
Investigation of MALDI-TOF Mass Spectrometry for Assessing the Molecular Diversity of Campylobacter jejuni and Comparison with MLST and cgMLST: A Luxembourg One-Health Study

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection
2.2. MALDI-TOF MS Analysis
2.2.1. Sample Preparation
2.2.2. Data Acquisition
2.2.3. Mass Spectra Analysis
2.3. Typing Methods Concordance
2.4. Machine Learning Approach
2.4.1. Data Pre-Processing
2.4.2. Prediction Models and Evaluation
2.4.3. Evaluation Metrics
2.4.4. Retro-Engineering
3. Results
3.1. Spectra Quality
3.2. Classification
3.3. Congruency: Proteomics vs. Genomics
3.4. Machine Learning for Automatic Attribution of ST
3.5. Features of Importance: Beyond Biomarkers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Butzler, J.-P. Campylobacter, from obscurity to celebrity. Clin. Microbiol. Infect. 2004, 10, 868–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EFSA. ECDC the European Union One Health 2019 Zoonoses Report. EFSA J. 2021, 19, e06406. [Google Scholar]
- Devleesschauwer, B.; Bouwknegt, M.; Mangen, M.-J.J.; Havelaar, A.H. Health and economic burden of Campylobacter. In Campylobacter: Features, Detection, and Prevention of Foodborne Disease; Klein, G., Ed.; Elsevier BV: Amsterdam, The Netherlands, 2017; pp. 27–40. [Google Scholar]
- Murray, C.J.L.; Vos, T.; Lozano, R.; Naghavi, M.; Flaxman, A.D.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2013, 380, 2197–2223. [Google Scholar] [CrossRef]
- Joensen, K.G.; Schjørring, S.; Gantzhorn, M.R.; Vester, C.T.; Nielsen, H.L.; Engberg, J.H.; Holt, H.M.; Ethelberg, S.; Müller, L.; Sandø, G.; et al. Whole genome sequencing data used for surveillance of Campylobacter infections: Detection of a large continuous outbreak, Denmark, 2019. Eurosurveillance 2021, 26, 2001396. [Google Scholar] [CrossRef] [PubMed]
- Llarena, A.-K.; Taboada, E.; Rossi, M. Whole-Genome Sequencing in Epidemiology of Campylobacter jejuni Infections. J. Clin. Microbiol. 2017, 55, 1269–1275. [Google Scholar] [CrossRef] [Green Version]
- ISO 23418:2018(E): Microbiology of the Food Chain—Whole Genome Sequencing for Typing and Genomic Characterization of Foodborne Bacteria—General Requirements and Guidance. Available online: https://www.iso.org/standard/75509.html (accessed on 2 August 2021).
- European Food Safety Authority (EFSA). ECDC The European Union Summary Report on Antimicrobial Resistance in zoonotic and indicator bacteria from humans, animals and food in 2017/2018 European. EFSA J. 2020, 18, e06007. [Google Scholar]
- Ragimbeau, C.; Schneider, F.; Losch, S.; Even, J.; Mossong, J. Multilocus Sequence Typing, Pulsed-Field Gel Electrophoresis, and fla Short Variable Region Typing of Clonal Complexes of Campylobacter jejuni Strains of Human, Bovine, and Poultry Origins in Luxembourg. Appl. Environ. Microbiol. 2008, 74, 7715–7722. [Google Scholar] [CrossRef] [Green Version]
