
Differences between Acute Exacerbations of Idiopathic Pulmonary Fibrosis and Other Interstitial Lung Diseases

 , , , , ,
, , , , ,  ,
,
Abstract
:1. Introduction
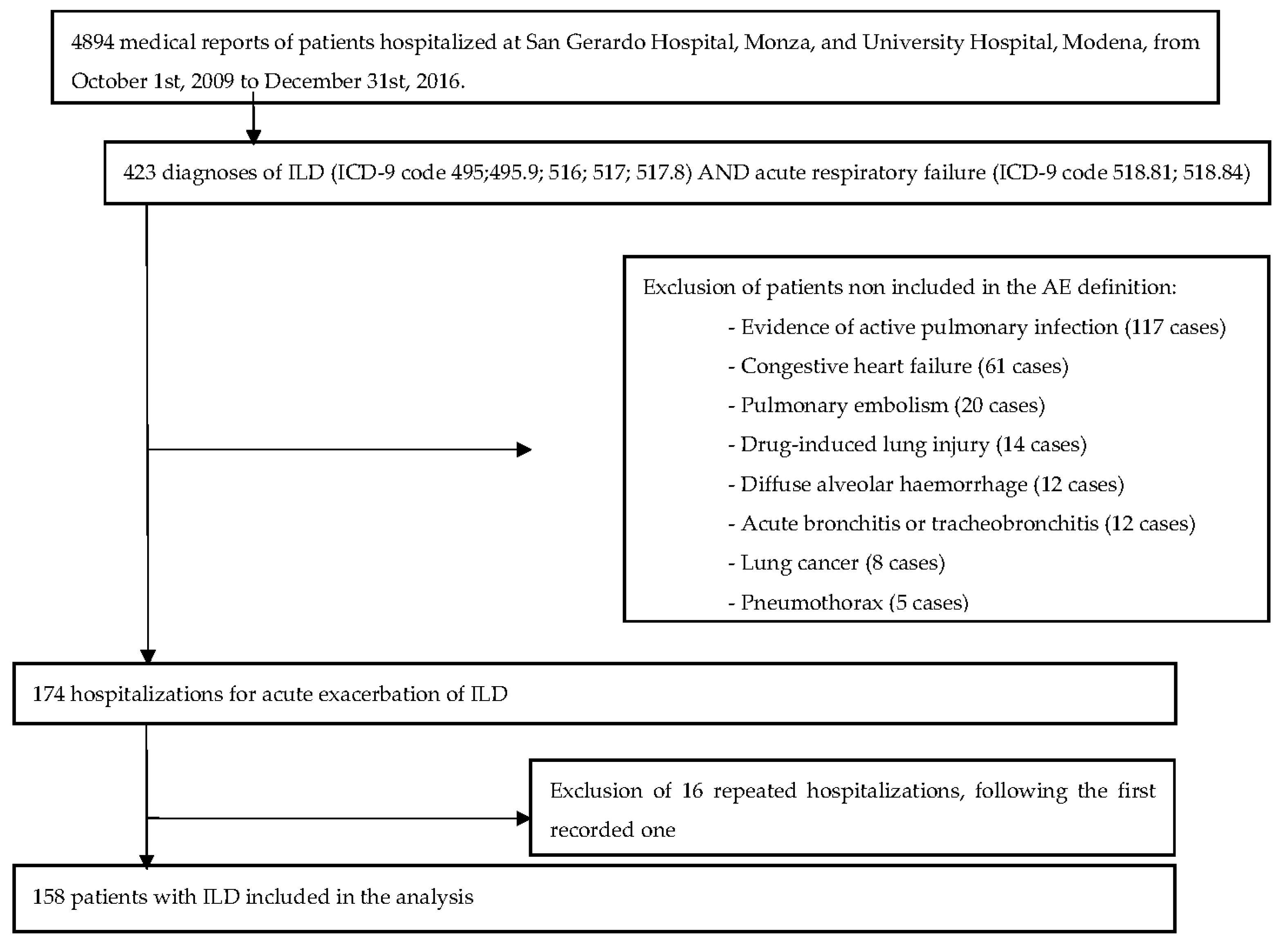
2. Material and Methods
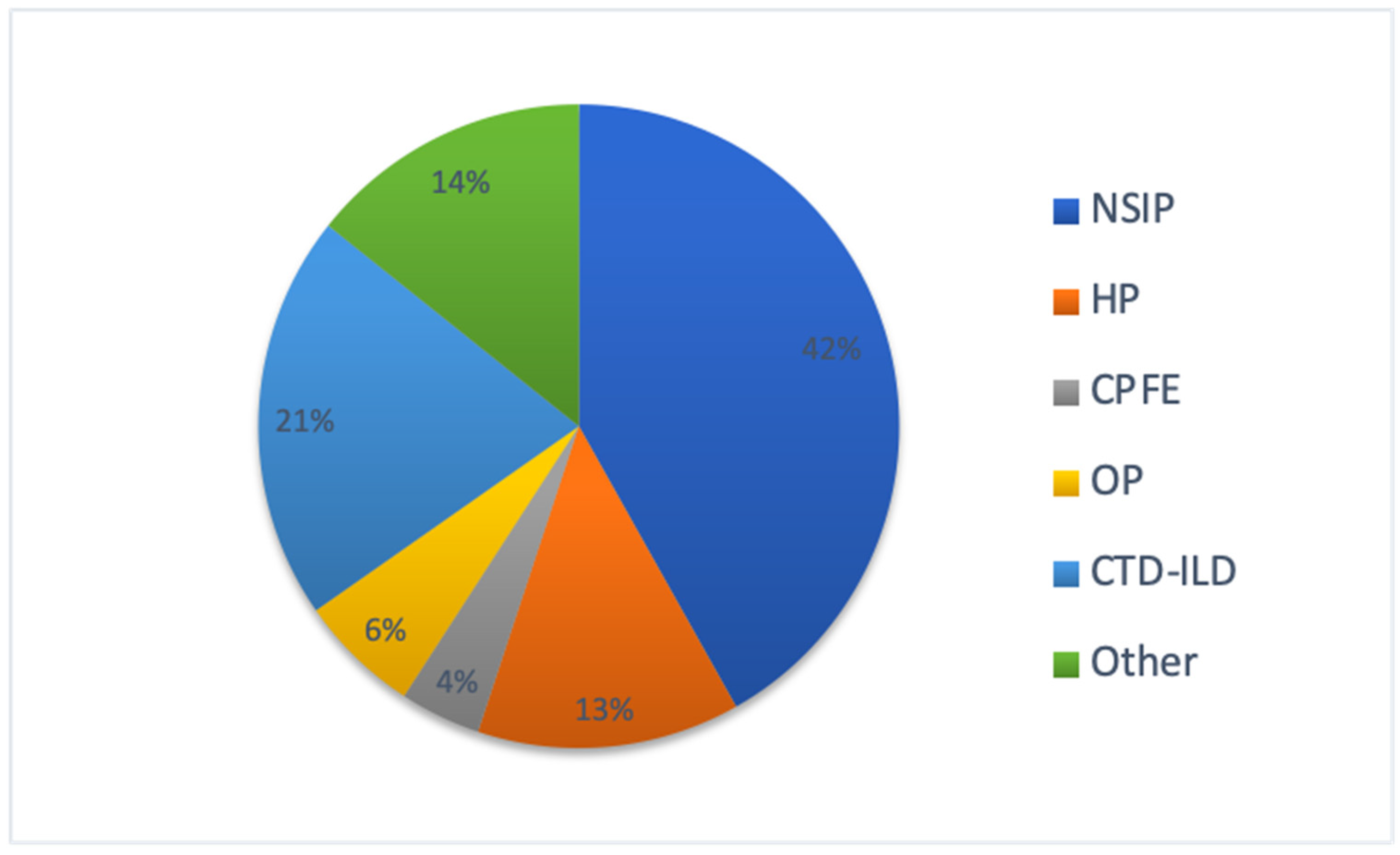
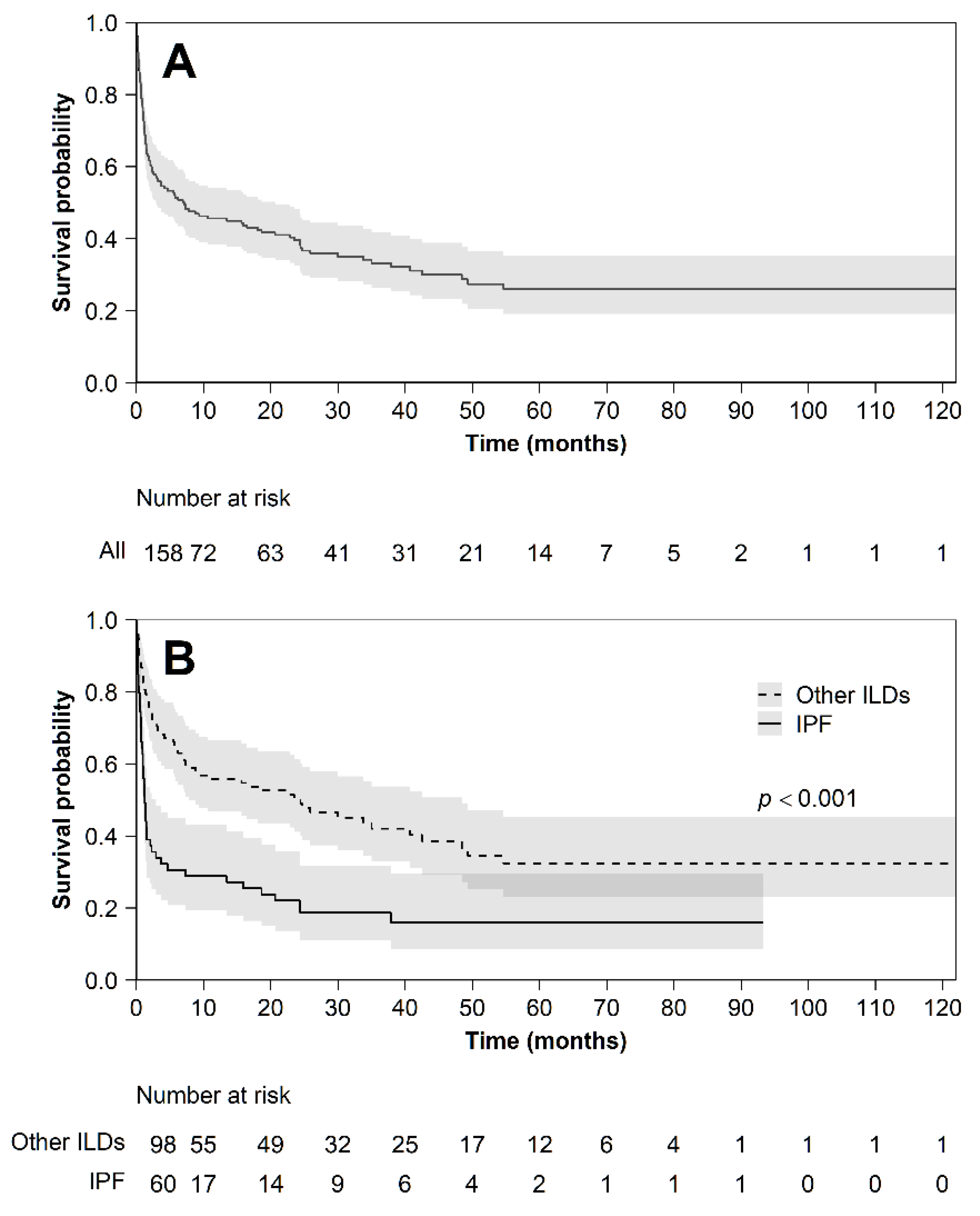
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IPF | Idiopathic pulmonary fibrosis |
| ILD | Interstitial lung diseases |
| AE | Acute exacerbation |
| AE-IPF | Acute exacerbation of IPF |
| AE-ILD | Acute exacerbation of ILD |
| HP | Hypersensitivity pneumonia |
| CTD-ILD | Connective-tissue-disease-related ILD |
| INSIP | Idiopathic non-specific interstitial pneumonia |
| ICD-9-CM | International Classification of Diseases, Ninth Revision, Clinical Modification |
| CHF | Congestive heart failure |
| PE | Pulmonary embolism |
| CT | Computed tomography |
| BAL | Broncho-alveolar lavage |
| STD | Standard deviation |
| FVC | Forced vital capacity |
| TLC | Total lung capacity |
| CRP | C-reactive protein |
| IQR | Interquartile range |
| WBC | White blood cells |
| OR | Odds ratio |
| CI | Confidence interval |
| PH | Pulmonary hypertension |
| HR | Hazard ratio |
| GPS | Glasgow Prognostic Score |
| SIRS | Systemic inflammatory response syndrome |
| LDH | Lactate dehydrogenase |
References
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef]
- Luppi, F.; Spagnolo, P.; Cerri, S.; Richeldi, L. The big clinical trials in idiopathic pulmonary fibrosis. Curr. Opin. Pulm. Med. 2012, 18, 428–432. [Google Scholar] [CrossRef]
- Collins, B.F.; Luppi, F. Diagnosis and Management of Fibrotic Interstitial Lung Diseases. Clin. Chest Med. 2021, 42, 321–335. [Google Scholar] [CrossRef]
- Kolb, M.; Bondue, B.; Pesci, A.; Miyazaki, Y.; Song, J.W.; Bhatt, N.Y.; Huggins, J.T.; Oldham, J.M.; Padilla, M.L.; Roman, J.; et al. Acute exacerbations of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180071. [Google Scholar] [CrossRef]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute exacerbation of idiopathic pulmonary fibrosis an international working group report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Ryerson, C.J.; Cottin, V.; Brown, K.K.; Collard, H.R. Acute exacerbation of idiopathic pulmonary fibrosis: Shifting the paradigm. Eur. Respir. J. 2015, 46, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Luppi, F.; Cerri, S.; Taddei, S.; Ferrara, G.; Cottin, V. Acute exacerbation of idiopathic pulmonary fibrosis: A clinical review. Intern. Emerg. Med. 2015, 10, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Wuyts, W. Acute exacerbations of interstitial lung disease. Curr. Opin. Pulm. Med. 2017, 23, 411–417. [Google Scholar] [CrossRef]
- Suzuki, A.; Kondoh, Y.; Brown, K.K.; Johkoh, T.; Kataoka, K.; Fukuoka, J.; Kimura, T.; Matsuda, T.; Yokoyama, T.; Fukihara, J.; et al. Acute exacerbations of fibrotic interstitial lung diseases. Respirology 2020, 25, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Collard, H.R.; Moore, B.B.; Flaherty, K.R.; Brown, K.K.; Kaner, R.J.; King, T.E., Jr.; Lasky, J.A.; Loyd, J.E.; Noth, I.; Olman, M.A.; et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, A.; Sebastiani, M.; Cerri, S.; Vacchi, C.; Tonelli, R.; Della Casa, G.; Cassone, G.; Spinella, A.; Fabrizio, P.; Luppi, F.; et al. Acute exacerbation of interstitial lung diseases secondary to systemic rheumatic diseases: A prospective study and review of the literature. J. Thorac. Dis. 2019, 11, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Salonen, J.; Purokivi, M.; Bloigu, R.; Kaarteenaho, R. Prognosis and causes of death of patients with acute exacerbation of fibrosing interstitial lung diseases. BMJ Open Respir. Res. 2020, 7, e000563. [Google Scholar] [CrossRef] [Green Version]
- Park, I.-N.; Kim, D.S.; Shim, T.S.; Lim, C.-M.; Lee, S.D.; Koh, Y.; Kim, W.S.; Kim, W.D.; Jang, S.J.; Colby, T.V. Acute Exacerbation of Interstitial Pneumonia Other Than Idiopathic Pulmonary Fibrosis. Chest 2007, 132, 214–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachikawa, R.; Tomii, K.; Ueda, H.; Nagata, K.; Nanjo, S.; Sakurai, A.; Otsuka, K.; Kaji, R.; Hayashi, M.; Katakami, N.; et al. Clinical Features and Outcome of Acute Exacerbation of Interstitial Pneumonia: Collagen Vascular Diseases-Related versus Idiopathic. Respiration 2012, 83, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Simon-Blancal, V.; Freynet, O.; Nunes, H.; Bouvry, D.; Naggara, N.; Brillet, P.-Y.; Denis, D.; Cohen, Y.; Vincent, F.; Valeyre, D.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis: Outcome and Prognostic Factors. Respiration 2012, 83, 28–35. [Google Scholar] [CrossRef]
