
Quantitation of a Urinary Profile of Biomarkers in Gaucher Disease Type 1 Patients Using Tandem Mass Spectrometry

 , ,
, ,
Abstract
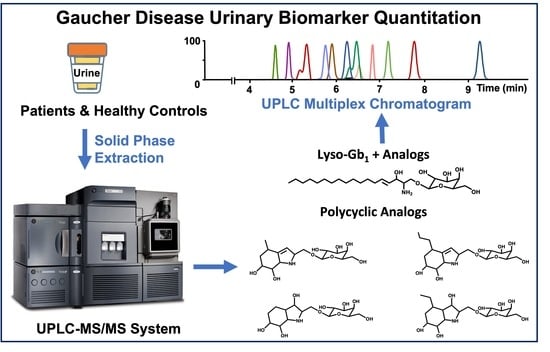
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Ethics Approval
2.3. Study Population
2.4. Calibration Curves and Quality Control Samples Preparation
2.5. Sample Preparation
2.6. Instrumentation and Parameters
2.7. Biomarker Quantitation
2.8. Method Validation
3. Results and Discussion
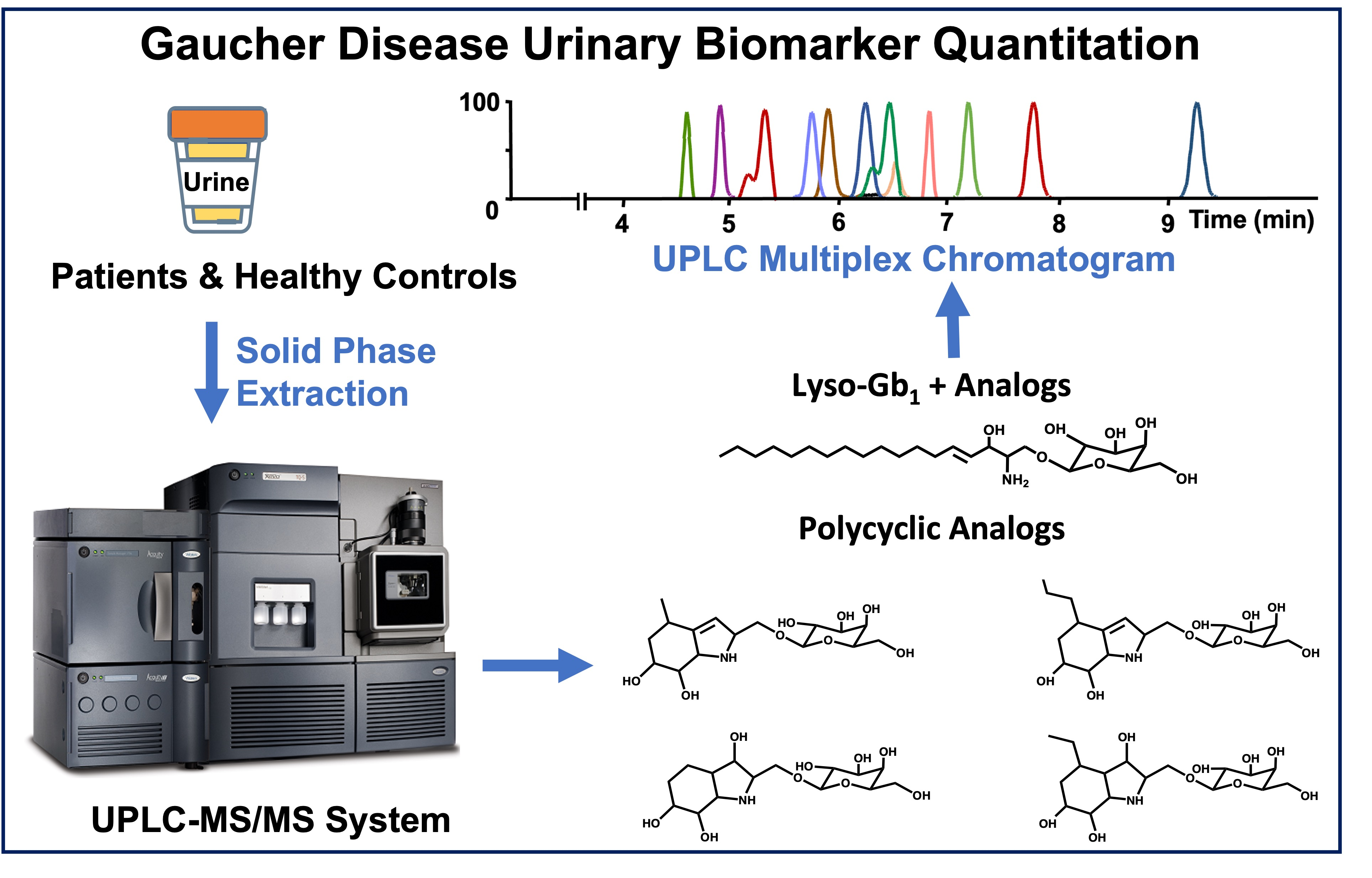
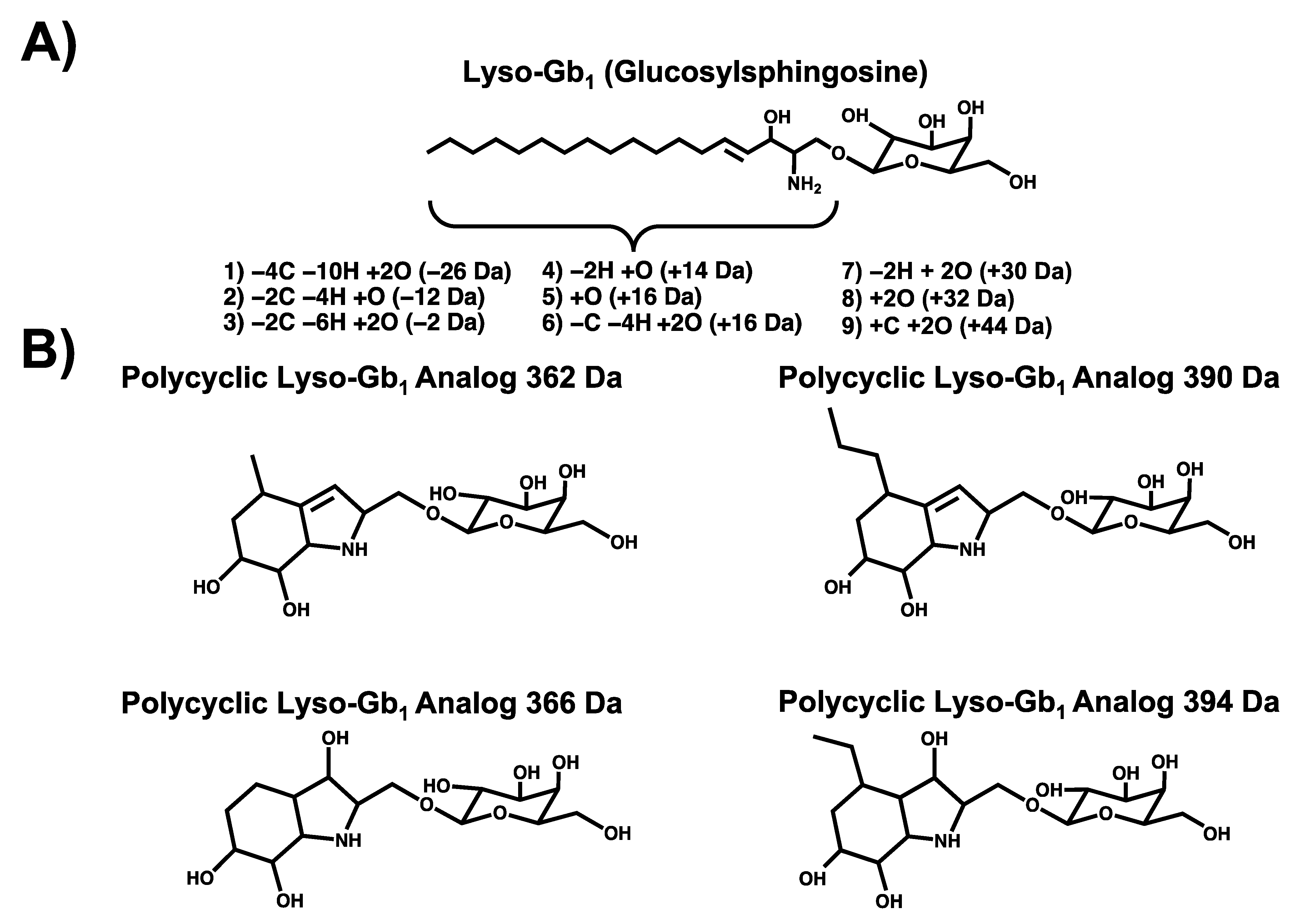
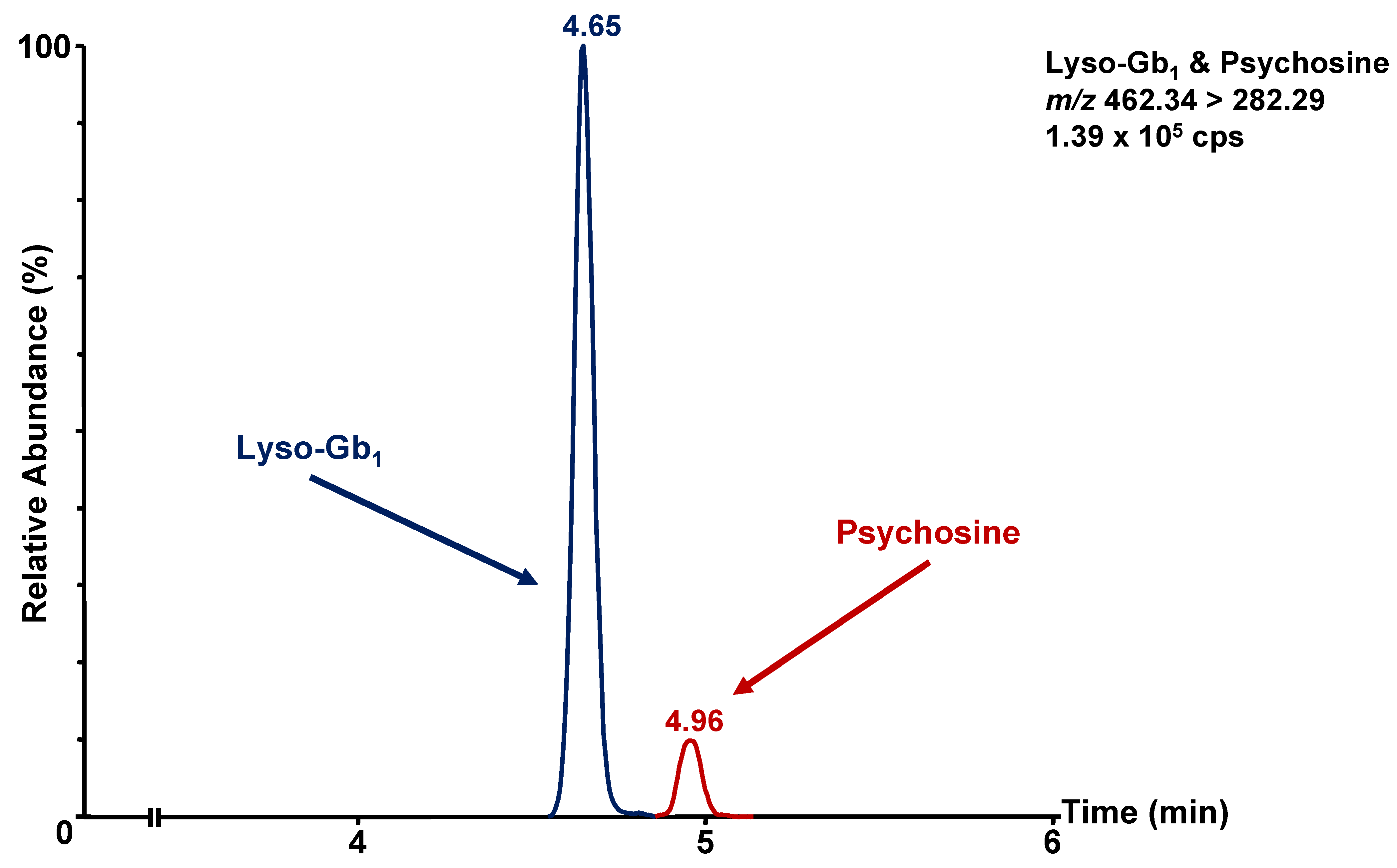
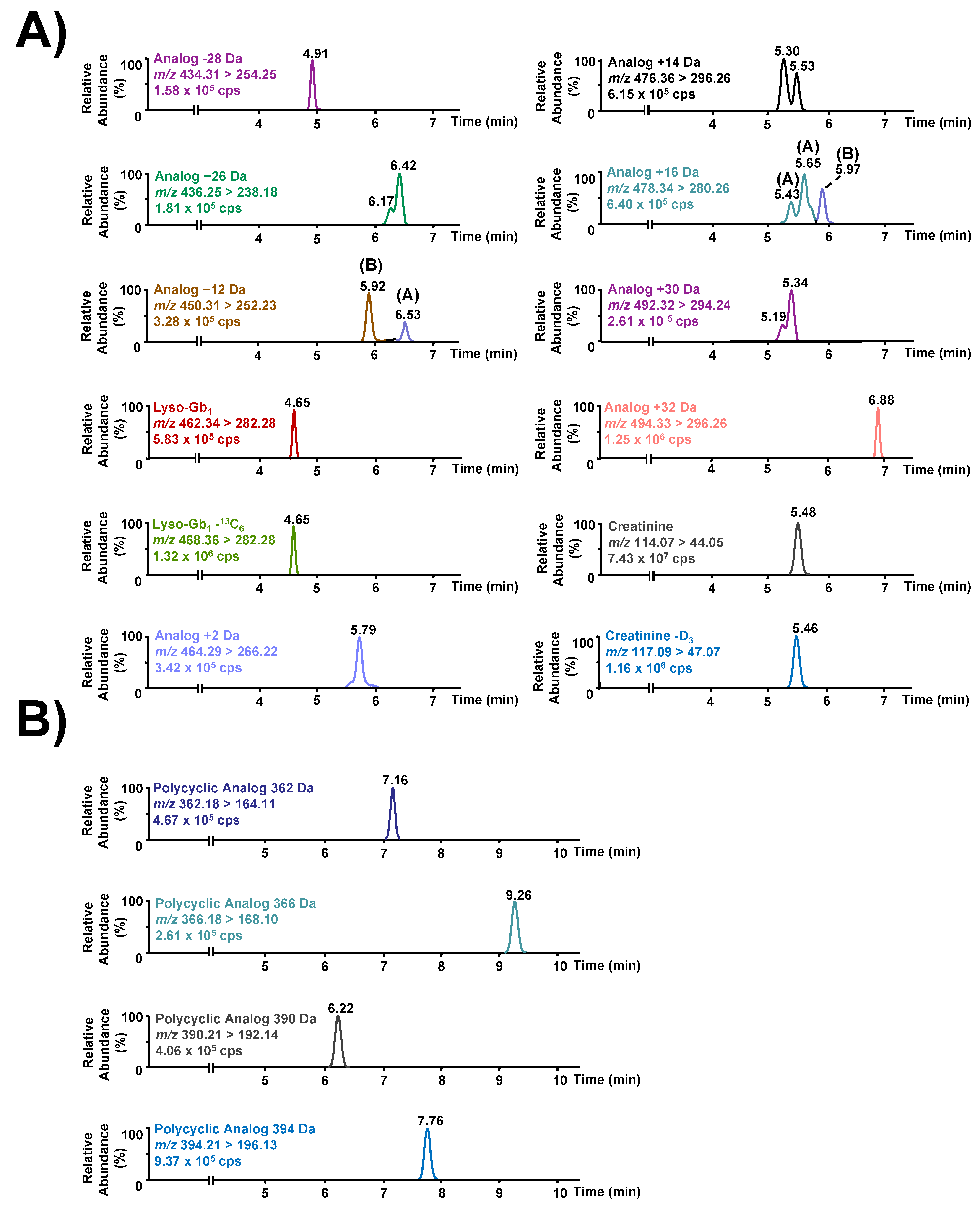
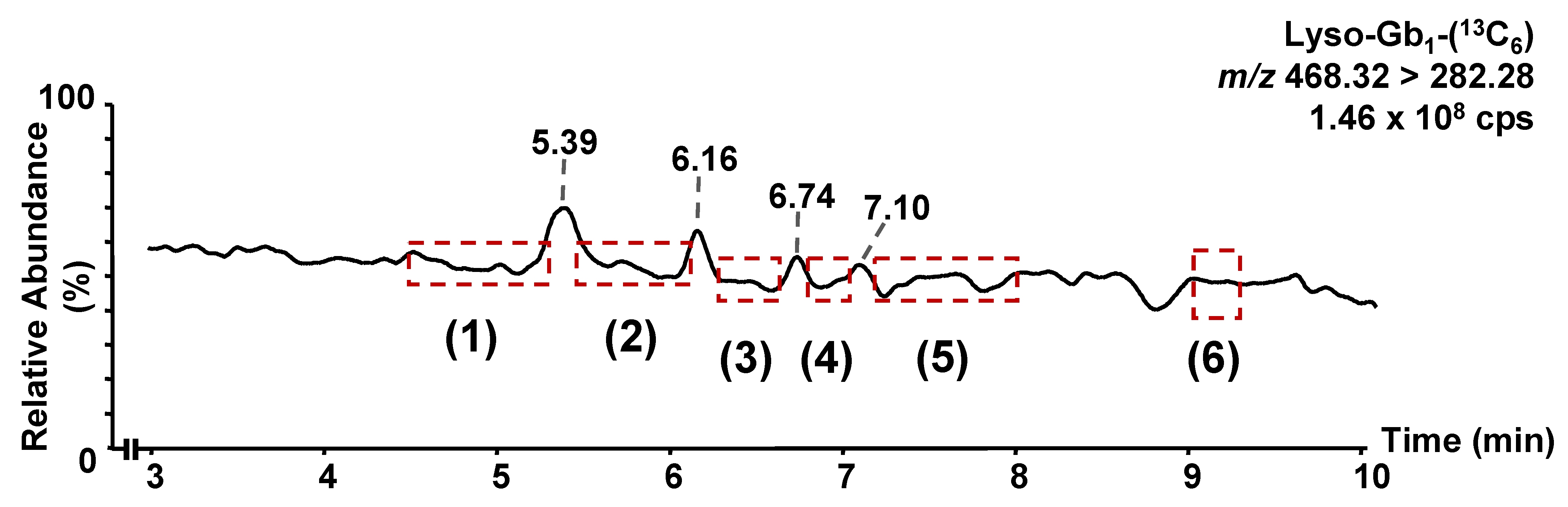
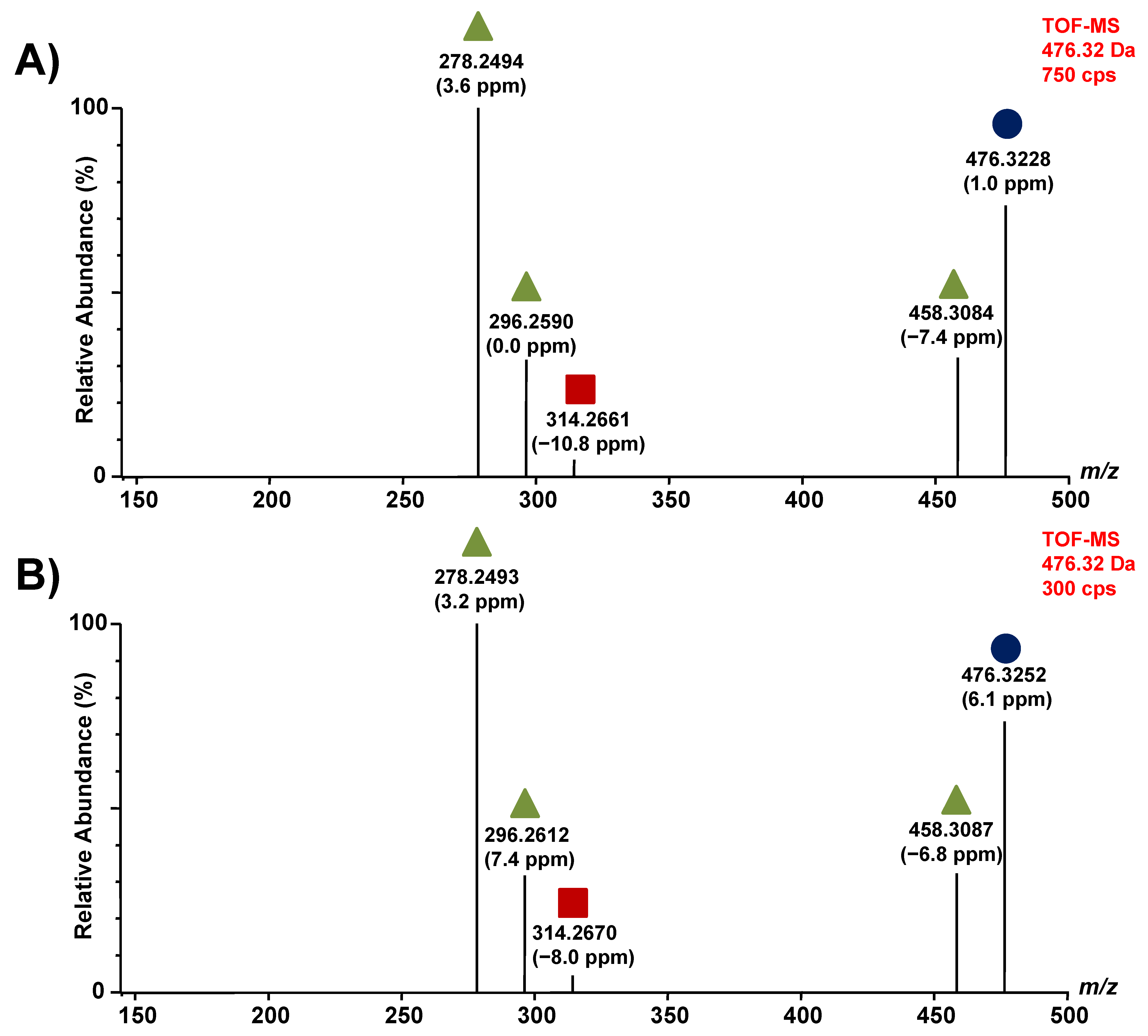
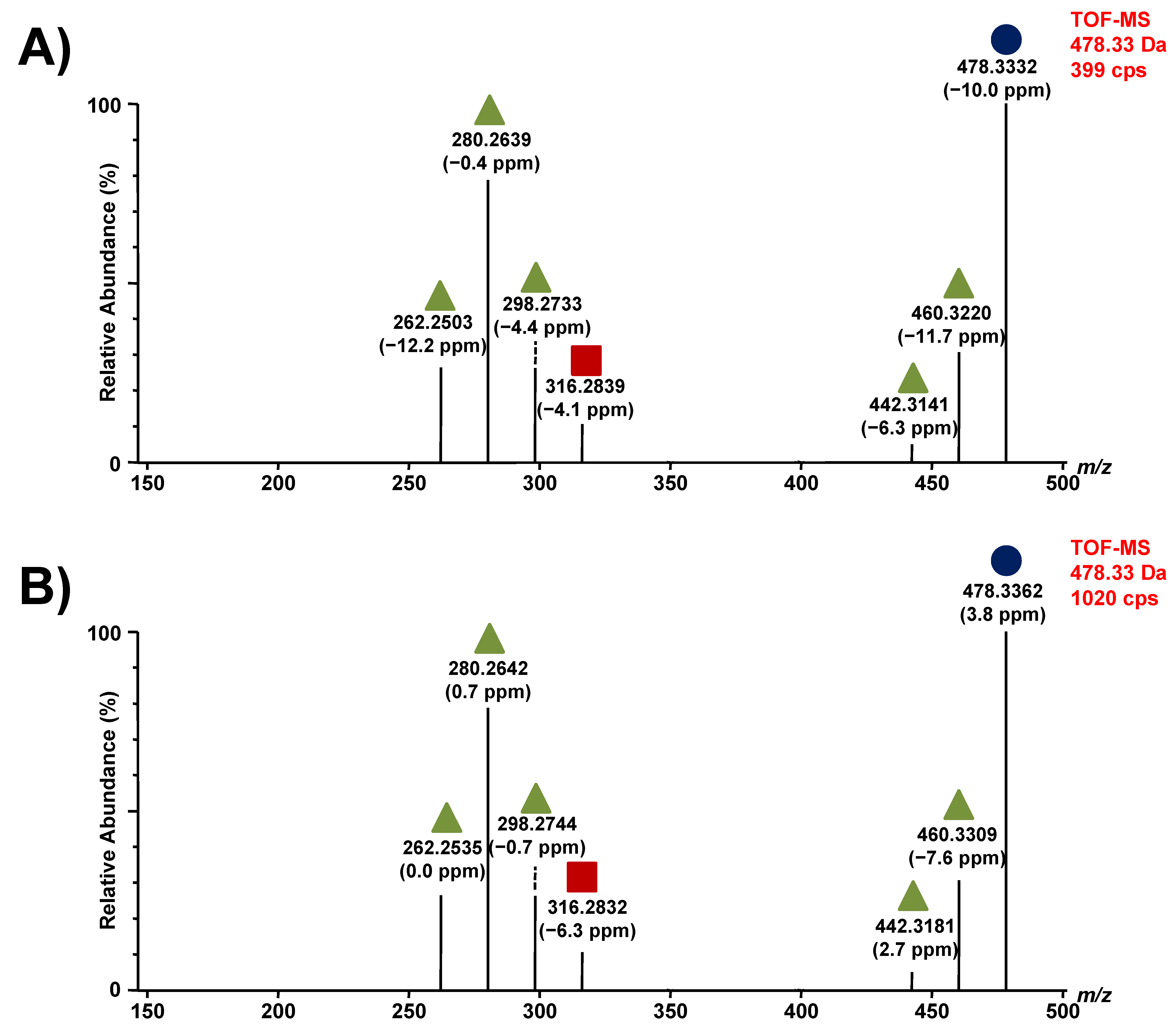
3.1. Chromatographic Separation
3.2. Method Validation
3.3. Biomarker Quantitation
3.4. Biomarker Concentrations upon Treatment
4. Conclusions
5. Future Perspectives
6. Study Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dandana, A.; Ben Khelifa, S.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2016, 83, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Baris, H.N.; Cohen, I.J.; Mistry, P.K. Gaucher disease: The metabolic defect, pathophysiology, phenotypes and natural history. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. 1), 72–81. [Google Scholar] [PubMed]
- Rowland, R.J.; Chen, Y.; Breen, I.; Wu, L.; Offen, W.A.; Beenakker, T.J.; Su, Q.; Nieuwendijk, A.M.C.H.V.D.; Aerts, J.M.F.G.; Artola, M.; et al. Design, Synthesis and Structural Analysis of Glucocerebrosidase Imaging Agents. Chem.–A Eur. J. 2021, 27, 16377–16388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zheng, M.-F.; Cui, S.-Y.; Zhang, W.; Gao, R.-P. Ambroxol chaperone therapy for Gaucher disease type I-associated liver cirrhosis and portal hypertension: A case report. Endocr. Metab. Immune Disord. Drug Targets 2021. [Google Scholar] [CrossRef]