- Ragimbeau, C.; Colin, S.; Devaux, A.; Decruyenaere, F.; Cauchie, H.-M.; Losch, S.; Penny, C.; Mossong, J. Investigating the host specificity of Campylobacter jejuni and Campylobacter coli by sequencing gyrase subunit A. BMC Microbiol. 2014, 14, 205. [Google Scholar] [CrossRef] [Green Version]
- Mossong, J.; Mughini-Gras, L.; Penny, C.; Devaux, A.; Olinger, C.; Losch, S.; Cauchie, H.-M.; Van Pelt, W.; Ragimbeau, C. Human Campylobacteriosis in Luxembourg, 2010–2013: A Case-Control Study Combined with Multilocus Sequence Typing for Source Attribution and Risk Factor Analysis. Sci. Rep. 2016, 6, 20939. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, S.K.; Jolley, K.A.; Maiden, M.C.J. A Gene-By-Gene Approach to Bacterial Population Genomics: Whole Genome MLST of Campylobacter. Genes 2012, 3, 261–277. [Google Scholar] [CrossRef] [Green Version]
- Dingle, K.E.; Colles, F.M.; Wareing, D.R.A.; Ure, R.; Fox, A.J.; Bolton, F.E.; Bootsma, H.J.; Willems, R.J.L.; Urwin, R.; Maiden, M.C.J. Multilocus Sequence Typing System for Campylobacter jejuni. J. Clin. Microbiol. 2001, 39, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, J.P.; Frank, K.M. Commentary: Next-Generation Epidemiology: Using Real-Time Core Genome Multilocus Sequence Typing To Support Infection Control Policy. J. Clin. Microbiol. 2016, 54, 2850–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gona, F.; Comandatore, F.; Battaglia, S.; Piazza, A.; Trovato, A.; Lorenzin, G.; Cichero, P.; Biancardi, A.; Nizzero, P.; Moro, M.; et al. Comparison of core-genome MLST, coreSNP and PFGE methods for Klebsiella pneumoniae cluster analysis. Microb. Genom. 2020, 6, 1–12. [Google Scholar] [CrossRef]
- Cody, A.J.; Bray, J.E.; Jolley, K.A.; McCarthy, N.D.; Maiden, M.C.J. Core Genome Multilocus Sequence Typing Scheme for Stable, Comparative Analyses of Campylobacter jejuni and C. coli Human Disease Isolates. J. Clin. Microbiol. 2017, 55, 2086–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Silva, M.; Gonçalves, B.; Silva, D.N.; Machado, M.P.; Oleastro, M.; Borges, V.; Isidro, J.; Viera, L.; Halkilahti, J.; et al. Innuendo Whole Genome and Core Genome Mlst Schemas and Datasets for Salmonella enterica. Available online: https://zenodo.org/record/1322564 (accessed on 11 August 2021).
- Llarena, A.; Ribeiro-Gonçalves, B.F.; Nuno Silva, D.; Halkilahti, J.; Machado, M.P.; Da Silva, M.S.; Jaakkonen, A.; Isidro, J.; Hämäläinen, C.; Joenperä, J.; et al. INNUENDO: A cross-sectoral platform for the integration of genomics in the surveillance of food-borne pathogens. EFSA Support. Publ. 2018, 15, 1498E. [Google Scholar] [CrossRef]
- Nennig, M.; Llarena, A.-K.; Herold, M.; Mossong, J.; Penny, C.; Losch, S.; Tresse, O.; Ragimbeau, C. Investigating Major Recurring Campylobacter jejuni Lineages in Luxembourg Using Four Core or Whole Genome Sequencing Typing Schemes. Front. Cell. Infect. Microbiol. 2021, 10, 8020. [Google Scholar] [CrossRef]