- Kawamura, K.; Ichikado, K.; Ichiyasu, H.; Anan, K.; Yasuda, Y.; Suga, M.; Sakagami, T. Acute exacerbation of chronic fibrosing interstitial pneumonia in patients receiving antifibrotic agents: Incidence and risk factors from real-world experience. BMC Pulm. Med. 2019, 19, 113. [Google Scholar] [CrossRef]
- Marchioni, A.; Tonelli, R.; Ball, L.; Fantini, R.; Castaniere, I.; Cerri, S.; Luppi, F.; Malerba, M.; Pelosi, P.; Clini, E. Acute exacerbation of idiopathic pulmonary fibrosis: Lessons learned from acute respiratory distress syndrome? Crit. Care 2018, 22, 80. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, K.; Kono, M.; Saito, G.; Koyanagi, Y.; Tsutsumi, A.; Kobayashi, T.; Miki, Y.; Hashimoto, D.; Nakamura, Y.; Suda, T.; et al. Prognosis after acute exacerbation in patients with interstitial lung disease other than idiopathic pulmonary fibrosis. Clin. Respir. J. 2020, 15, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Sheng, J.; Qiu, X.; Wang, D.; Wang, D.; Wang, Y.; Xiao, Y.; Cai, H. Acute exacerbations of fibrosing interstitial lung disease associated with connective tissue diseases: A population-based study. BMC Pulm. Med. 2019, 19, 215. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Oyama, Y.; Enomoto, Y.; Yasui, H.; Karayama, M.; Kono, M.; Hozumi, H.; Suzuki, Y.; Furuhashi, K.; Fujisawa, T.; et al. Differences in clinical features of acute exacerbation between connective tissue disease-associated interstitial pneumonia and idiopathic pulmonary fibrosis. Chron. Respir. Dis. 2019, 16, 147997231880947. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, G.; Behr, J. Acute exacerbation in interstitial lung disease. Front. Med. 2017, 4, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.W.; Hong, S.-B.; Lim, C.-M.; Koh, Y.; Kim, D.S. Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur. Respir. J. 2011, 37, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Cho, K.W.; Kwon, S.S.; Kim, Y.H. Prognostic significance of Glasgow prognostic score in patients with acute exacerbation of idiopathic pulmonary fibrosis. Respirology 2018, 23, 206–212. [Google Scholar] [CrossRef]
- Usui, Y.; Kaga, A.; Sakai, F.; Shiono, A.; Komiyama, K.; Hagiwara, K.; Kanazawa, M. A cohort study of mortality predictors in patients with acute exacerbation of chronic fibrosing interstitial pneumonia. BMJ Open 2013, 3, e002971. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, H.; Panlaqui, O.M. Systematic review and meta-analysis of prognostic factors of acute exacerbation of idiopathic pulmonary fibrosis. BMJ Open 2020, 10, e035420. [Google Scholar] [CrossRef]
