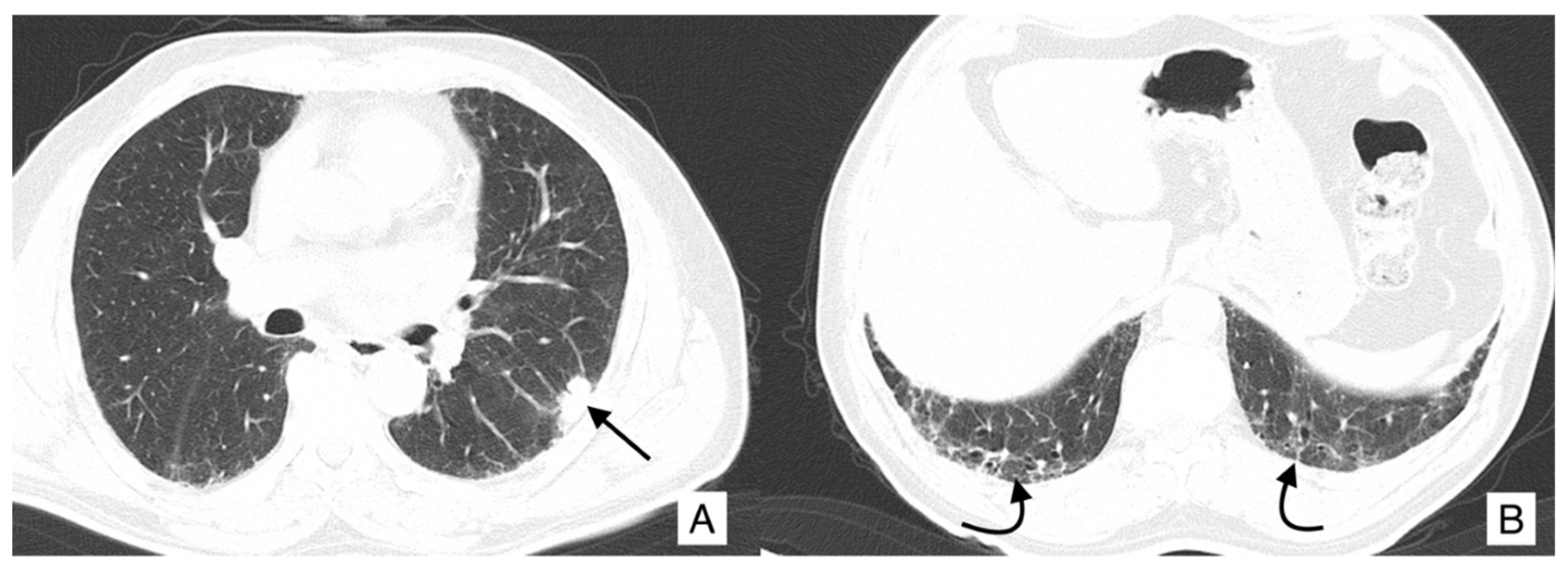
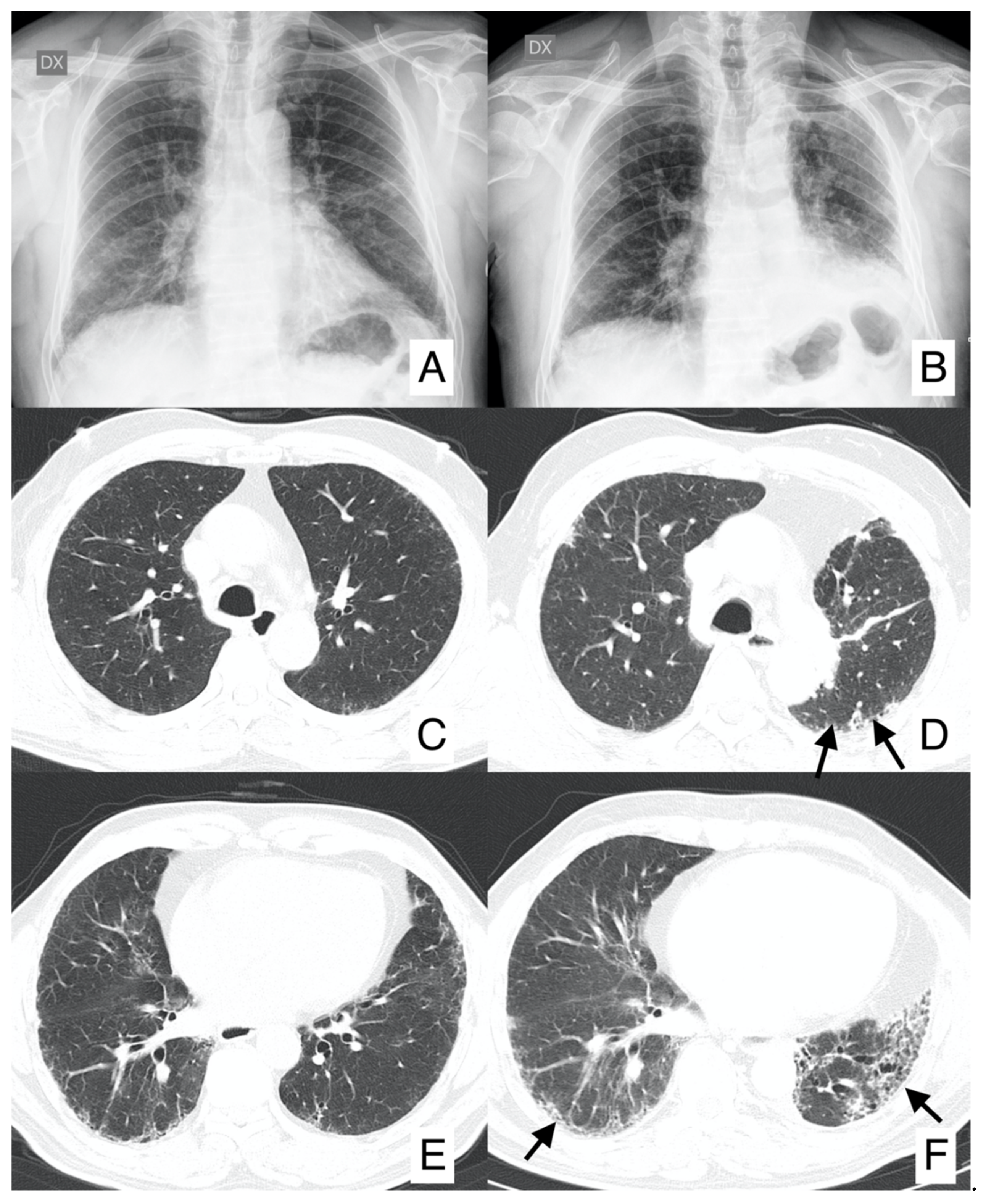
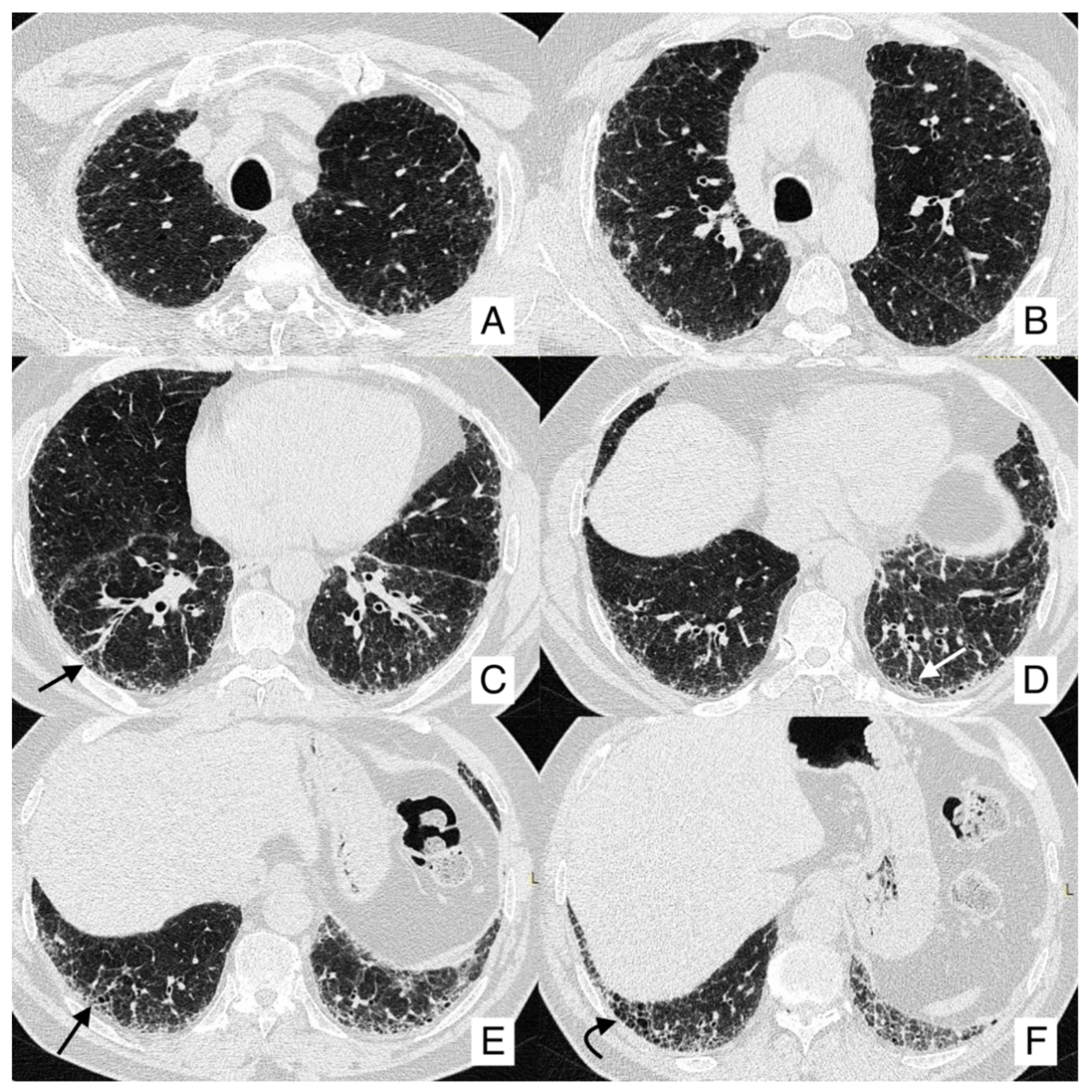
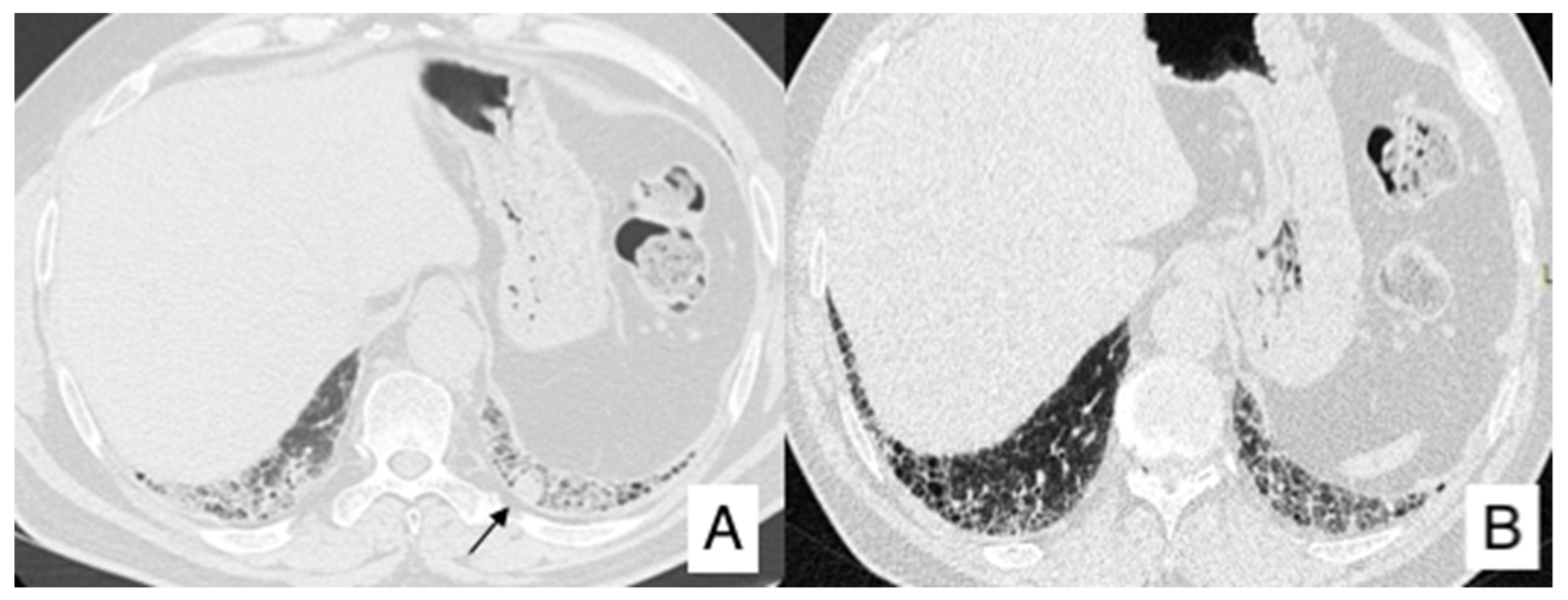
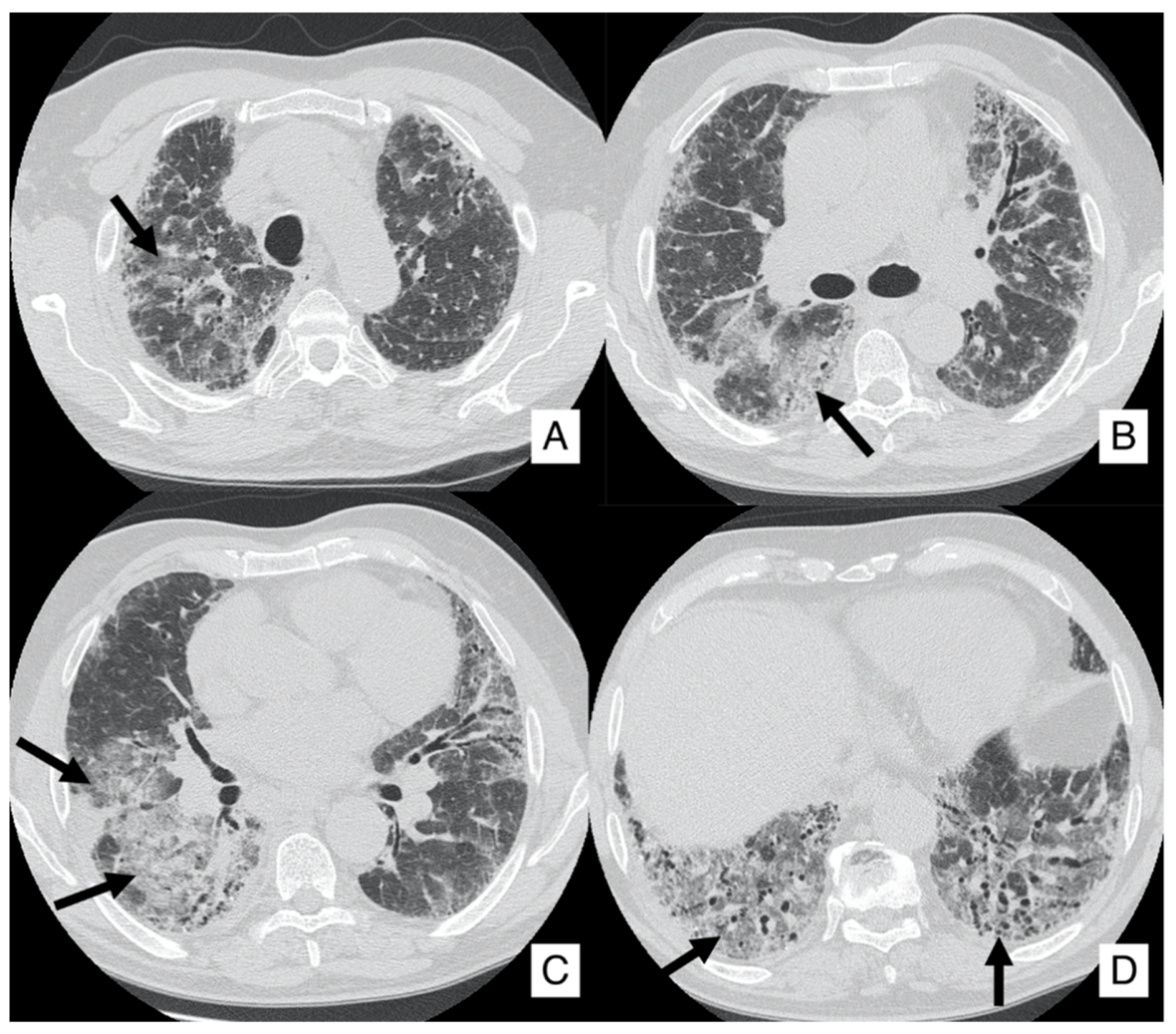
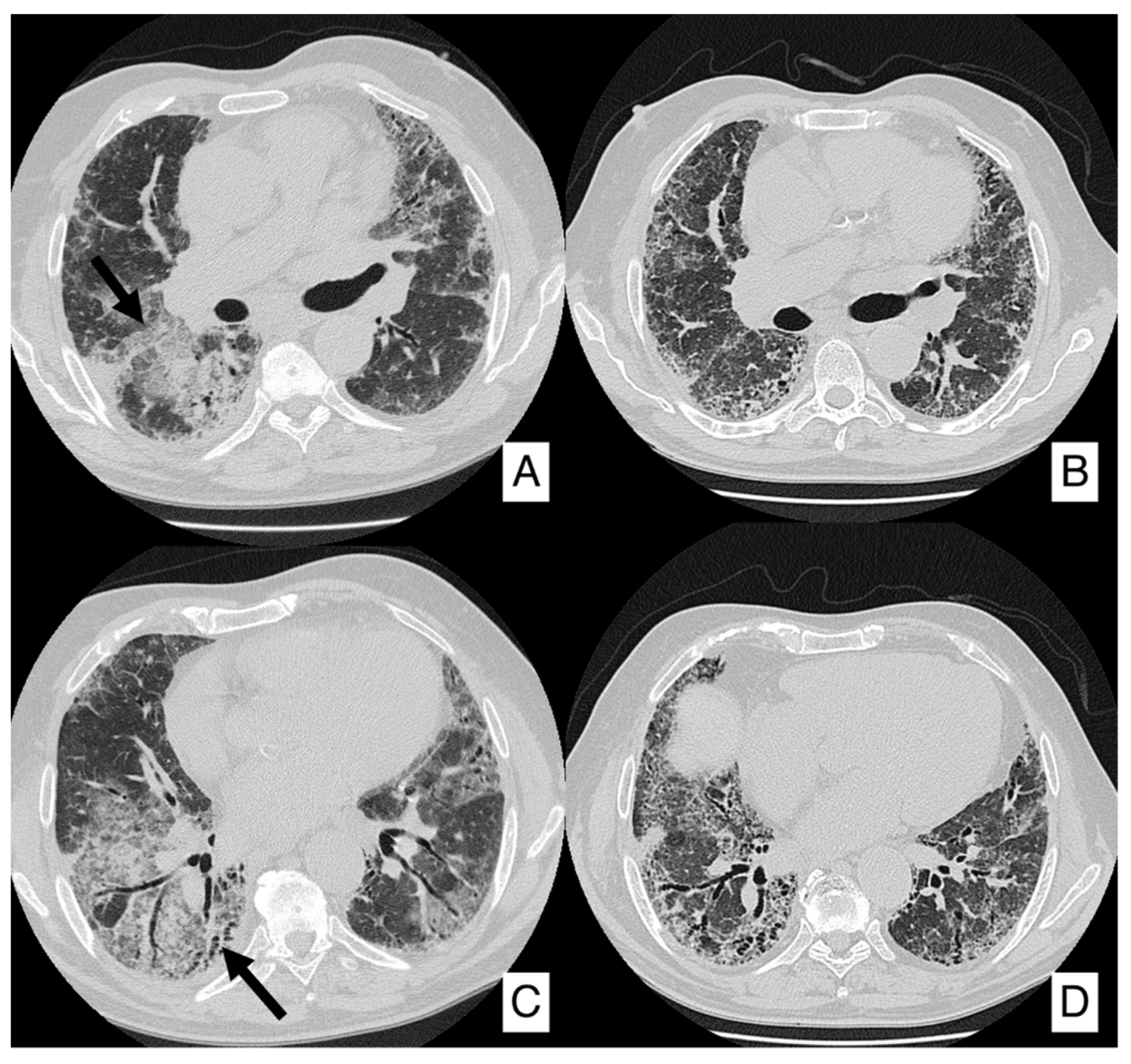
4.1. Lung Cancer
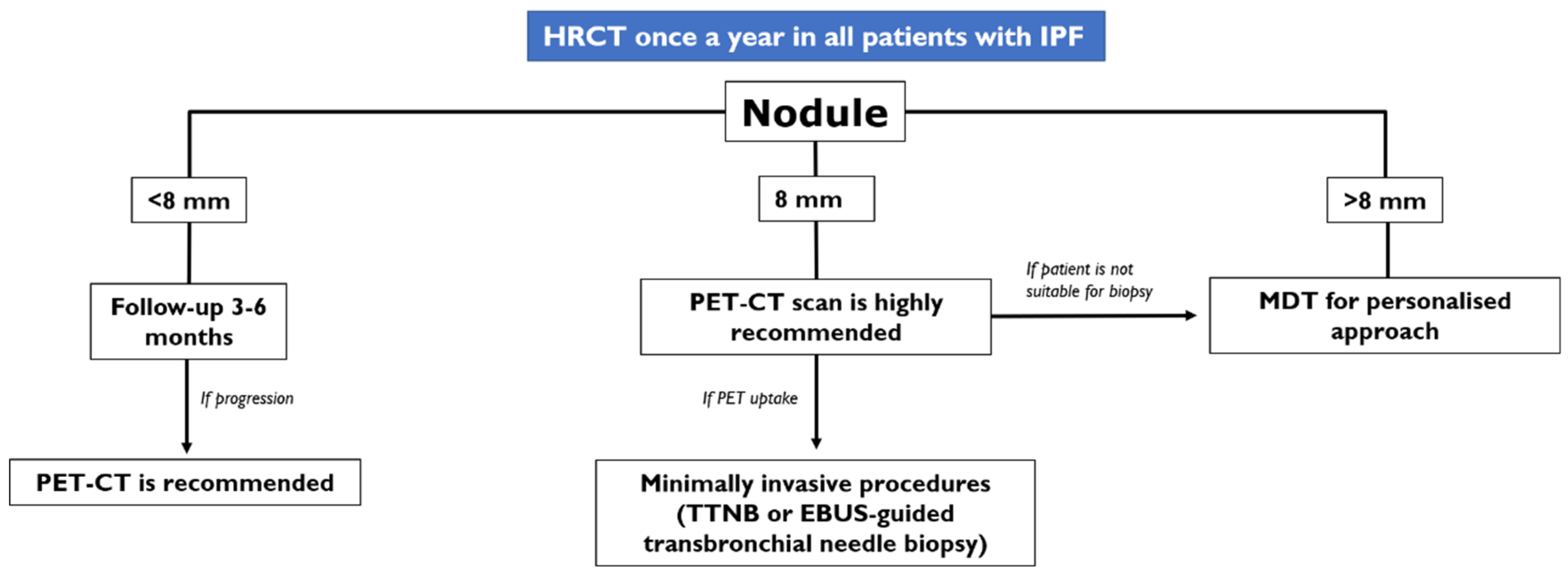
- In case of an IPF patient having a nodule of <8 mm, an HRCT is needed