- Magana, M.; Chatzipanagiotou, S.; Burriel, A.R.; Ioannidis, A. Inquiring into the Gaps of Campylobacter Surveillance Methods. Vet. Sci. 2017, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Angeletti, S.; Ciccozzi, M. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry in clinical microbiology: An updating review. Infect. Genet. Evol. 2019, 76, 104063. [Google Scholar] [CrossRef]
- Robert, M.-G.; Cornet, M.; Hennebique, A.; Rasamoelina, T.; Caspar, Y.; Pondérand, L.; Bidart, M.; Durand, H.; Jacquet, M.; Garnaud, C.; et al. MALDI-TOF MS in a Medical Mycology Laboratory: On Stage and Backstage. Microorganisms 2021, 9, 1283. [Google Scholar] [CrossRef]
- Alcalá, L.; Marín, M.; Ruiz, A.; Quiroga, L.; Zamora-Cintas, M.; Fernández-Chico, M.A.; Muñoz, P.; Rodríguez-Sánchez, B. Identifying Anaerobic Bacteria Using MALDI-TOF Mass Spectrometry: A Four-Year Experience. Front. Cell. Infect. Microbiol. 2021, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Sy, I.; Margardt, L.; Ngbede, E.O.; Adah, M.I.; Yusuf, S.T.; Keiser, J.; Rehner, J.; Utzinger, J.; Poppert, S.; Becker, S.L. Identification of Adult Fasciola spp. Using Matrix-Assisted Laser/Desorption Ionization Time-of-Flight (MALDI-TOF) Mass Spectrometry. Microorganisms 2021, 9, 82. [Google Scholar] [CrossRef]
- Bredtmann, C.M.; Krücken, J.; Murugaiyan, J.; Balard, A.; Hofer, H.; Kuzmina, T.A.; Von Samson-Himmelstjerna, G.; Samson-Himmelstjerna, G. Concurrent Proteomic Fingerprinting and Molecular Analysis of Cyathostomins. Proteomics 2019, 19, e1800290. [Google Scholar] [CrossRef]
- Marzano, V.; Pane, S.; Foglietta, G.; Mortera, S.L.; Vernocchi, P.; Muda, A.O.; Putignani, L. Mass Spectrometry Based-Proteomic Analysis of Anisakis spp.: A Preliminary Study towards a New Diagnostic Tool. Genes 2020, 11, 693. [Google Scholar] [CrossRef] [PubMed]
- Rybicka, M.; Miłosz, E.; Bielawski, K.P. Superiority of MALDI-TOF Mass Spectrometry over Real-Time PCR for SARS-CoV-2 RNA Detection. Viruses 2021, 13, 730. [Google Scholar] [CrossRef]
- Iles, R.K.; Zmuidinaite, R.; Iles, J.K.; Carnell, G.; Sampson, A.; Heeney, J.L. A clinical MALDI-ToF Mass spectrometry assay for SARS-CoV-2: Rational design and multi-disciplinary team work. Diagnostics 2020, 10, 746. [Google Scholar] [CrossRef]
- Hernandez, M.M.; Banu, R.; Shrestha, P.; Patel, A.; Chen, F.; Cao, L.; Fabre, S.; Tan, J.; Lopez, H.; Chiu, N.; et al. RT-PCR/MALDI-TOF mass spectrometry-based detection of SARS-CoV-2 in saliva specimens. J. Med. Virol. 2021, 93, 5481–5486. [Google Scholar] [CrossRef]
- Yan, L.; Yi, J.; Huang, C.; Zhang, J.; Fu, S.; Li, Z.; Lyu, Q.; Xu, Y.; Wang, K.; Yang, H.; et al. Rapid Detection of COVID-19 Using MALDI-TOF-Based Serum Peptidome Profiling. Anal. Chem. 2021, 93, 4782–4787. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, C.; Rehnstam-Holm, A.