- Mistry, P.K.; Lopez, G.; Schiffmann, R.; Barton, N.W.; Weinreb, N.J.; Sidransky, E. Gaucher disease: Progress and ongoing challenges. Mol. Genet. Metab. 2016, 120, 8–21. [Google Scholar] [CrossRef] [Green Version]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Schiffmann, R.; Sevigny, J.; Rolfs, A.; Davies, E.H.; Goker-Alpan, O.; Abdelwahab, M.; Vellodi, A.; Mengel, E.; Lukina, E.; Yoo, H.; et al. The definition of neuronopathic Gaucher disease. J. Inherit. Metab. Dis. 2020, 43, 1056–1059. [Google Scholar] [CrossRef]
- Deegan, P.; Fernandez-Sasso, D.; Giraldo, P.; Lau, H.; Panahloo, Z.; Zimran, A. Treatment patterns from 647 patients with Gaucher disease: An analysis from the Gaucher Outcome Survey. Blood Cells Mol. Dis. 2018, 68, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Gumus, E.; Karhan, A.N.; Hizarcioglu-Gulsen, H.; Demir, H.; Ozen, H.; Temizel, I.N.S.; Emre, S.D.; Yuce, A. Clinical-genetic characteristics and treatment outcomes of Turkish children with Gaucher disease type 1 and type 3: A sixteen year single-center experience. Eur. J. Med Genet. 2021, 64, 104339. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef] [Green Version]
- Ramadža, D.P.; Zekušić, M.; Žigman, T.; Škaričić, A.; Bogdanić, A.; Mustać, G.; Bošnjak-Nađ, K.; Ozretić, D.; Ohno, K.; Fumić, K.; et al. Early initiation of ambroxol treatment diminishes neurological manifestations of type 3 Gaucher disease: A long-term outcome of two siblings. Eur. J. Paediatr. Neurol. 2021, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Gonzalez-Rodriguez, D.E.; Abrahamov, A.; Elstein, D.; Paz, A.; Brill-Almon, E.; Chertkoff, R. Safety and efficacy of two dose levels of taliglucerase alfa in pediatric patients with Gaucher disease. Blood Cells Mol. Dis. 2015, 54, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sam, R.; Ryan, E.; Daykin, E.; Sidransky, E. Current and emerging pharmacotherapy for Gaucher disease in pediatric populations. Expert Opin. Pharmacother. 