every three–six months with continuous surveillance. If the HRCT demonstrates progression of the nodule, a PET-CT is recommended;
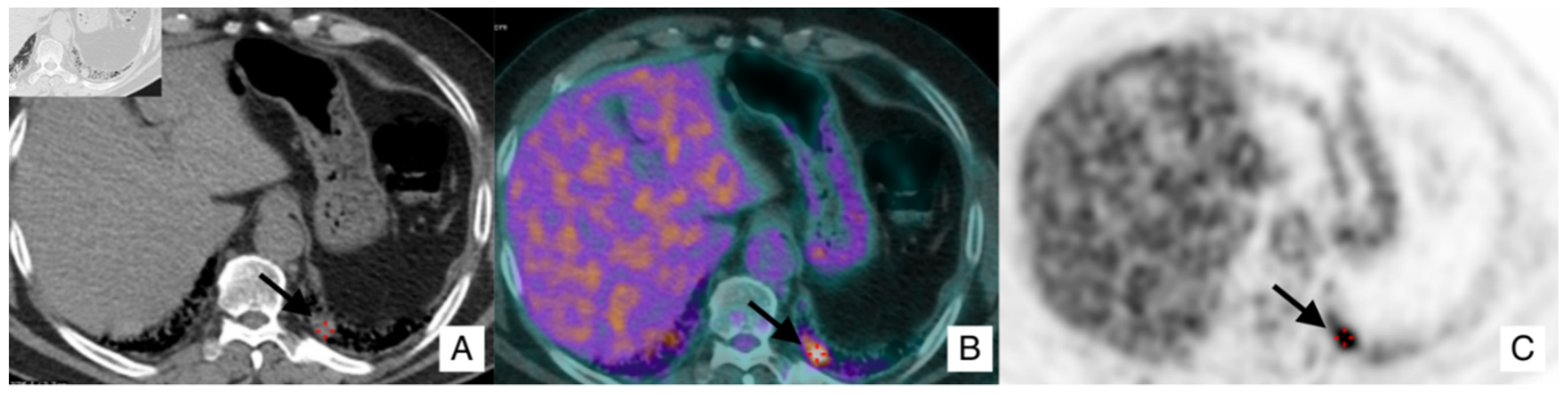
- In case of an IPF patient having a nodule of at least 8 mm, a PET-CT scan is highly recommended. If the PET reports a great uptake, the panel expert state that “minimally invasive procedures are needed, such as TTNB (Transthoracic needle biopsy) or a CT-TTNB for peripheral lesions or endobronchial ultrasound-guided transbronchial needle biopsy (EBUS-guided transbronchial needle biopsy) if there are pathological lymph nodes (≥8 mm)” [35]. If biopsy could be unsafe for the patient, or not indicated for the clinical context, “it is suggested a multidisciplinary and personalized approach”;
- In case of a patient having an advanced lesion on the HRCT (mass or nodule greater than 8 mm), a multidisciplinary and personalized approach is needed; based on the clinical conditions of the patient, in these cases, the panel experts state that “no further diagnostic procedures could be adopted” [35]—planning a personalized approach, based also on palliative support.