-S.; Nilson, B. Rapid detection of antibiotic resistance in positive blood cultures by MALDI-TOF MS and an automated and optimized MBT-ASTRA protocol for Escherichia coli and Klebsiella pneumoniae. Infect. Dis. 2020, 52, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Sparbier, K.; Schubert, S.; Kostrzewa, M. MBT-ASTRA: A suitable tool for fast antibiotic susceptibility testing? Methods 2016, 104, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Oviaño, M.; Bou, G. Matrix-Assisted Laser Desorption Ionization–Time of Flight Mass Spectrometry for the Rapid Detection of Antimicrobial Resistance Mechanisms and Beyond. Clin. Microbiol. Rev. 2018, 32, e00037-18. [Google Scholar] [CrossRef] [Green Version]
- Wolters, M.; Rohde, H.; Maier, T.; Belmar-Campos, C.; Franke, G.; Scherpe, S.; Aepfelbacher, M.; Christner, M. MALDI-TOF MS fingerprinting allows for discrimination of major methicillin-resistant Staphylococcus aureus lineages. Int. J. Med. Microbiol. 2011, 301, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Lee, T.-Y.; Tseng, Y.-J.; Liu, T.-P.; Huang, K.-Y.; Chang, Y.-T.; Chen, C.-H.; Lu, J.-J. A new scheme for strain typing of methicillin-resistant Staphylococcus aureus on the basis of matrix-assisted laser desorption ionization time-of-flight mass spectrometry by using machine learning approach. PLoS ONE 2018, 13, e0194289. [Google Scholar] [CrossRef] [PubMed]
- Egli, A.; Tschudin-Sutter, S.; Oberle, M.; Goldenberger, D.; Frei, R.; Widmer, A.F. Matrix-Assisted Laser Desorption/Ionization Time of Flight Mass-Spectrometry (MALDI-TOF MS) Based Typing of Extended-Spectrum β-Lactamase Producing E. coli—A Novel Tool for Real-Time Outbreak Investigation. PLoS ONE 2015, 10, e0120624. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Yang, J.; Duan, J.; Liu, S.; Huang, X.; Wen, X.; Huang, X.; Fu, C.; Li, J.; Dou, Q.; et al. Assessing Molecular Epidemiology of Carbapenem-resistant Klebsiella pneumoniae (CR-KP) with MLST and MALDI-TOF in Central China. Sci. Rep. 2019, 9, 2271. [Google Scholar] [CrossRef]
- Giacometti, F.; Piva, S.; Vranckx, K.; De Bruyne, K.; Drigo, I.; Lucchi, A.; Manfreda, G.; Serraino, A. Application of MALDI-TOF MS for the subtyping of Arcobacter butzleri strains and comparison with their MLST and PFGE types. Int. J. Food Microbiol. 2018, 277, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Zautner, A.E.; Masanta, W.O.; Tareen, A.M.; Weig, M.; Lugert, R.; Groß, U.; Bader, O. Discrimination of multilocus sequence typing-based Campylobacter jejuni subgroups by MALDI-TOF mass spectrometry. BMC Microbiol. 2013, 13, 247. [Google Scholar] [CrossRef] [Green Version]
- Emele, M.F.; Karg, M.; Hotzel, H.; Bloois, L.G.-V.; Groß, U.; Bader, O.; Zautner, A.E. Differentiation of Campylobacter fetus subspecies by proteotyping. Eur. J. Microbiol. Immunol. 2019, 9, 62–71. [Google Scholar] [CrossRef]
- Zautner, A.E.; Masanta, W.O.; Weig, M.; Groß, U.; Bader, O. Mass Spectrometry-based PhyloProteomics (MSPP): A novel microbial typing Method. Sci. Rep. 2015, 5, 13431. [Google Scholar] [CrossRef] [Green Version]