2021, 22, 1489–1503. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Mehta, A.; Hughes, D. Diagnosing Gaucher disease: An on-going need for increased awareness amongst haematologists. Blood Cells Mol. Dis. 2013, 50, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Gary, S.; Ryan, E.; Steward, A.; Sidransky, E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev. Endocrinol. Metab. 2018, 13, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, P.; López de Frutos, L.; Cebolla, J.J. Biomarker combination is necessary for the assessment of Gaucher disease? Ann. Transl. Med. 2018, 6 (Suppl. 1), S81. [Google Scholar] [CrossRef] [Green Version]
- Danilov, S.M.; Tikhomirova, V.; Metzger, R.; Naperova, I.A.; Bukina, T.M.; Goker-Alpan, O.; Tayebi, N.; Gayfullin, N.M.; Schwartz, D.E.; Samokhodskaya, L.M.; et al. ACE phenotyping in Gaucher disease. Mol. Genet. Metab. 2018, 123, 501–551. [Google Scholar] [CrossRef]
- Van Dussen, L.; Lips, P.; Everts, V.E.; Bravenboer, N.; Jansen, I.D.C.; Groener, J.E.M.; Maas, M.; Blokland, J.A.K.; Aerts, J.M.F.G.; Hollak, C.E.M. Markers of Bone Turnover in Gaucher Disease: Modeling the Evolution of Bone Disease. J. Clin. Endocrinol. Metab. 2011, 96, 2194–2205. [Google Scholar] [CrossRef] [Green Version]
- Mistry, P.K.; Cappellini, M.D.; Lukina, E.; Özsan, H.; Pascual, S.M.; Rosenbaum, H.; Solano, M.H.; Spigelman, Z.; Villarrubia, J.; Watman, N.P.; et al. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am. J. Hematol. 2010, 86, 110–115. [Google Scholar] [CrossRef]