- Kreuter, M.; Polke, M.; Walsh, S.L.F.; Krisam, J.; Collard, H.R.; Chaudhuri, N.; Avdeev, S.; Behr, J.; Calligaro, G.; Corte, T.; et al. Acute exacerbation of idiopathic pulmonary fibrosis: International survey and call for harmonisation. Eur. Respir. J. 2020, 55, 1901760. [Google Scholar] [CrossRef]
- Scott, M.K.D.; Quinn, K.; Li, Q.; Carroll, R.; Warsinske, H.; Vallania, F.; Chen, S.; Carns, M.A.; Aren, K.; Sun, J.; et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: A retrospective, multicentre cohort study. Lancet Respir. Med. 2019, 7, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.; Chen, Y.; Ye, Q. Risk factors for acute exacerbation of idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Clin. Respir. J. 2018, 12, 1084–1092. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| IPF | Total (N = 158) | p-Value | ||
|---|---|---|---|---|
| No (N = 98) | Yes (N = 60) | |||
| Baseline characteristics | ||||
| Men—N (%) | 58 (59.18) | 48 (80.00) | 106 (67.09) | 0.0069 |
| Age at exacerbation—median (Q1–Q3) | 70 (62–75) | 70 (65–76.5) | 70 (64–76) | 0.1930 |
| Age at diagnosis—median (Q1–Q3) | 67.5 (59–74) | 68 (60–74) | 68 (60–74) | 0.7972 |
| Duration PF—median (Q1–Q3) | 9 (0–36) | 24 (8–48) | 12 (1–43) | 0.0072 |
| Smoker—N (%) | 0.0003 | |||
| Never | 40 (40.82) | 11 (18.33) | 51 (32.28) | |
| Current/ex | 52 (53.06) | 48 (80.00) | 100 (63.29) | |
| Comorbidities—N (%) | ||||
| Lung cancer | 0 (0.00) | 7 (11.67) | 7 (4.43) | <0.0001 |
| Other cancers | 17 (17.35) | 0 (0.00) | 17 (10.76) | <0.0001 |
| Arterial hypertension | 47 (47.96) | 25 (41.67) | 72 (45.57) | 0.4408 |
| Diabetes | 23 (23.47) | 15 (25.00) | 38 (24.05) | 0.8271 |
| Tachyarrhythmia | 15 (15.31) | 9 (15.00) | 24 (15.19) | 0.9374 |
| Heart failure and/or coronary artery disease | 26 (26.53) | 20 (33.33) | 46 (29.11) | 0.3610 |
| Gastro-esophageal reflux disease | 30 (30.61) | 11 (18.33) | 41 (25.95) | 0.0875 |
| Osteoporosis | 15 (15.31) | 8 (13.33) | 23 (14.56) | 0.7137 |
| Hypothyroidism | 8 (8.16) | 5 (8.33) | 13 (8.23) | 1.0000 |
| Pulmonary hypertension | 21 (21.43) | 13 (21.67) | 34 (21.52) | 0.9718 |