- Emele, M.F.; Možina, S.S.; Lugert, R.; Bohne, W.; Masanta, W.O.; Riedel, T.; Groß, U.; Bader, O.; Zautner, A.E. Proteotyping as alternate typing method to differentiate Campylobacter coli clades. Sci. Rep. 2019, 9, 4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zautner, A.E.; Lugert, R.; Masanta, W.O.; Weig, M.; Groß, U.; Bader, O. Subtyping of Campylobacter jejuni ssp. doylei Isolates Using Mass Spectrometry-based PhyloProteomics (MSPP). J. Vis. Exp. 2016, e54165. [Google Scholar] [CrossRef] [PubMed]
- Weis, C.; Jutzeler, C.; Borgwardt, K.M. Machine learning for microbial identification and antimicrobial susceptibility testing on MALDI-TOF mass spectra: A systematic review. Clin. Microbiol. Infect. 2020, 26, 1310–1317. [Google Scholar] [CrossRef]
- Weis, C.; Cuénod, A.; Rieck, B.; Llinares-López, F.; Dubuis, O.; Graf, S.; Lang, C.; Oberle, M.; Soegaard, K.K.; Osthoff, M.; et al. Direct Antimicrobial Resistance Prediction from MALDI-TOF mass spectra profile in clinical isolates through Machine Learning. bioRxiv 2020, 1, 1–35. [Google Scholar]
- Delavy, M.; Cerutti, L.; Croxatto, A.; Prod’Hom, G.; Sanglard, D.; Greub, G.; Coste, A.T. Machine Learning Approach for Candida albicans Fluconazole Resistance Detection Using Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Front. Microbiol. 2020, 10, 3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.; Wang, H.; Li, G.; Feng, Z.; Song, Y.; Wang, P.; Shao, H.; Zhou, H.; Chen, G. A novel short-term high-lactose culture approach combined with a matrix-assisted laser desorption ionization-time of flight mass spectrometry assay for differentiating Escherichia coli and Shigella species using artificial neural networks. PLoS ONE 2019, 14, e0222636. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Matsumura, Y.; Ito, Y.; Fujisawa, T.; Chang, B.; Suga, S.; Kato, K.; Yunoki, T.; Hotta, G.; Noguchi, T.; et al. Development and evaluation of MALDI-TOF MS-based serotyping for Streptococcus pneumoniae. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Camoez, M.; Sierra, J.M.; Dominguez, M.; Ferrer-Navarro, M.; Vila, J.; Roca, I. Automated categorization of methicillin-resistant Staphylococcus aureus clinical isolates into different clonal complexes by MALDI-TOF mass spectrometry. Clin. Microbiol. Infect. 2016, 22, 161.e1–161.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Ding, J.; Rao, X.; Yu, J.; Chu, M.; Ren, W.; Wang, L.; Xue, W. Analysis of methicillin-resistant Staphylococcus aureus major clonal lineages by Matrix-Assisted Laser Desorption Ionization–Time of Flight Mass Spectrometry (MALDI–TOF MS). J. Microbiol. Methods 2015, 117, 122–127. [Google Scholar] [CrossRef]
- Severiano, A.; Pinto, F.; Ramirez, M.; Carriço, J.A. Adjusted Wallace Coefficient as a Measure of Congruence between Typing Methods. J. Clin. Microbiol. 2011, 49, 3997–4000. [Google Scholar] [CrossRef] [Green Version]
- Hunter, P.R.; Gaston, M.A. Numerical index of the discriminatory ability of typing systems: An application of Simpson’s index of diversity. J. Clin. Microbiol. 1988, 26, 2465–2466. [Google Scholar] [CrossRef] [Green Version]