- Arkadir, D.; Dinur, T.; Revel-Vilk, S.; Becker Cohen, M.; Cozma, C.; Hovakimyan, M.; Eichler, S.; Rolfs, A.; Zimran, A. Glucosylsphingosine is a reliable response biomarker in Gaucher disease. Am. J. Hematol. 2018, 93, E140–E142. [Google Scholar] [CrossRef] [Green Version]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef] [PubMed]
- Menkovic, I.; Boutin, M.; Alayoubi, A.; Curado, F.; Bauer, P.; Mercier, F.E.; Auray-Blais, C. Metabolomic Study Using Time-of-Flight Mass Spectrometry Reveals Novel Urinary Biomarkers for Gaucher Disease Type 1. J. Proteome Res. 2022, 21, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Menkovic, I.; Boutin, M.; Alayoubi, A.; Mercier, F.E.; Rivard, G.É.; Auray-Blais, C. Identification of a Reliable Biomarker Profile for the Diagnosis of Gaucher Disease Type 1 Patients Using a Mass Spectrometry-Based Metabolomic Approach. Int. J. Mol. Sci. 2020, 21, 7869. [Google Scholar] [CrossRef] [PubMed]
- Mirzaian, M.; Wisse, P.; Ferraz, M.J.; Gold, H.; Donker-Koopman, W.E.; Verhoek, M.; Overkleeft, H.S.; Boot, R.G.; Kramer, G.; Dekker, N.; et al. Mass spectrometric quantification of glucosylsphingosine in plasma and urine of type 1 Gaucher patients using an isotope standard. Blood Cells Mol. Dis. 2015, 54, 307–314. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Boutin, M.; Gagnon, R.; Dupont, F.O.; Lavoie, P.; Clarke, J.T. Urinary globotriaosylsphingosine-related biomarkers for Fabry disease targeted by metabolomics. Anal. Chem. 2012, 84, 2745–2753. [Google Scholar] [CrossRef]
- Yazd, H.S.; Bazargani, S.F.; Vanbeek, C.A.; King-Morris, K.; Heldermon, C.; Segal, M.S.; Clapp, W.L.; Garrett, T.J. LC-MS lipidomics of renal biopsies for the diagnosis of Fabry disease. J. Mass Spectrom. Adv. Clin. Lab 2021, 22, 71–78. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Lavoie, P.; Boutin, M.; Ntwari, A.; Hsu, T.R.; Huang, C.K.; Niu, D.M. Biomarkers associated with clinical manifestations in Fabry disease patients with a late-onset cardiac variant mutation. Clin. Chim. Acta 2017, 466, 185–193. [Google Scholar] [CrossRef]