4.2. Acute Exacerbation
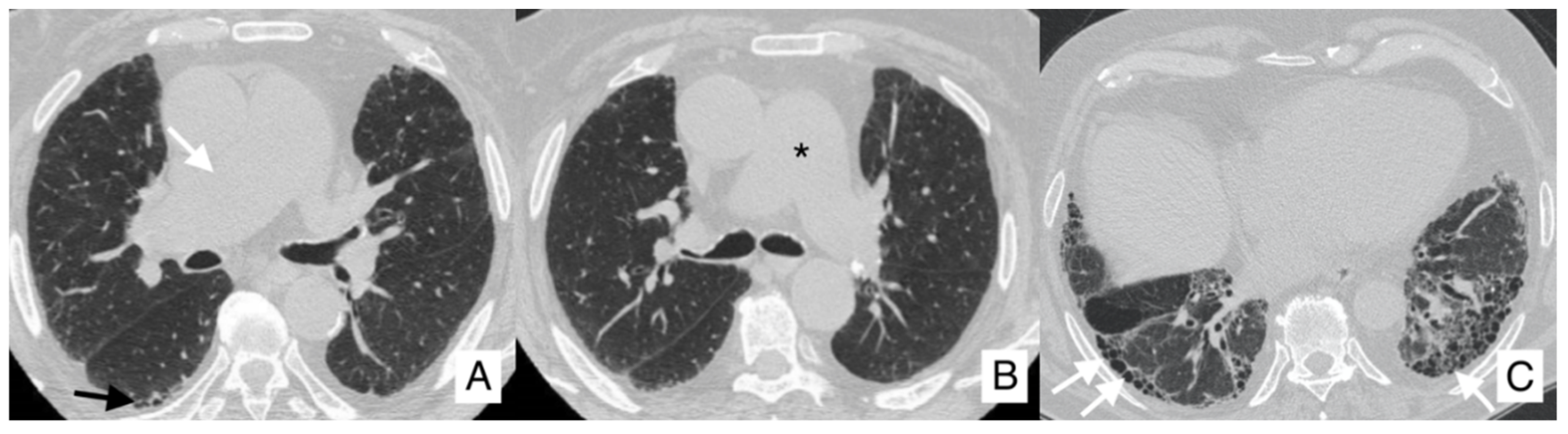
4.3. Pulmonary Hypertension
- Group I—which includes idiopathic or hereditable pulmonary arterial hypertension due to the affection of lung vasculature;
- Group II—related to left heart disease;
- Group III—PH associated with chronic lung disease and hypoxemia;
- Group IV—represented by thromboembolic pulmonary hypertension (CTEPH);
- Group V—PH caused by unclear and multifactorial mechanisms.
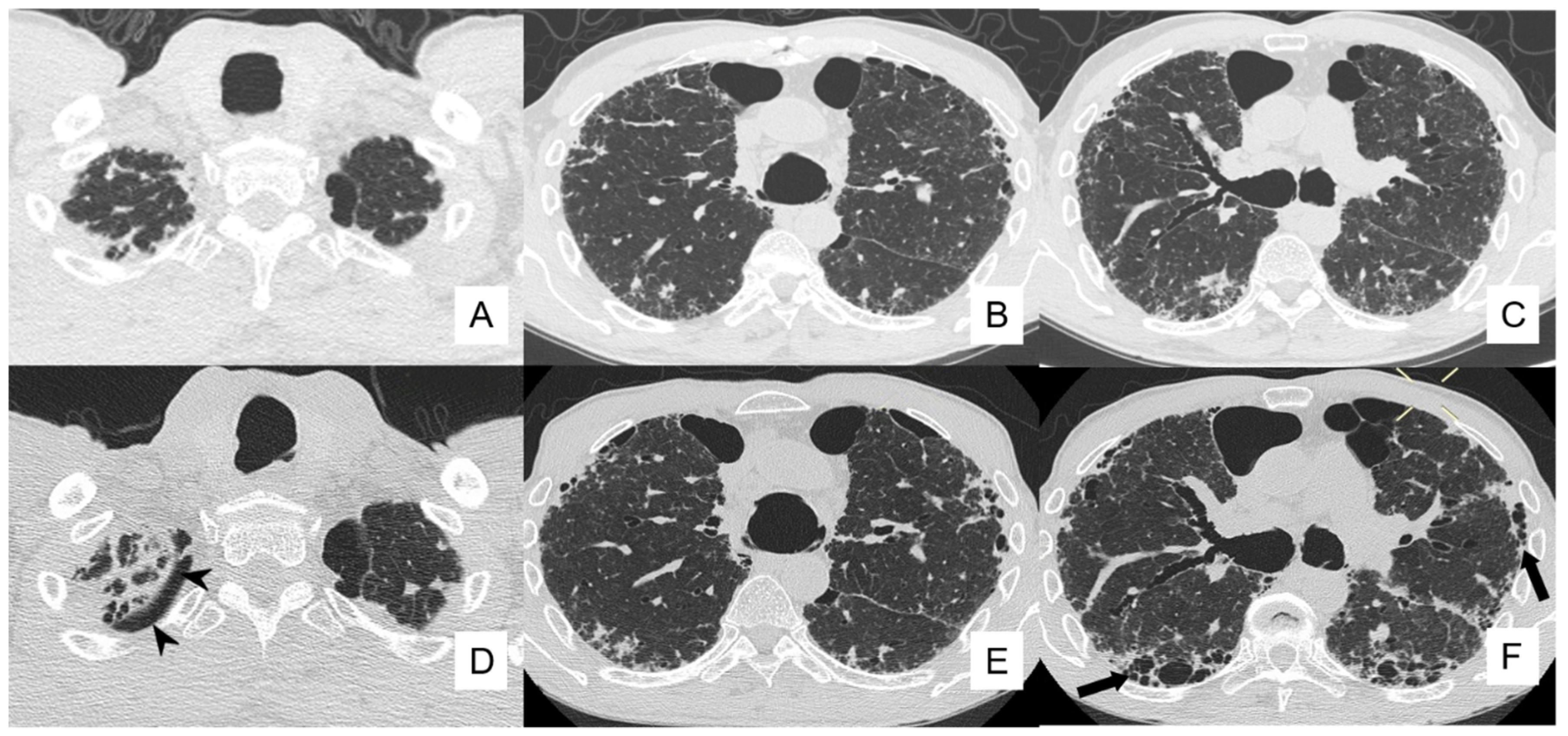
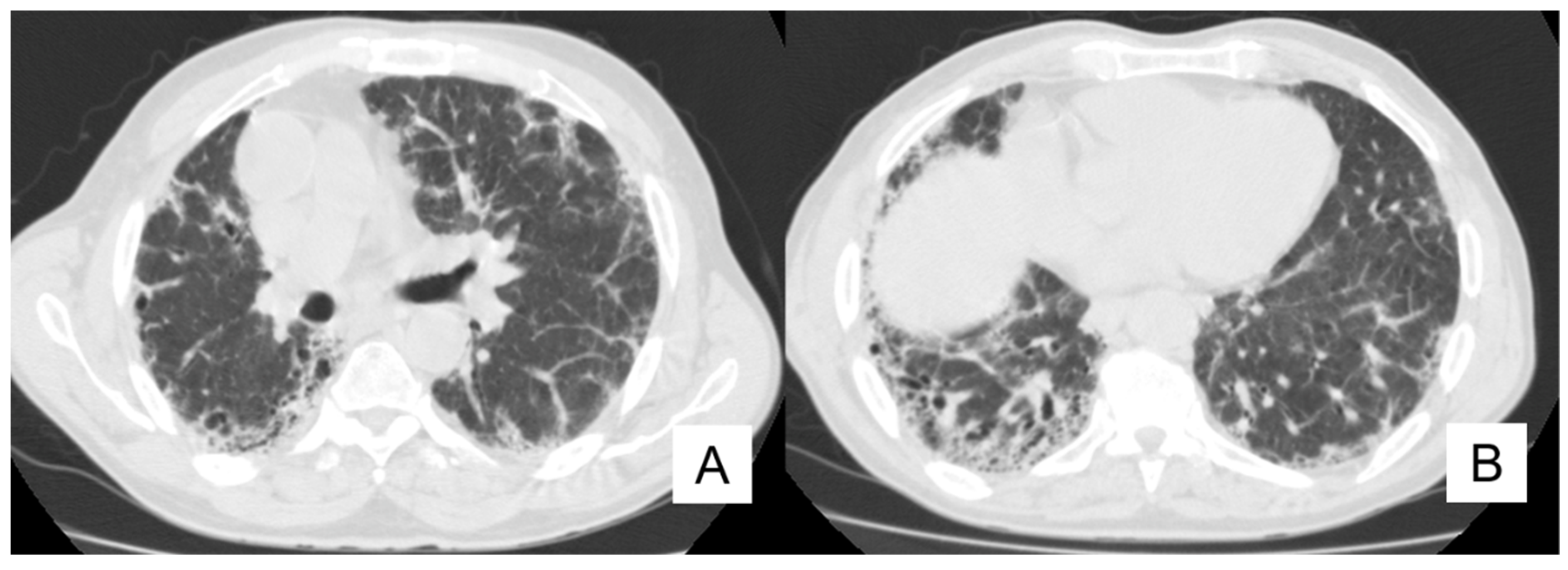
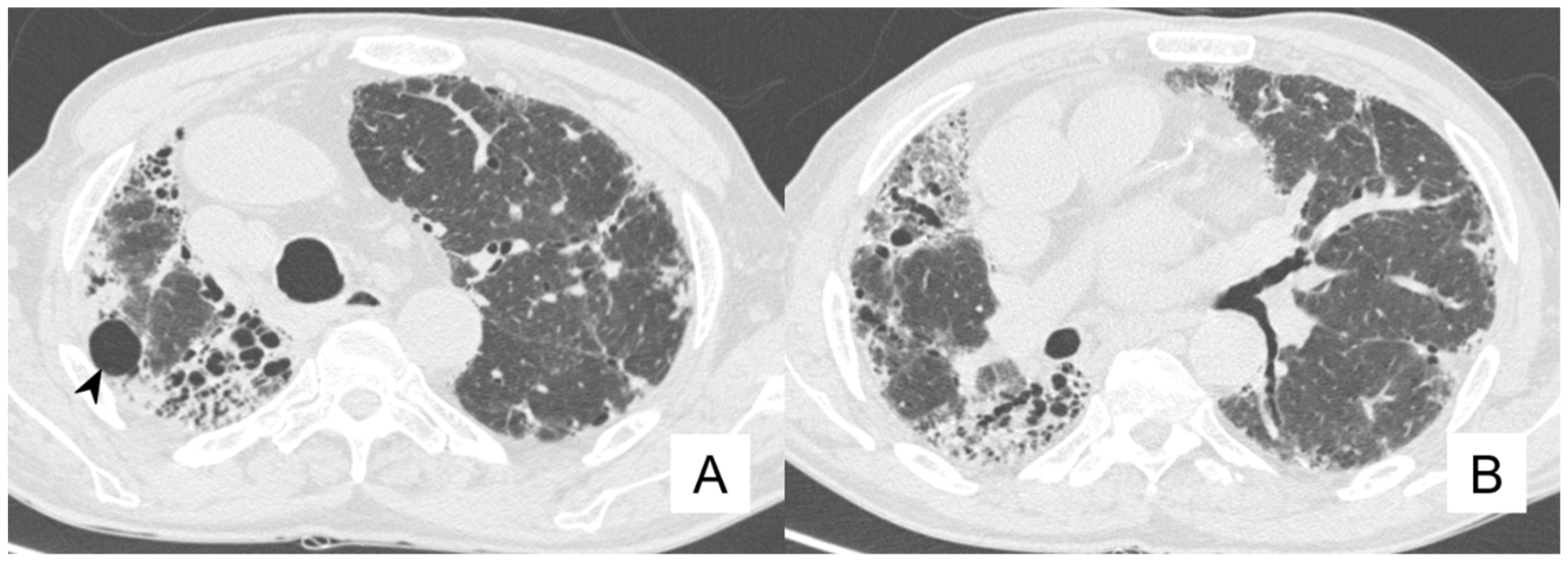
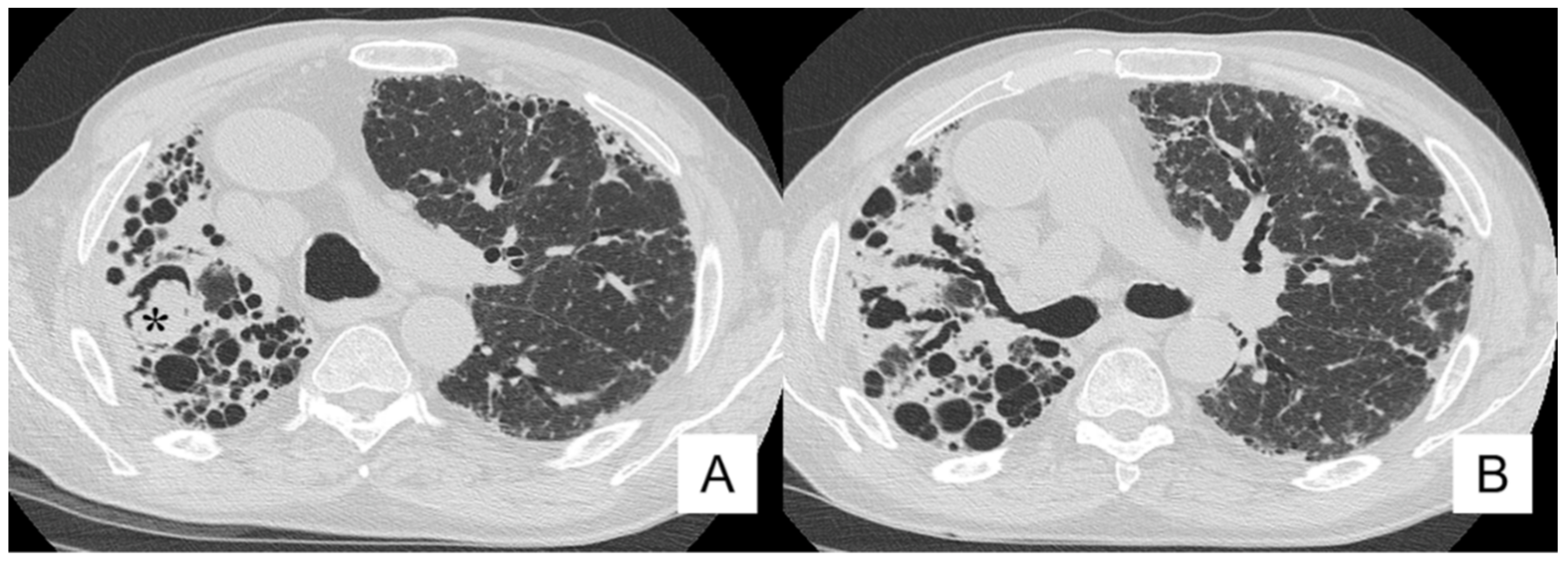
4.4. Pneumothorax
4.5. Pulmonary Infection
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raghu, G.; Remy-Jardin, M. Diagnosis of idiopathic pulmonary fibrosis An Official ATS/ERS/JRS/ALAT Clinical practice guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Hammer, G.; Mcphee, S. Pathophysiology of Disease: An Introduction to Clinical Medicine, 8th ed.; McGraw-Hill Education: Columbus, OH, USA, 2018. [Google Scholar]
- Mohning, M.P.; Richards, J.C. Idiopathic pulmonary fibrosis: The radiologist’s role in making the diagnosis. Br. J. Radiol. 2019, 92, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gruden, J.F. CT in idiopathic pulmonary fibrosis: Diagnosis and beyond. Am. J. Roentgenol. 2016, 206, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Panos, R.J.; Mortenson, R.L. Clinical deterioration in patients with idiopathic pulmonary fibrosis: Causes and assessment. Am. J. Med. 1990, 88, 396–404. [Google Scholar] [CrossRef]
- Fernández Fabrellas, E.; Peris Sánchez, R. Prognosis and Follow-Up of Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- ACR-STR Collaborative Committee. ACR–STR Practice Parameter for the Performance of High-Resolution Computed Tomography (HRCT) of the Lungs in Adults (PDF); American College of Radiology: Richmond, VA, USA, 2015. [Google Scholar]
- American Thoracic Society. Idiopathic Pulmonary Fibrosis: Diagnosis and Treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Perlman, D. Natural history of idiopathic pulmonary fibrosis. Respir. Med. 2015, 109, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, C.R.; Walsh, S.L. High-resolution CT of complications of idiopathic fibrotic lung disease. Br. J. Radiol. 2011, 84, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, R.; Venn, A. Lung cancer and cryptogenic fibrosing alveolitis: A population-based cohort study. Am. J. Respir. Crit. Care Med. 2000, 161, 5–8. [Google Scholar] [CrossRef]
- Kuwano, K.; Kunitake, R. P21Waf1Cip1/Sdi1 and p53 Expression in Association with DNA Strand Breaks in Idiopathic Pulmonary Fibrosis. Pneumologie 1997, 51, 870. [Google Scholar]
- Sanders, Y.Y.; Pardo, A. Thy-1 promoter hypermethylation: A novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Vancheri, C.; Failla, M. Idiopathic pulmonary fibrosis: A disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner-Warwick, M.; Lebowitz, M. Cryptogenic fibrosing alveolitis and lung cancer. Thorax 1980, 35, 496–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubry, M.-C.; Myers, J.L. Primary Pulmonary Carcinoma in Patients With Idiopathic Pulmonary Fibrosis. Mayo Clin. Proc. 2002, 77, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, D.S. Lung cancer in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2001, 17, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Daniels, C.E.; Jett, J.R. Does interstitial lung disease predispose to lung cancer? Curr. Opin. Pulm. Med. 2005, 11, 431–437. [Google Scholar] [CrossRef]
- Kishi, K.; Homma, S. High-Resolution Computed Tomography Findings of Lung Cancer Associated With Idiopathic Pulmonary Fibrosis. J. Comput. Assist. Tomogr. 2006, 30, 95–99. [Google Scholar] [CrossRef]