| Anxious depressive syndrome | 12 (12.24) | 3 (5.00) | 15 (9.49) | 0.1316 |
| Therapies before hospitalization | ||||
| Pirfenidone—N (%) | 2 (2.04) | 12 (20.00) | 14 (8.86) | 0.0001 |
| Nintedanib—N (%) | 1 (1.02) | 1 (1.67) | 2 (1.27) | 1.0000 |
| Clinical presentation at admission | ||||
| Symptoms | ||||
| Chest pain—N (%) | 4 (4.08) | 3 (5.00) | 7 (4.43) | 1.0000 |
| Dyspnea—N (%) | 95 (96.94) | 59 (98.33) | 154 (97.47) | 0.2918 |
| Cough—N (%) | 40 (40.82) | 16 (26.67) | 56 (35.44) | 0.0827 |
| Fever—N (%) | 28 (28.57) | 12 (20.00) | 40 (25.32) | 0.2516 |
| Sputum—N (%) | 0.5435 | |||
| No | 75 (76.53) | 51 (85.00) | 126 (79.75) | |
| Clear | 13 (13.27) | 6 (10.00) | 19 (12.03) | |
| Mucoid | 2 (2.04) | 2 (3.33) | 4 (2.53) | |
| Mucopurulent | 3 (3.06) | 0 (0.00) | 3 (1.90) | |
| Bloody | 2 (2.04) | 0 (0.00) | 2 (1.27) | |
| Duration (days)—median (Q1–Q3) | 10 (3–30) | 7 (4–15) | 10 (3–25) | 0.3415 |
| Physical examination | ||||
| Bilateral inspiratory crackles—N (%) | 74 (75.51) | 56 (93.33) | 130 (82.28) | 0.0003 |
| Reduced vesicular murmur—N (%) | 18 (18.37) | 8 (13.33) | 26 (16.46) | 0.7313 |
| Peripheral edema—N (%) | 17 (17.35) | 12 (20.00) | 29 (18.35) | 0.5944 |
| Blood exams at admission | ||||
| White blood cell count (103/µL)—median (Q1–Q3) | 9.91 (7.25–13.53) | 10.9 (8.9–13.6) | 10.28 (7.59–13.53) | 0.1129 |
| Neutrophilia—N (%) | 0.0283 | |||
| Normal | 48 (48.98) | 19 (31.67) | 67 (42.41) | |
| Pathological | 49 (50.00) | 41 (68.33) | 90 (56.96) | |
| Neutrophilia (103/µL)—median (Q1–Q3) | 7.18 (4.68–10.78) | 8.90 (6.18–11.00) | 8.09 (5.40–10.88) | 0.0372 |
| Neutrophilia %—median (Q1–Q3) | 75.4 (64.3–85.2) | 80.3 (68.9–86.7) | 77 (65.4–85.9) | 0.2200 |
| Lymphocytes (103/µL)—median (Q1–Q3) | 1.34 (0.96–1.93) | 1.4 (1–2.03) | 1.39 (0.99–1.97) | 0.2127 |
| Lymphocytes %—median (Q1–Q3) | 14.5 (9.6–23.25) | 16.1 (9.7–20.2) | 14.6 (9.6–22.1) | 0.8728 |
| Monocytes (103/µL)—median (Q1–Q3) | 0.63 (0.45–0.85) | 0.68 (0.5–0.8) | 0.65 (0.49–0.85) | 0.6019 |
| Monocytes %—median (Q1–Q3) | 6.55 (4.55–9) | 6 (4.3–7.9) | 6.3 (4.5–8.3) | 0.2486 |
| Eosinophils (103/µL)—median (Q1–Q3) | 0.13 (0.03–0.3) | 0.06 (0.03–0.18) | 0.1 (0.03–0.28) | 0.1572 |
| Eosinophils %—median (Q1–Q3) | 1.35 (0.2–3.2) | 0.7 (0.2–1.5) | 0.9 (0.2–2.7) | 0.0663 |
| Basophils (103/µL)—median (Q1–Q3) | 0.02 (0.01–0.04) | 0.02 (0.02–0.03) | 0.02 (0.01–0.04) | 0.8109 |
