
Practical Approach to Histological Diagnosis of Peripheral Nerve Sheath Tumors: An Update

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Classification of the Peripheral Nerve Sheath Tumors
3. Neurofibromas
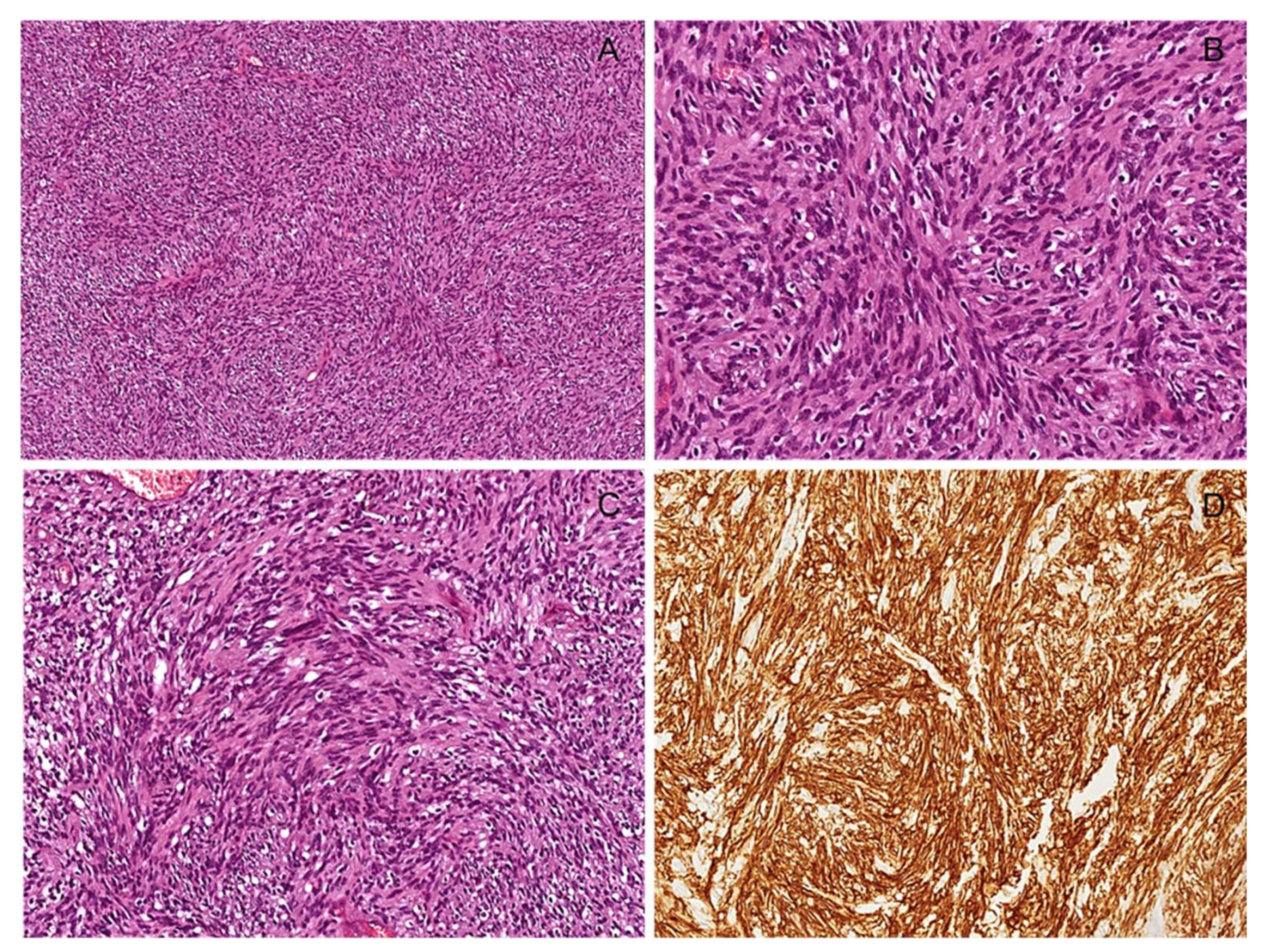
3.1. Localized/Nodular Neurofibroma
Immunohistochemical Features
3.2. Diffuse Neurofibroma
Immunohistochemical Features
3.3. Plexiform Neurofibroma
3.4. Potential Morphological Pitfalls of Malignancy in Neurofibromas: Cytological Atypia and Hypercellularity
Immunohistochemical Features
4. Atypical Neurofibromatous Neoplasm with Uncertain Biologic Potential (ANNUBP)
Immunohistochemical Features
5. Malignant Transformation in Neurofibromas
6. Schwannoma
6.1. Classic Schwannoma
Immunohistochemical Features
6.2. Schwannoma with Degenerative/Ancient Changes (“Ancient Schwannoma”)
6.3. Cellular Schwannoma
Immunohistochemical Features
6.4. Plexiform Schwannoma
Immunohistochemical Features
6.5. Epithelioid Cell Schwannoma
Immunohistochemical Features
6.6. Unusual Features in Schwannomas
6.7. Malignant Transformation in Schwannoma
7. Perineuriomas
7.1. Intraneural Perineurioma
7.1.1. Immunohistochemical Features
7.1.2. Molecular Features
7.2. Soft Tissue (Extraneural) Perineurioma
7.2.1. Immunohistochemical Features
7.2.2. Molecular Features
8. Hybrid Tumors
Molecular Features
9. Malignant Peripheral Nerve Sheath Tumors (MPNSTs)
9.1. Classic MPNST
Immunohistochemical Features
9.2. Epithelioid Cell MPNST
Immunohistochemical Features
9.3. Perineurial Malignant Peripheral Nerve Sheath Tumors (Malignant Perineurioma)
10. Malignant Melanotic Schwannian Tumor (So-Called “Melanotic Schwannoma”)
Molecular Features
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goldblum, J.R.; Folpe, A.L.; Weiss, S.W. Enzinger & Weiss’s Soft Tissue Tumors, 7th ed.; Elsevier: Philadelphia, PA, USA, 2020. [Google Scholar]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Huson, S.M.; Evans, D.G. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet. Med. 2022, in press. [Google Scholar]
- Rodriguez, F.J.; Folpe, A.L.; Giannini, C.; Perry, A. Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012, 123, 295–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, A.P.; Fritchie, K.J. Update on Peripheral Nerve Sheath Tumors. Surg. Pathol. Clin. 2019, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.; Kim, A.; Lazar, A.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1—A consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Reuss, D.E.; Rodriguez, F. Neurofibroma. In WHO Classification of Tumours- Soft Tissue and Bone Tumours, 5th ed.; WHO Classification of Tumours Editorial Board, International Angency for Research on Cancer: Lyon, France, 2020; Volume 3, pp. 232–236. [Google Scholar]