- Menkovic, I.; Lavoie, P.; Boutin, M.; Auray-Blais, C. Distribution of heparan sulfate and dermatan sulfate in mucopolysaccharidosis type II mouse tissues pre- and post-enzyme-replacement therapy determined by UPLC-MS/MS. Bioanalysis 2019, 11, 727–740. [Google Scholar] [CrossRef]
- Boutin, M.; Lavoie, P.; Menkovic, I.; Toupin, A.; Abaoui, M.; Elidrissi-Elawad, M.; Arthus, M.F.; Fortier, C.; Ménard, C.; Maranda, B.; et al. Diurnal Variation of Urinary Fabry Disease Biomarkers during Enzyme Replacement Therapy Cycles. Int. J. Mol. Sci. 2020, 21, 6114. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Cyr, D.; Mills, K.; Giguère, R.; Drouin, R. Development of a filter paper method potentially applicable to mass and high-risk urinary screenings for Fabry disease. J. Inherit. Metab. Dis. 2007, 30, 106. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Lavoie, P.; Boutin, M.; Abaoui, M. High-Risk Screening for Fabry Disease: Analysis by Tandem Mass Spectrometry of Globotriaosylceramide (Gb3) in Urine Collected on Filter Paper. Curr. Protoc. Hum. Genet. 2017, 93, 17.26.1–17.26.12. [Google Scholar] [CrossRef] [PubMed]
- Auray-Blais, C.; Collerette-Tremblay, J.; Lavoie, P. UPLC–MS/MS analysis of keratan sulfate from urine samples collected on filter paper for monitoring & follow-up of Morquio A patients. Bioanalysis 2018, 10, 1181–1192. [Google Scholar] [PubMed]
- Auray-Blais, C.; Lavoie, P.; Zhang, H.; Gagnon, R.; Clarke, J.T.; Maranda, B.; Young, S.P.; Millington, D.S. An improved method for glycosaminoglycan analysis by LC-MS/MS of urine samples collected on filter paper. Clin. Chim. Acta 2012, 413, 771–778. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