| Basophils %—median (Q1–Q3) | 0.2 (0.1–0.4) | 0.2 (0.1–0.3) | 0.2 (0.1–0.4) | 0.3359 |
| CRP-N(%) | 0.0072 | |||
| Normal | 25 (25.51) | 5 (8.33) | 30 (18.99) | |
| Pathological | 71 (72.45) | 54 (90.00) | 125 (79.11) | |
| CRP (mg/dL)—median (Q1–Q3) | 1.18 (0.49–4.06) | 3.55 (1.29–5) | 1.8 (0.57–4.8) | 0.0123 |
| IPF | Total (N = 158) | p-Value | ||
|---|---|---|---|---|
| No (N = 98) | Yes (N = 60) | |||
| FVC (L)—median (Q1–Q3) | 1.86 (1.18–2.35) | 1.98 (1.46–2.48) | 1.895 (1.26–2.36) | 0.1010 |
| FVC %—median (Q1–Q3) | 62 (49–74) | 69 (53–82) | 62 (50–79) | 0.2432 |
| FEV1 (L)—median (Q1–Q3) | 1.59 (1.04–1.98) | 1.78 (1.43–2.19) | 1.69 (1.19–2.09) | 0.0063 |
| FEV1 %—median (Q1–Q3) | 68 (56–86) | 78.775 (61–91) | 72 (57–88) | 0.0707 |
| DLCO (mmol/min/kPa)—median (Q1–Q3) | 2.48 (1.32–3.88) | 3.32 (1.88–7.09) | 2.91 (1.74–6.00) | 0.0752 |
| DLCO %—median (Q1–Q3) | 36 (23–45) | 27 (20–38) | 32 (22- 42) | 0.1260 |
| TLC (L)—median (Q1–Q3) | 3.49 (2.79–4.44) | 3.53 (3.03–4.22) | 3.50 (2.82–4.42) | 0.6133 |
| TLC %—median (Q1–Q3) | 64 (52–76) | 65.4 (50–76) | 64 (52–76) | 0.6665 |
| IPF | Total (N = 158) | p-Value | ||
|---|---|---|---|---|
| No (N = 98) | Yes (N = 60) | |||
| Therapies during hospitalization | ||||
| Oxygen supplementation—N (%) | 87 (88.78) | 56 (93.33) | 143 (90.51) | 0.3727 |
| Anticoagulant—N (%) | 48 (48.98) | 32 (53.33) | 80 (50.63) | 0.4622 |
| Intravenous steroids—N (%) | 66 (67.35) | 48 (80.00) | 114 (72.15) | 0.0566 |
| Antibiotics—N (%) | 62 (63.27) | 48 (80.00) | 110 (69.62) | 0.0165 |
| Immunosuppressives—N (%) | 0.3756 | |||
| Cyclophosphamide | 11 (11.22) | 6 (10.00) | 17 (10.76) | |
| Azathioprine | 3 (3.06) | 0 (0.00) | 3 (1.90) | |
| Methotrexate | 0 (0.00) | 1 (1.67) | 1 (0.63) | |
| NIV during hospitalization—N (%) | 35 (35.71) | 15 (25.00) | 50 (31.65) | 0.1801 |
| IMV during hospitalization—N (%) | 8 (8.16) | 3 (5.00) | 11 (6.96) | 0.4642 |
| Hospitalization length (days)—median (Q1–Q3) | 12 (8–18) | 14.5 (9–23) | 13 (9–20) | 0.1539 |
| In-hospital death—N (%) | 21 (21.43) | 28 (46.67) | 49 (31.01) | 0.0003 |
| IPF | Total | ||
|---|---|---|---|
| No | Yes | ||
| Baseline characteristics | |||
| Gender | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Diagnosis | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Age at exacerbation | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Age at diagnosis | 4 (4.08) | 9 (15.00) | 13 (8.23) |