- Ruggieri, M.; Praticò, A.D.; Caltabiano, R.; Polizzi, A. Early history of the different forms of neurofibromatosis from ancient Egypt to the British Empire and beyond: First descriptions, medical curiosities, misconceptions, landmarks, and the persons behind the syndromes. Am. J. Med Genet. Part A 2018, 176, 515–550. [Google Scholar] [CrossRef]
- Ruggieri, M.; Huson, S.M. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology 2001, 56, 1433–1443. [Google Scholar] [CrossRef]
- Ruggieri, M.; Praticò, A.D. Mosaic neurocutaneous disorders and their causes. Semin. Pediatr. Neurol. 2015, 22, 207–233. [Google Scholar] [CrossRef]
- Ruggieri, M.; Polizzi, A.; Catanzaro, S.; Lo Bianco, M.; Praticò, A.D.; Di Rocco, C. Introduction to phacomatoses (neurocutaneous disorders) in childhood. Childs Nerv. Syst. 2020, 36, 2229–2268. [Google Scholar] [CrossRef]
- Magro, G.; Amico, P.; Vecchio, G.M.; Caltabiano, R.; Castaing, M.; Kacerovska, D.; Kazakov, D.; Michal, M. Multinucleated floret-like giant cells in sporadic and NF1-associated neurofibromas: A clinicopathologic study of 94 cases. Virchows Arch. 2010, 456, 71–76. [Google Scholar] [CrossRef]
- Schaefer, I.-M.; Fletcher, C.D. Malignant Peripheral Nerve Sheath Tumor (MPNST) Arising in Diffuse-type Neurofibroma. Am. J. Surg. Pathol. 2015, 39, 1234–1241. [Google Scholar] [CrossRef]
- Bernthal, N.M.; Putnam, A.; Jones, K.B.; Viskochil, D.; Randall, R.L. The effect of surgical margins on outcomes for low grade MPNSTs and atypical neurofibroma. J. Surg. Oncol. 2014, 110, 813–816. [Google Scholar] [CrossRef]
- Evans, D.G.R.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Uusitalo, E.; Leppävirta, J.; Koffert, A.; Suominen, S.B.; Vahtera, J.; Vahlberg, T.; Pöyhönen, M.; Peltonen, J.; Peltonen, S. Incidence and Mortality of Neurofibromatosis: A Total Population Study in Finland. J. Investig. Dermatol. 2015, 135, 904–906. [Google Scholar] [CrossRef] [Green Version]
- Karamchandani, J.R.; Nielsen, T.O.; van de Rijn, M.; West, R.B. Sox10 and S100 in the Diagnosis of Soft-tissue Neoplasms. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Fanburg-Smith, J.C.; Majidi, M.; Miettinen, M. Keratin expression in schwannoma; a study of 115 retroperitoneal and 22 peripheral schwannomas. Mod. Pathol. 2006, 19, 115–121. [Google Scholar] [CrossRef]