- Grandini, M.; Bagli, E.; Visani, G. Metrics for Multi-Class Classification: An Overview. arXiv 2020, arXiv:2008.05756. [Google Scholar]
- Uelze, L.; Grützke, J.; Borowiak, M.; Hammerl, J.A.; Juraschek, K.; Deneke, C.; Tausch, S.H.; Malorny, B. Typing methods based on whole genome sequencing data. One Health Outlook 2020, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, N.D.; Colles, F.M.; Dingle, K.; Bagnall, M.C.; Manning, G.; Maiden, M.C.J.; Falush, D. Host-associated Genetic Import in Campylobacter jejuni. Emerg. Infect. Dis. 2007, 13, 267–272. [Google Scholar] [CrossRef]
- Denis, M.; Rose, V.; Nagard, B.; Serghine, J.; Meunier, M.; Benoit, F.; Rince, A.; Cauvin, E.; Gourmelon, M. Comparison of Campylobacter jejuni using MALDI-TOF to identify relationships between different origins: Poultry, bovine, sheep, shorebird, river and shellfish. In Proceedings of the Conference: MICROBE, 15th Congress of the French Society of Microbiology (SFM), Paris, France, 30 September–2 October 2019. [Google Scholar]
- Lawton, S.J.; Weis, A.M.; Byrne, B.A.; Fritz, H.; Taff, C.C.; Townsend, A.K.; Weimer, B.; Mete, A.; Wheeler, S.; Boyce, W.M. Comparative analysis of Campylobacter isolates from wild birds and chickens using MALDI-TOF MS, biochemical testing, and DNA sequencing. J. Vet. Diagn. Investig. 2018, 30, 354–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadhav, S.; Gulati, V.; Fox, E.M.; Karpe, A.; Beale, D.J.; Sevior, D.; Bhave, M.; Palombo, E.A. Rapid identification and source-tracking of Listeria monocytogenes using MALDI-TOF mass spectrometry. Int. J. Food Microbiol. 2015, 202, 1–9. [Google Scholar] [CrossRef]
- Cuénod, A.; Egli, A. Advanced Applications of MALDI-TOF MS—Typing and Beyond. In Application and Integration of Omics-powered Diagnostics in Clinical and Public Health Microbiology; Moran-Gilad, J., Yagel, Y., Eds.; Springer: Heidelebrg/Berlin, Germany, 2021; pp. 153–173. [Google Scholar]
- Josten, M.; Reif, M.; Szekat, C.; Al-Sabti, N.; Roemer, T.; Sparbier, K.; Kostrzewa, M.; Rohde, H.; Sahl, H.-G.; Bierbaum, G. Analysis of the Matrix-Assisted Laser Desorption Ionization–Time of Flight Mass Spectrum of Staphylococcus aureus Identifies Mutations That Allow Differentiation of the Main Clonal Lineages. J. Clin. Microbiol. 2013, 51, 1809–1817. [Google Scholar] [CrossRef] [Green Version]
- Kuhns, M.; Zautner, A.E.; Rabsch, W.; Zimmermann, O.; Weig, M.; Bader, O.; Groß, U. Rapid Discrimination of Salmonella enterica Serovar Typhi from Other Serovars by MALDI-TOF Mass Spectrometry. PLoS ONE 2012, 7, e40004. [Google Scholar] [CrossRef]