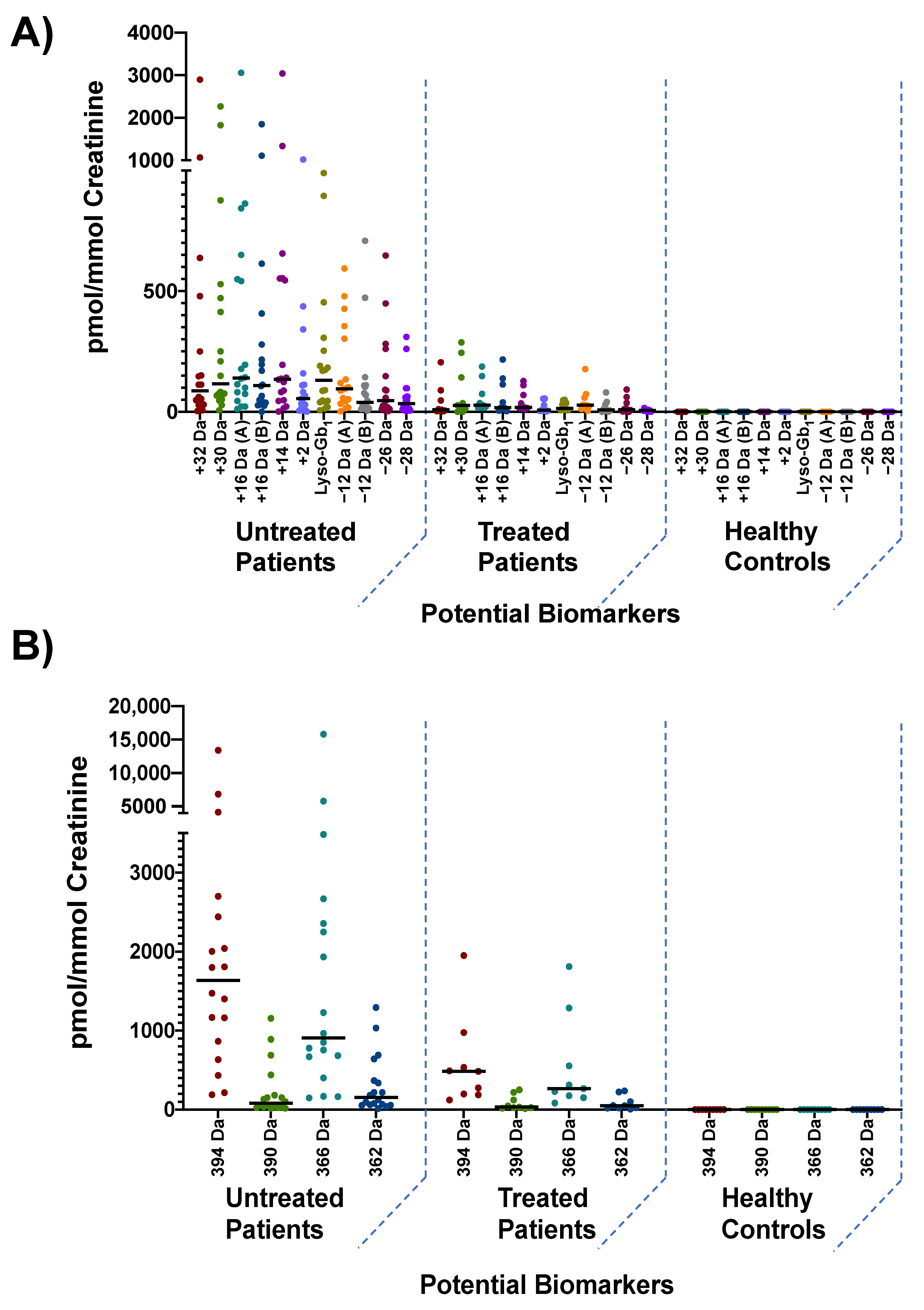{kind=link}
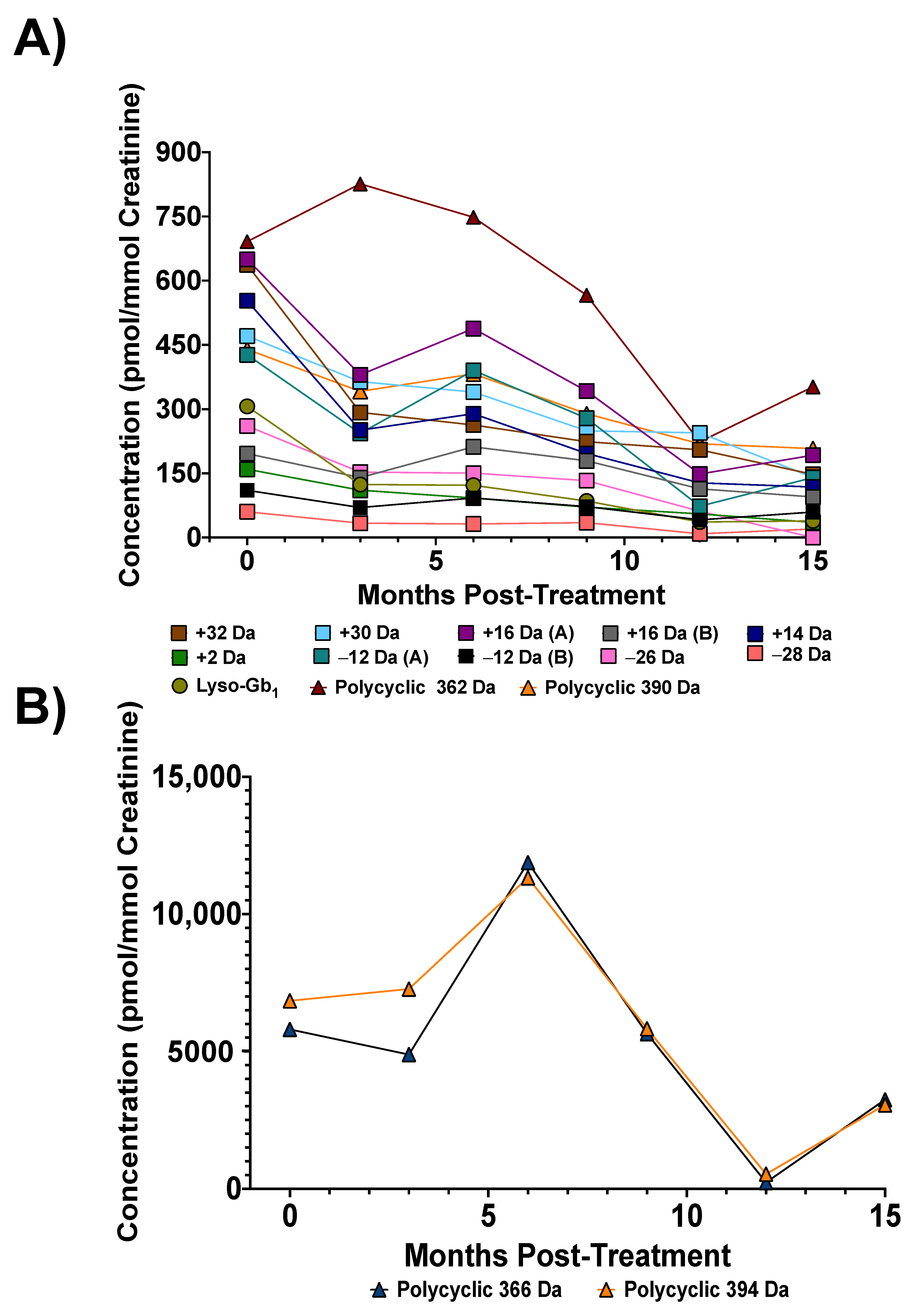{kind=link}
| Parameters | Description | |||
| Chromatographic parameters | ||||
| HPLC Column | Halo HILIC 2.7 Advanced Materials Technology (Wilmington, DE, USA) | |||
| Column Dimension | 4.6 × 1500 mm | |||
| Particle Size | 2.7 μm | |||
| Column Temperature | 24 °C | |||
| Weak Wash Solvent | ACN | |||
| Strong Wash Solvent | ACN | |||
| Injection Mode | Partial Loop | |||
| Injection Volume | 4 μL | |||
| Mobile Phase A | 94.5:2.5:2.5:0.5 ACN: MeOH: H2O: FA + 5 mM Amm. Form. | |||
| Mobile Phase B | 10:89.5:0.5 ACN: H2O: FA + 5 mM Amm. Form. | |||
| Flow rate | 0.85 mL/min | |||
| Gradient (% Mobile Phase B) | 0.0 → 9.5 min: 12.5% | |||
| 9.5 → 11.0 min: 70.0% | ||||
| 11.0 → 12.0 min: 12.5% | ||||
| Mass spectrometry parameters | ||||
| Ionization Mode | Electrospray Ionization (ESI) | |||
| Polarity | Positive | |||
| Acquisition Mode | Multiple Reaction Monitoring (MRM) | |||
| Capillary Voltage | 3.2 kV | |||
| Desolvation Temperature | 550 °C | |||
| Desolvation Gas Flow | 750 L/h | |||
| Cone Gas Flow | 150 L/h | |||
| Source Temperature | 150 °C | |||
| Analytes | ||||
| Compound | Transitions (m/z) | Cone Voltage (V) | Collision Energy (V) | Dwell Time (s) |
| Creatinine | 144.07 > 44.05 | 10 | 5 | 0.200 |
| Creatinine-(D3) | 117.09 > 47.07 | 10 | 5 | 0.200 |
| Lyso-Gb1 −28 Da | 434.31 > 254.25 | 38 | 18 | 0.021 |
| Lyso-Gb1 −26 Da | 436.25 > 238.18 | 38 | 18 | 0.021 |
| Lyso-Gb1 −12 Da | 450.31 > 252.23 | 38 | 18 | 0.021 |
| Lyso-Gb1 | 462.34 > 282.28 | 38 | 18 | 0.021 |
| Lyso-Gb1 +2 Da | 464.29 > 284.22 | 38 | 18 | 0.021 |
| Lyso-Gb1 −(13C6) | 468.36 > 282.28 | 38 | 18 | 0.021 |
| Lyso-Gb1 +14 Da | 476.36 > 296.26 | 38 | 18 | 0.021 |