- Matsushita, H.; Tanaka, S. Lung cancer associated with usual interstitial pneumonia. Pathol. Int. 1995, 45, 925–932. [Google Scholar] [CrossRef]
- Yoshida, R.; Arakawa, H. Lung Cancer in Chronic Interstitial Pneumonia: Early Manifestation From Serial CT Observations. Am. J. Roentgenol. 2012, 199, 85–90. [Google Scholar] [CrossRef]
- Ballester, B.; Milara, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [Green Version]
- Hironaka, M.; Fukayama, M. Pulmonary fibrosis and lung carcinoma: A comparative study of metaplastic epithelia in honeycombed areas of usual interstitial pneumonia with or without lung carcinoma. Pathol. Int. 1999, 49, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Im, J.G. Lung Cancer in Patients with Idiopathic Pulmonary Fibrosis: CT Findings. J. Comput. Assist. Tomogr. 1996, 20, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Ono, M. Lung Cancer Associated With Diffuse Pulmonary Fibrosis: CT–Pathologic Correlation. J. Thorac. Imaging 2003, 18, 67–71. [Google Scholar] [CrossRef]
- Mizushima, Y.; Kobayashi, M. Clinical Characteristics of Synchronous Multiple Lung Cancer Associated With Idiopathic Pulmonary Fibrosis: A Review of Japanese Cases. Chest 1995, 108, 1272–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomassetti, S.; Gurioli, C. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest 2015, 147, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Suda, T. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology 2009, 14, 723–728. [Google Scholar] [CrossRef]
- Kato, E.; Takayanagi, N. Incidence and predictive factors of lung cancer in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2018, 4, 111–2016. [Google Scholar] [CrossRef]
- Jang, H.J.; Lee, K.S. Bronchioloalveolar carcinoma: Focal area of ground-glass attenuation at thin-section CT as an early sign. Radiology 1996, 199, 485–488. [Google Scholar] [CrossRef]
- Caliò, A.; Lever, V. Increased frequency of bronchiolar histotypes in lung carcinomas associated with idiopathic pulmonary fibrosis. Histopathology 2017, 71, 725–735. [Google Scholar] [CrossRef]
- Karampitsakos, T.; Tzilas, V. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2017, 45, 1–10. [Google Scholar] [CrossRef]
- Reck, M.; Kaiser, R. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014, 15, 143–155. [Google Scholar] [CrossRef]
- Mediavilla-Varela, M.; Boateng, K. The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer 2016, 16, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzouvelekis, A.; Spagnolo, P. Patients with IPF and lung cancer: Diagnosis and management. Lancet Respir. Med. 2018, 6, 86–88. [Google Scholar] [CrossRef]
- Collard, H.R.; Ryerson, C.J. Acute exacerbation of idiopathic pulmonary fibrosis an international working group report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, Y.; Cottin, V. Recent lessons learned in the management of acute exacerbation of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2017, 26, 170050. [Google Scholar] [CrossRef]
- Kumar, P.; Goldstraw, P. Pulmonary fibrosis and lung cancer: Risk and benefit analysis of pulmonary resection. J. Thorac. Cardiovasc. Surg. 2003, 125, 1321–1327. [Google Scholar] [CrossRef] [Green Version]
- Yüksel, M.; Özyurtkan, M.O. Acute Exacerbation of Interstitial Fibrosis After Pulmonary Resection. Ann. Thorac. Surg. 2006, 82, 336–338. [Google Scholar] [CrossRef]
- Utz, J.P.; Ryu, J.H. High short-term mortality following lung biopsy for usual interstitial pneumonia. Eur. Respir. J. 2001, 17, 175–179. [Google Scholar] [CrossRef]