- Reil, M.; Erhard, M.; Kuijper, E.; Kist, M.; Zaiss, H.; Witte, W.; Gruber, H.; Borgmann, S. Recognition of Clostridium difficile PCR-ribotypes 001, 027 and 126/078 using an extended MALDI-TOF MS system. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 1431–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djordjevic, S.P.; Unicomb, L.E.; Adamson, P.J.; Mickan, L.; Rios, R.; Adamson, P.; Cheung, K.; Combs, B.; Dalton, C.; Doyle, R.; et al. Clonal Complexes of Campylobacter jejuni Identified by Multilocus Sequence Typing Are Reliably Predicted by Restriction Fragment Length Polymorphism Analyses of the flaA Gene. J. Clin. Microbiol. 2007, 45, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Mohan, V.; Habib, I. Multilocus sequence typing (MLST), porA and flaA typing of Campylobacter jejuni isolated from cats attending a veterinary clinic. BMC Res. Notes 2019, 12, 76. [Google Scholar] [CrossRef]
- Dingle, K.E.; McCarthy, N.D.; Cody, A.J.; Peto, T.E.A.; Maiden, M.C.J. Extended Sequence Typing of Campylobacter spp. Emerg. Infect. Dis. 2008, 14, 1620–1622. [Google Scholar] [CrossRef]
- Harrington, C.S.; Thomson-Carter, F.M.; Carter, P.E. Evidence for recombination in the flagellin locus of Campylobacter jejuni: Implications for the flagellin gene typing scheme. J. Clin. Microbiol. 1997, 35, 2386–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, S.H.; Nachamkin, I. Common and variable domains of the flagellin gene, flaA, in Campylobacter jejuni. Mol. Microbiol. 1991, 5, 1151–1158. [Google Scholar] [CrossRef]
- Giraud-gatineau, A. Using MALDI-TOF spectra in epidemiological surveillance for the detection of bacterial subgroups with a possible epidemic potential. Res. Sq. 2021, 1, 1–16. [Google Scholar]
- Oberle, M.; Wohlwend, N.; Jonas, D.; Maurer, F.; Jost, G.; Tschudin-Sutter, S.; Vranckx, K.; Egli, A. The Technical and Biological Reproducibility of Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry (MALDI-TOF MS) Based Typing: Employment of Bioinformatics in a Multicenter Study. PLoS ONE 2016, 11, e0164260. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
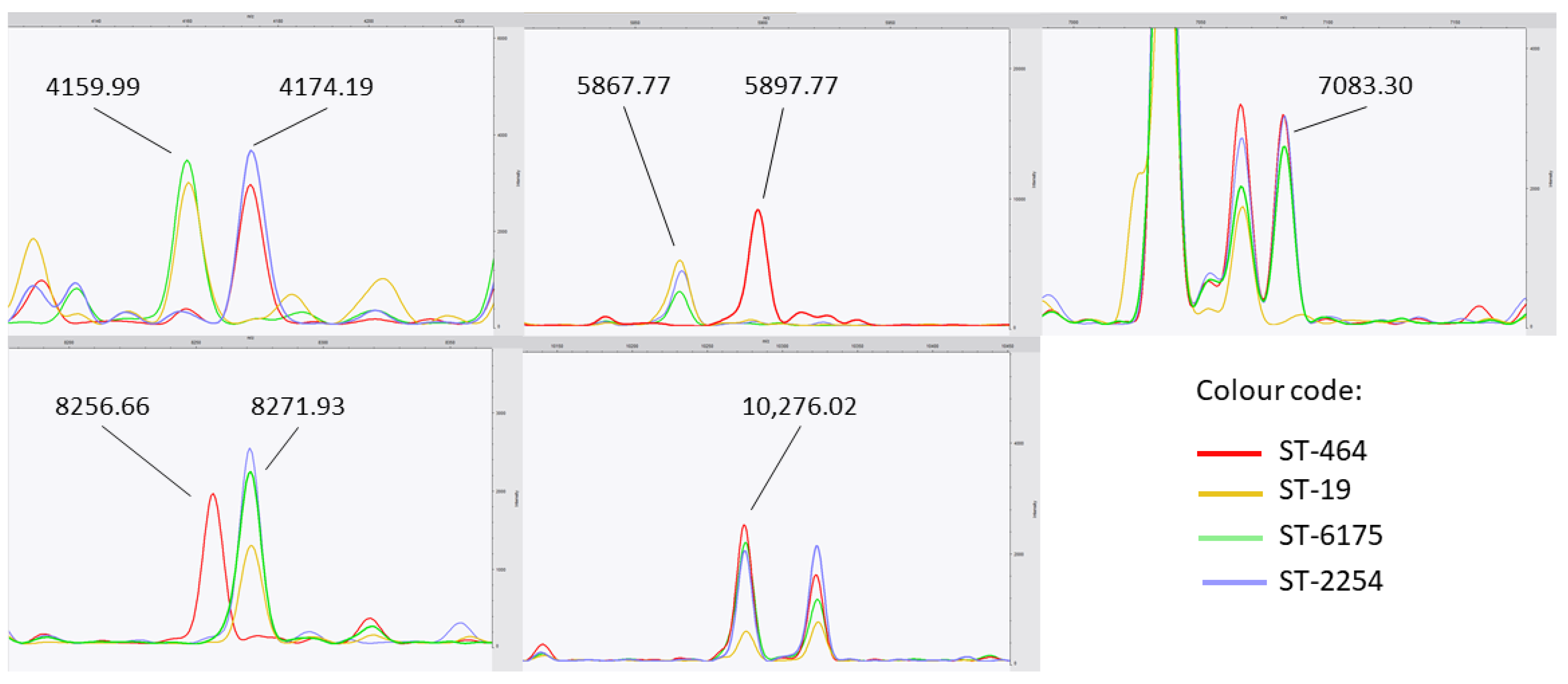{kind=link}
| Clusters | SID | CI (95%) | |
|---|---|---|---|
| Complex Clonal (CC) | 10 | 0.579 | 0.495–0.664 |
| Sequence Type (ST) | 21 | 0.829 | 0.785–0.873 |
| Complex Type (CT) | 42 | 0.887 | 0.849–0.926 |
| MALDI-TOF MS (Cut-off = 92%) | 12 | 0.830 | 0.800–0.861 |
| MALDI-TOF MS (Cut-off = 93%) | 20 | 0.862 | 0.828–0.897 |
| MALDI-TOF MS (Cut-off = 94%) | 40 | 0.939 | 0.918–0.960 |
| Adjusted Wallace Coefficient | MLST (CC) | MLST (ST) | cgMLST (CT) | MALDI (94%) | MALDI (93%) | MALDI (92%) |
|---|---|---|---|---|---|---|
| MLST (CC) | 0.284 (0.171–0.396) | 0.175 (0.079–0.270) | 0.090 (0.040–0.140) | 0.212 (0.132–0.293) | 0.248 (0.179–0.317) | |
| MLST (ST) | 1.000 (1.000–1.000) | 0.616 (0.474–0.758) | 0.297 (0.197–0.396) | 0.563 (0.427–0.699) | 0.567 (0.447–0.686) | |
| cgMLST (CT) | 1.000 (1.000–1.000) | 1.000 (1.000–1.000) | 0.439 (0.317–0.561) | 0.829 (0.703–0.955) | 0.824 (0.696–0.951) | |
| MALDI-TOF MS (94%) | 1.000 (1.000–1.000) | 0.933 (0.916–0.949) | 0.851 (0.830–0.872) | 1.000 (1.000–1.000) | 1.000 (1.000–1.000) | |