| Lyso-Gb1 +16 Da | 478.34 > 280.26 | 38 | 18 | 0.021 |
| Lyso-Gb1 +30 Da | 492.32 > 294.24 | 38 | 18 | 0.021 |
| Lyso-Gb1 +32 Da | 494.33 > 296.26 | 38 | 18 | 0.021 |
| Polycyclic Analog 362 Da | 362.18 > 164.11 | 38 | 18 | 0.021 |
| Polycyclic Analog 366 Da | 366.18 > 168.10 | 38 | 18 | 0.021 |
| Polycyclic Analog 390 Da | 390.21 > 192.14 | 38 | 18 | 0.021 |
| Polycyclic Analog 394 Da | 394.21 > 196.13 | 38 | 18 | 0.021 |
| Validation Parameter | Results | |
|---|---|---|
| Lyso-Gb1 | Creatinine | |
| Average intraday precision (RSD *) (n = 15) | 2.9% | 2.0% |
| Average intraday accuracy (Bias) (n = 15) | 3.8% | 3.0% |
| Average interday precision (RSD *) (n = 15) | 5.3% | 3.9% |
| Average interday accuracy (Bias) (n = 15) | 4.2% | 4.3% |
| Calibration curve ** (n = 5) | r2 > 0.998 | r2 > 0.998 |
| Stability *** at − 30 °C | At least a month | At least a month |
| Stability *** at 4 °C | 7 Days | 7 Days |
| Stability *** at room temperature | 48 h | 72 h |
| Stability in the sample organizer (10 °C) | 48 h | 48 h |
| Freeze/Thaw (3 cycles) (Bias) | 7.6% | 5.3% |
| Glassware adhesion (Bias) | 4.2% | 2.2% |
| Plasticware adhesion (Bias) | 5.8% | 3.1% |
| Biomarker | Parameters | ||||||
|---|---|---|---|---|---|---|---|
| LOD (pM) | LOQ (pM) | Intraday Precision (%RSD) | Interday Precision (%RSD) | Recovery (%) | |||
| LQC | HQC | LQC | HQC | ||||
| Lyso-Gb1 −28 Da | 56 | 186 | ND * | 9.5 | ND * | 14.3 | 82.5 |
| Lyso-Gb1 −26 Da | 108 | 360 | 5.3 | 10.9 | 11.9 | 11.3 | 87.6 |
| Lyso-Gb1 −12 Da | 106 | 354 | 1.8 | 6.5 | 10.6 | 8.2 | 78.9 |
| Lyso-Gb1 −12 Da (B) | 98 | 328 | 11.2 | 5.3 | 14.8 | 11.3 | 81.3 |
| Lyso-Gb1 +2 Da | 101 | 337 | 7.8 | 6.9 | 12.2 | 8.5 | 82.3 |
| Lyso-Gb1 +14 Da | 84 | 283 | 6.2 | 7.6 | 8.5 | 7.2 | 72.1 |
| Lyso-Gb1 +16 Da | 105 | 351 | 6.9 | 2.1 | 15.9 | 11.1 | 78.9 |