- Cano-Jiménez, E.; Hernández González, F. Comorbidities and Complications in Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Ohshimo, S.; Ishikawa, N. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir. Med. 2014, 108, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Egan, J.J.; Stewart, J.P. Epstein-Barr virus replication within pulmonary epithelial cells in cryptogenic fibrosing alveolitis. Thorax 1995, 50, 1234–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, K.; Hayakawa, H. Involvement of Epstein-Barr virus latent membrane protein 1 in disease progression in patients with idiopathic pulmonary fibrosis. Thorax 2000, 55, 958–961. [Google Scholar] [CrossRef] [Green Version]
- Yonemaru, M.; Kasuga, I. Elevation of antibodies to cytomegalovirus and other herpes viruses in pulmonary fibrosis. Eur. Respir. J. 1997, 10, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.P.; Egan, J.J. The detection of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1999, 159, 1336–1341. [Google Scholar] [CrossRef]
- Tang, Y.-W.; Johnson, J.E. Herpesvirus DNA Is Consistently Detected in Lungs of Patients with Idiopathic Pulmonary Fibrosis. J. Clin. Microbiol. 2003, 41, 2633–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Freudenberger, T.D. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur. Respir. J. 2006, 27, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Yang, S.T.-Y. Sole Treatment of Acid Gastroesophageal Reflux in Idiopathic Pulmonary Fibrosis: A Case Series. Chest 2006, 129, 794–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweet, M.P.; Patti, M.G. Gastroesophageal reflux in patients with idiopathic pulmonary fibrosis referred for lung transplantation. J. Thorac. Cardiovasc. Surg. 2007, 133, 1078–1084. [Google Scholar] [CrossRef] [Green Version]
- Ware, L.B.; Matthay, M.A. The Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Selman, M.; King, T.E., Jr. Idiopathic Pulmonary Fibrosis: Prevailing and Evolving Hypotheses about Its Pathogenesis and Implications for Therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef]
- Ambrosini, V.; Cancellieri, A. Acute exacerbation of idiopathic pulmonary fibrosis: Report of a series. Eur. Respir. J. 2003, 22, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Park, J.H. Acute exacerbation of idiopathic pulmonary fibrosis: Frequency and clinical features. Eur. Respir. J. 2006, 27, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Parambil, J.G.; Myers, J.L. Histopathologic Features and Outcome of Patients With Acute Exacerbation of Idiopathic Pulmonary Fibrosis Undergoing Surgical Lung Biopsy. Chest 2005, 128, 3310–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishaba, T. Acute exacerbation of idiopathic pulmonary fibrosis. Medicina 2019, 55, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collard, H.R.; Moore, B.B. Acute exacerbation of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, M.; Hamada, H. CT findings during phase of accelerated deterioration in patients with idiopathic pulmonary fibrosis. Am. J. Roentgenol. 1997, 168, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Hoeper, M.M.; Bogaard, H.J. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62 (Suppl. S25), D42–D50. [Google Scholar] [CrossRef] [Green Version]
- Lettieri, C.J.; Nathan, S.D. Prevalence and Outcomes of Pulmonary Arterial Hypertension in Advanced Idiopathic Pulmonary Fibrosis. Chest 2006, 129, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Harari, S.; Elia, D. Pulmonary Hypertension in Parenchymal Lung Diseases: Any Future for New Therapies? Chest 2018, 153, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Le Pavec, J. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur. Respir. J. 2010, 35, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Collum, S.D.; Amione-Guerra, J. Pulmonary Hypertension Associated with Idiopathic Pulmonary Fibrosis: Current and Future Perspectives. Can. Respir. J. 2017, 2017, 1430350. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F.; Wainright, J.L. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur. Respir. J. 2007, 30, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonneau, G.; Montani, D. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Minai, O.A.; Santacruz, J.F. Impact of pulmonary hemodynamics on 6-min walk test in idiopathic pulmonary fibrosis. Respir. Med. 2012, 106, 1613–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galiè, N.; Humbert, M. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Taniguchi, H. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration 2013, 85, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Shlobin, O.A. Pulmonary Hypertension and Pulmonary Function Testing in Idiopathic Pulmonary Fibrosis. Chest 2007, 131, 657–663. [Google Scholar] [CrossRef]
- Arcasoy, S.M.; Christie, J.D. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am. J. Respir. Crit. Care Med. 2003, 167, 735–740. [Google Scholar] [CrossRef] [Green Version]
- Alhamad, E.H.; Al-boukai, A.A. Prediction of pulmonary hypertension in patients with or without interstitial lung disease: Reliability of CT findings. Radiology 2011, 260, 875–883. [Google Scholar] [CrossRef]
- Olschewski, H.; Simonneau, G. Inhaled iloprost for severe pulmonary hypertension. N. Engl. J. Med. 2002, 347, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Ghofrani, H.A. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimoto, K.; Fujisawa, T. The prognostic significance of pneumothorax in patients with idiopathic pulmonary fibrosis. Respirology 2018, 23, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, T.; Ogura, T. Pneumothorax and idiopathic pulmonary fibrosis. Jpn. J. Radiol. 2010, 28, 672–679. [Google Scholar] [CrossRef]
- Picado, C.; Gómez de Almeida, R. Spontaneous Pneumothorax in Cryptogenic Fibrosing Alveolitis. Respiration 1985, 48, 77–80. [Google Scholar] [CrossRef]
- Chung, M.J.; Goo, J.M. Pulmonary tuberculosis in patients with idiopathic pulmonary fibrosis. Eur. J. Radiol. 2004, 52, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Shachor, Y.; Schindler, D. Increased incidence of pulmonary tuberculosis in chronic interstitial lung disease. Thorax 1989, 44, 151–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraceno, J.L.; Phelps, D.T. Chronic Necrotizing Pulmonary Aspergillosis*: Approach to Management. Chest 1997, 112, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.; de la Horra, C. Pneumocystis jirovecii colonisation in patients with interstitial lung disease. Clin. Microbiol. Infect. 2006, 12, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.M.; Citron, K.M. Intrathoracic aspergilloma: Role of CT in diagnosis and treatment. Radiology 1987, 165, 123–128. [Google Scholar] [CrossRef]
- Reittner, P.; Ward, S. Pneumonia: High-resolution CT findings in 114 patients. Eur. Radiol. 2003, 13, 515–521. [Google Scholar] [CrossRef]















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galioto, F.; Palmucci, S.; Astuti, G.M.; Vancheri, A.; Distefano, G.; Tiralongo, F.; Libra, A.; Cusumano, G.; Basile, A.; Vancheri, C. Complications in Idiopathic Pulmonary Fibrosis: Focus on Their Clinical and Radiological Features. Diagnostics 2020, 10, 450. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10070450
Galioto F, Palmucci S, Astuti GM, Vancheri A, Distefano G, Tiralongo F, Libra A, Cusumano G, Basile A, Vancheri C. Complications in Idiopathic Pulmonary Fibrosis: Focus on Their Clinical and Radiological Features. Diagnostics. 2020; 10(7):450. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10070450
Chicago/Turabian StyleGalioto, Federica, Stefano Palmucci, Giovanna M. Astuti, Ada Vancheri, Giulio Distefano, Francesco Tiralongo, Alessandro Libra, Giacomo Cusumano, Antonio Basile, and Carlo Vancheri. 2020. "Complications in Idiopathic Pulmonary Fibrosis: Focus on Their Clinical and Radiological Features" Diagnostics 10, no. 7: 450. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics10070450