| Duration PF | 1 (1.02) | 10 (16.67) | 11 (6.96) |
| Smoker | 6 (6.12) | 1 (1.67) | 7 (4.43) |
| Comorbidities | |||
| Lung cancer | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Other cancers | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Arterial hypertension | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Diabetes | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Tachyarrhythmia | 1 (1.02) | 0 (0.00) | 1 (0.63) |
| Heart failure and/or coronary artery disease | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Gastro-esophageal reflux disease | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Osteoporosis | 1 (1.02) | 0 (0.00) | 1 (0.63) |
| Hypothyroidism | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Pulmonary hypertension | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Anxious depressive syndrome | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Therapies before hospitalization | |||
| Pirfenidone | 1 (1.02) | 0 (0.00) | 1 (0.63) |
| Nintedanib | 1 (1.02) | 0 (0.00) | 1 (0.63) |
| Clinical presentation at admission | |||
| Symptoms | |||
| Chest pain | 1 (1.02) | 1 (1.67) | 2 (1.27) |
| Dyspnea | 0 (0.00) | 1 (1.67) | 1 (0.63) |
| Cough | 0 (0.00) | 1 (1.67) | 1 (0.63) |
| Fever | 0 (0.00) | 1 (1.67) | 1 (0.63) |
| Sputum | 3 (3.06) | 1 (1.67) | 4 (2.53) |
| Duration (days) | 18 (18.37) | 15 (25.00) | 33 (20.89) |
| Physical examination | |||
| Bilateral inspiratory crackles | 1 (1.02) | 3 (5.00) | 4 (2.53) |
| Reduced vesicular murmur | 15 (15.31) | 18 (30.00) | 33 (20.89) |
| Peripheral edema | 3 (3.06) | 4 (6.67) | 7 (4.43) |
| Blood exams at admission | |||
| White blood cell count 103/µL | 3 (3.06) | 3 (5.00) | 6 (3.80) |
| Neutrophilia | 1 (1.02) | 0 (0.00) | 1 (0.63) |
| Neutrophilia 103/µl | 6 (6.12) | 3 (5.00) | 9 (5.70) |
| Neutrophilia % | 6 (6.12) | 3 (5.00) | 9 (5.70) |
| Lymphocytes 103/µl | 6 (6.12) | 7 (11.67) | 13 (8.23) |
| Lymphocytes % | 6 (6.12) | 7 (11.67) | 13 (8.23) |
| Monocytes 103/µl | 6 (6.12) | 7 (11.67) | 13 (8.23) |
| Monocytes % | 6 (6.12) | 7 (11.67) | 13 (8.23) |
| Eosinophils 103/µl | 6 (6.12) | 7 (11.67) | 13 (8.23) |
| Eosinophils % | 6 (6.12) | 7 (11.67) | 13 (8.23) |
| Basophils 103/µl | 6 (6.12) | 7 (11.67) | 13 (8.23) |