- Wang, D.-Z.; Liu, P.; Yao, L.; Hao, Y.-H.; Zhu, R.-J.; Zhang, T.; Tang, X.-B. Aberrant expression of thyroid transcription factor–1 in schwannomas. Hum. Pathol. 2018, 71, 84–90. [Google Scholar] [CrossRef]
- Caltabiano, R.; Magro, G.; Polizzi, A.; Praticò, A.D.; Ortensi, A.; D’Orazi, V.; Panunzi, A.; Milone, P.; Maiolino, L.; Nicita, F.; et al. A mosaic pattern of INI1/SMARCB1 protein expression distinguishes Schwannomatosis and NF2-associated peripheral schwannomas from solitary peripheral schwannomas and NF2-associated vestibular schwannomas. Childs Nerv. Syst. 2017, 33, 933–940. [Google Scholar] [CrossRef]
- Harazono, Y.; Kayamori, K.; Sakamoto, J.; Akaike, Y.; Kurasawa, Y.; Tsushima, F.; Sasaki, Y.; Harada, H.; Yoda, T. Retrospective analysis of schwannoma in the oral and maxillofacial region: Clinicopathological characteristics and specific pathology of ancient change. Br. J. Oral Maxillofac. Surg. 2022, 60, 326–331. [Google Scholar] [CrossRef]
- Ratnagiri, R.; Mallikarjun, S. Retroperitoneal ancient schwannoma: Two cases and review of literature. J. Cancer Res. Ther. 2014, 10, 368. [Google Scholar] [CrossRef]
- White, W.; Shiu, M.H.; Rosenblum, M.K.; Erlandson, R.A.; Woodruff, J.M. Cellular schwannoma. A clinicopathologic study of 57 patients and 58 tumors. Cancer 1990, 66, 1266–1275. [Google Scholar] [CrossRef]
- Woodruff, J.M.; Marshall, M.L.; Godwin, T.A.; Funkhouser, J.W.; Thompson, N.J.; Erlandson, R.A. Plexiform (multinodular) schwannoma. Am. J. Surg. Pathol. 1983, 7, 691–698. [Google Scholar] [CrossRef]
- Fletcher, C.D.; Davies, S.E. Benign plexiform (multinodular) schwannoma: A rare tumour unassociated with neurofibromatosis. Histopathology 1986, 10, 971–980. [Google Scholar] [CrossRef]
- Megahed, M. Plexiform schwannoma. Am. J. Dermatopathol. 1994, 16, 288–293. [Google Scholar] [CrossRef]
- Agaram, N.P.; Prakash, S.; Antonescu, C.R. Deep-seated plexiform schwannoma: A pathologic study of 16 cases and comparative analysis with the superficial variety. Am. J. Surg. Pathol. 2005, 29, 1042–1048. [Google Scholar] [CrossRef]
- Hébert-Blouin, M.N.; Amrami, K.K.; Scheithauer, B.W.; Spinner, R.J. Multinodular/plexiform (multifascicular) schwannomas of major peripheral nerves: An underrecognized part of the spectrum of schwannomas. J. Neurosurg. 2010, 112, 372–382. [Google Scholar] [CrossRef]
- Sun, M.; Shao, M.; Liu, J.; Zhao, L.; Weng Lao, I.; Yu, L.; Wang, J. Plexiform Cellular Schwannoma in Infancy and Childhood: A Clinicopathological Study of Seven Cases of an Underrecognized Nerve Sheath Tumor with a Tendency Toward Local Recurrence. Int. J. Surg. Pathol. 2022, 30, 265–272. [Google Scholar] [CrossRef]