| MALDI-TOF MS (93%) | 0.965 (0.934–0.996) | 0.725 (0.608–0.843) | 0.658 (0.551–0.765) | 0.410 (0.309–0.511) | 1.000 (1.000–1.000) | |
| MALDI-TOF MS (92%) | 0.881 (0.830–0.932) | 0.572 (0.470–0.673) | 0.512 (0.423–0.602) | 0.321 (0.236–0.406) | 0.783 (0.724–0.841) |
| Mass Observed (Da) | Theoretical Average Molecular Weight (Da) | Gene Names | Protein Name | UniProt ID |
|---|---|---|---|---|
| 4159.99 | 4158.55 | APU78_09005 | Flagellin subunit protein FlaA | A0A690Z7F7 |
| 4174.19 | 4173.56 | CDX23_07240, FQZ36_04085, FV854_03335 | Uncharacterized protein | A0A5Z0CYS5 |
| 5867.60 | 5868.02 5867.95 5867.84 | FH034_10320 F1576_10330 FDW21_07355 | Sulfurtransferase-like selenium metabolism protein YedF Magnesium transporter CorA family protein Motility accessory factor | A0A5C4YC48 A0A698D3Z1 A0A3Z8JXU3 |
| 5897.77 | 5897.85 5895.96 | GSG42_09710 FXB36_09400 | Polysaccharide deacetylase DNA adenine methylase | A0A7I9U468 A0A7I9S1R5 |
| 7083.30 | 7081.66 | B9Q65_09070, E7P40_09640, F0N82_09625, FC283_09220, FW424_09040 | Uncharacterized protein | A0A400EER0 |
| 8256.66 | 8256.27 8256.39 8255.77 8256.96 | JJD26997_1194 GD714_06815 EJC82_07015 TM42_09010 | Conserved domain protein Uncharacterized protein Uncharacterized protein Membrane protein | A7H434 A0A6W1IK17 A0A6C7UKG7 A0A0D7V4A9 |
| 8271.93 | 8270.56 8271.37 8271.52 | N/A FW192_09775 B7Q70_09720 | Uncharacterized protein Integrase Terminase small subunit | Q4VRA4 A0A7I9QCT5 A0A5T0PDL9 |
| 10,276.02 | 10,276.22 10,276.17 10,274.87 | JJD26997_0928 AT778_09125, B7Q70_06195, C3H43_07780, C3H69_07590, C3H86_07890 A2E15_06760 | Uncharacterized protein Uncharacterized protein Glycosyltransferase Family 9 protein | A7H3G2 A0A2U0QNA2 A0A5T0CX51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feucherolles, M.; Nennig, M.; Becker, S.L.; Martiny, D.; Losch, S.; Penny, C.; Cauchie, H.-M.; Ragimbeau, C. Investigation of MALDI-TOF Mass Spectrometry for Assessing the Molecular Diversity of Campylobacter jejuni and Comparison with MLST and cgMLST: A Luxembourg One-Health Study. Diagnostics 2021, 11, 1949. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11111949
Feucherolles M, Nennig M, Becker SL, Martiny D, Losch S, Penny C, Cauchie H-M, Ragimbeau C. Investigation of MALDI-TOF Mass Spectrometry for Assessing the Molecular Diversity of Campylobacter jejuni and Comparison with MLST and cgMLST: A Luxembourg One-Health Study. Diagnostics. 2021; 11(11):1949. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11111949
Chicago/Turabian StyleFeucherolles, Maureen, Morgane Nennig, Sören L. Becker, Delphine Martiny, Serge Losch, Christian Penny, Henry-Michel Cauchie, and Catherine Ragimbeau. 2021. "Investigation of MALDI-TOF Mass Spectrometry for Assessing the Molecular Diversity of Campylobacter jejuni and Comparison with MLST and cgMLST: A Luxembourg One-Health Study" Diagnostics 11, no. 11: 1949. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11111949