| Basophils % | 6 (6.12) | 7 (11.67) | 13 (8.23) |
| CRP | 2 (2.04) | 1 (1.67) | 3 (1.90) |
| CRP mg/dL | 7 (7.14) | 13 (21.67) | 20 (12.66) |
| Lung function tests in stability | |||
| FVC (L) | 19 (19.39) | 11 (18.33) | 30 (18.99) |
| FVC % | 19 (19.39) | 11 (18.33) | 30 (18.99) |
| FEV1(L) | 25 (25.51) | 12 (20.00) | 37 (23.42) |
| FEV1 % | 27 (27.55) | 12 (20.00) | 39 (24.68) |
| DLCO (mmoL/min/kPa) | 60 (61.22) | 24 (40.00) | 84 (53.16) |
| DLCO % | 43 (43.88) | 19 (31.67) | 62 (39.24) |
| TLC (L) | 33 (33.67) | 14 (23.33) | 47 (29.75) |
| TLC % | 34 (34.69) | 15 (25.00) | 49 (31.01) |
| Therapies during hospitalization | |||
| Oxygen supplementation | 1 (1.02) | 1 (1.67) | 2 (1.27) |
| Anticoagulants | 2 (2.04) | 3 (5.00) | 5 (3.16) |
| Intravenous steroids | 0 (0.00) | 1 (1.67) | 1 (0.63) |
| Antibiotics | 0 (0.00) | 1 (1.67) | 1 (0.63) |
| Immunosuppressives | 1 (1.02) | 0 (0.00) | 1 (0.63) |
| NIV during hospitalization | 0 (0.00) | 1 (1.67) | 1 (0.63) |
| IMV during hospitalization | 0 (0.00) | 1 (1.67) | 1 (0.63) |
| Hospitalization length, days | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| In-hospital death | 0 (0.00) | 0 (0.00) | 0 (0.00) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faverio, P.; Stainer, A.; Conti, S.; Madotto, F.; De Giacomi, F.; Della Zoppa, M.; Vancheri, A.; Pellegrino, M.R.; Tonelli, R.; Cerri, S.; et al. Differences between Acute Exacerbations of Idiopathic Pulmonary Fibrosis and Other Interstitial Lung Diseases. Diagnostics 2021, 11, 1623. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11091623
Faverio P, Stainer A, Conti S, Madotto F, De Giacomi F, Della Zoppa M, Vancheri A, Pellegrino MR, Tonelli R, Cerri S, et al. Differences between Acute Exacerbations of Idiopathic Pulmonary Fibrosis and Other Interstitial Lung Diseases. Diagnostics. 2021; 11(9):1623. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11091623
Chicago/Turabian StyleFaverio, Paola, Anna Stainer, Sara Conti, Fabiana Madotto, Federica De Giacomi, Matteo Della Zoppa, Ada Vancheri, Maria Rosaria Pellegrino, Roberto Tonelli, Stefania Cerri, and et al. 2021. "Differences between Acute Exacerbations of Idiopathic Pulmonary Fibrosis and Other Interstitial Lung Diseases" Diagnostics 11, no. 9: 1623. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics11091623