- Kindblom, L.G.; Meis-Kindblom, J.M.; Havel, G.; Busch, C. Benign epithelioid schwannoma. Am. J. Surg. Pathol. 1998, 22, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.; Chappell, M.E.; Weiss, S.W. Neuroblastoma-like epithelioid schwannoma. Histopathology 1995, 26, 193–194. [Google Scholar] [CrossRef] [PubMed]
- Hart, J.; Gardner, J.M.; Edgar, M.; Weiss, S.W. Epithelioid Schwannomas: An Analysis of 58 Cases Including Atypical Variants. Am. J. Surg. Pathol. 2016, 40, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Suchak, R.; Luzar, B.; Bacchi, C.E.; Maguire, B.; Calonje, E. Cutaneous neuroblastoma-like schwannoma: A report of two cases, one with a plexiform pattern, and a review of the literature. J. Cutan. Pathol. 2010, 37, 997–1001. [Google Scholar] [CrossRef]
- Goldblum, J.R.; Beals, T.F.; Weiss, S.W. Neuroblastoma-like neurilemoma. Am. J. Surg. Pathol. 1994, 18, 266–273. [Google Scholar] [CrossRef]
- Lewis, Z.T.; Geisinger, K.R.; Pichardo, R.; Sangueza, O.P. Schwannoma with neuroblastoma-like rosettes: An unusual morphologic variant. Am. J. Dermatopathol. 2005, 27, 243–246. [Google Scholar] [CrossRef]
- Jo, V.Y.; Fletcher, C.D.M. SMARCB1/INI1 Loss in Epithelioid Schwannoma: A Clinicopathologic and Immunohistochemical Study of 65 Cases. Am. J. Surg. Pathol. 2017, 41, 1013–1022. [Google Scholar] [CrossRef]
- Brooks, J.J.; Draffen, R.M. Benign glandular schwannoma. Arch. Pathol. Lab. Med. 1992, 116, 192–195. [Google Scholar]
- Ud Din, N.; Ahmad, Z.; Ahmed, A. Schwannomas with pseudoglandular elements: Clinicopathologic study of 61 cases. Ann. Diagn. Pathol. 2016, 20, 24–28. [Google Scholar] [CrossRef]
- Mourmouras, V.; Taddeucci, P.; Ambrosio, M.R.; Miracco, C. Neuroblastoma-like neurilemoma: An additional case. J. Dermatol. 2008, 35, 548–549. [Google Scholar] [CrossRef]
- Koubaa Mahjoub, W.; Jouini, R.; Khanchel, F.; Ben Brahim, E.; Llamas-Velasco, M.; Helel, I.; Khayat, O.; Chadli, A.; Badri, T.; Mentzel, T. Neuroblastoma-like schwannoma with giant rosette: A potential diagnostic pitfall for hyalinizing spindle cell tumor. J. Cutan. Pathol. 2019, 46, 234–237. [Google Scholar] [CrossRef]
- Vecchio, G.M.; Amico, P.; Leone, G.; Salvatorelli, L.; Magro, G. Lipoblast-like signet-ring cells in neurofibroma: A potential diagnostic pitfall of malignancy. Pathologica 2010, 102, 108–111. [Google Scholar]
- Plaza, J.A.; Wakely, P.E., Jr.; Suster, S. Lipoblastic nerve sheath tumors: Report of a distinctive variant of neural soft tissue neoplasm with adipocytic differentiation. Am. J. Surg. Pathol. 2006, 30, 337–344. [Google Scholar] [CrossRef]