| Lyso-Gb1 +16 Da (B) | 89 | 296 | 7.7 | 10.9 | 6.7 | 10.5 | 85.0 |
| Lyso-Gb1 +30 Da | 95 | 317 | 9.7 | 6.1 | 11.1 | 6.5 | 91.2 |
| Lyso-Gb1 +32 Da | 90 | 301 | 5.1 | 8.6 | 8.2 | 13.0 | 80.0 |
| Polycyclic Analog 362 Da | 105 | 305 | 3.3 | 5.6 | 6.9 | 4.7 | 98.3 |
| Polycyclic Analog 366 Da | 94 | 314 | 21.1 | 12.7 | 37.9 | 33.7 | 51.8 |
| Polycyclic Analog 390 Da | 109 | 364 | 5.2 | 4.2 | 21.6 | 15.3 | 82.8 |
| Polycyclic Analog 394 Da | 91 | 303 | 18.3 | 9.2 | 23.3 | 21.4 | 65.5 |
| Months Post-ERT | Platelet Count (×103 mm3) | Hemoglobin (g/dL) | Liver Volume (by MRI *) | Spleen Volume (by MRI *) |
|---|---|---|---|---|
| 0 | 94 | 12.9 | 1666 | 976 |
| 3 | 105 | 13.3 | - | - |
| 6 | 136 | 13.0 | 1200 | 750 |
| 9 | 185 | 13.8 | - | - |
| 12 | 184 | 13.9 | 1302 | 570 |
| 15 | 124 | 14.5 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menkovic, I.; Boutin, M.; Alayoubi, A.; Curado, F.; Bauer, P.; Mercier, F.E.; Auray-Blais, C. Quantitation of a Urinary Profile of Biomarkers in Gaucher Disease Type 1 Patients Using Tandem Mass Spectrometry. Diagnostics 2022, 12, 1414. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12061414
Menkovic I, Boutin M, Alayoubi A, Curado F, Bauer P, Mercier FE, Auray-Blais C. Quantitation of a Urinary Profile of Biomarkers in Gaucher Disease Type 1 Patients Using Tandem Mass Spectrometry. Diagnostics. 2022; 12(6):1414. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12061414
Chicago/Turabian StyleMenkovic, Iskren, Michel Boutin, Abdulfatah Alayoubi, Filipa Curado, Peter Bauer, François E. Mercier, and Christiane Auray-Blais. 2022. "Quantitation of a Urinary Profile of Biomarkers in Gaucher Disease Type 1 Patients Using Tandem Mass Spectrometry" Diagnostics 12, no. 6: 1414. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12061414