- Hanada, M.; Tanaka, T.; Kanayama, S.; Takami, M.; Kimura, M. Malignant transformation of intrathoracic ancient neurilemoma in a patient without von Recklinghausen’s disease. Acta Pathol. Jpn. 1982, 32, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, J.M.; Selig, A.M.; Crowley, K.; Allen, P.W. Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am. J. Surg. Pathol. 1994, 18, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Nayler, S.J.; Leiman, G.; Omar, T.; Cooper, K. Malignant transformation in a schwannoma. Histopathology 1996, 29, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Hidaka, T.; Akisada, T.; Takemoto, T.; Irei, I.; Manabe, T. Malignant peripheral nerve sheath tumor arising in benign ancient schwannoma: A case report with an immunohistochemical study. Pathol. Int. 2000, 50, 156–161. [Google Scholar] [CrossRef] [PubMed]
- McMenamin, M.E.; Fletcher, C.D. Expanding the spectrum of malignant change in schwannomas: Epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: A study of 17 cases. Am. J. Surg. Pathol. 2001, 25, 13–25. [Google Scholar] [CrossRef]
- Carter, J.M.; O’Hara, C.; Dundas, G.; Gilchrist, D.; Collins, M.S.; Eaton, K.; Judkins, A.R.; Biegel, J.A.; Folpe, A.L. Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma-like” schwannomatosis and a novel germline SMARCB1 mutation. Am. J. Surg. Pathol. 2012, 36, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Boyanton, B.L., Jr.; Jones, J.K.; Shenaq, S.M.; Hicks, M.J.; Bhattacharjee, M.B. Intraneural perineurioma: A systematic review with illustrative cases. Arch. Pathol. Lab. Med. 2007, 131, 1382–1392. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D. Soft tissue perineurioma: Clinicopathologic analysis of 81 cases including those with atypical histologic features. Am. J. Surg. Pathol. 2005, 29, 845–858. [Google Scholar] [CrossRef]
- Emory, T.S.; Scheithauer, B.W.; Hirose, T.; Wood, M.; Onofrio, B.M.; Jenkins, R.B. Intraneural perineurioma. A clonal neoplasm associated with abnormalities of chromosome 22. Am. J. Clin. Pathol. 1995, 103, 696–704. [Google Scholar] [CrossRef]
- Klein, C.J.; Wu, Y.; Jentoft, M.E.; Mer, G.; Spinner, R.J.; Dyck, P.J.; Dyck, P.J.; Mauermann, M.L. Genomic analysis reveals frequent TRAF7 mutations in intraneural perineuriomas. Ann. Neurol. 2017, 81, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Manasseh, R.G.; Edis, R.H.; Page, R.; Keith-Rokosh, J.; Walsh, P.; Song, S.; Laycock, A.; Griffiths, L.; Fabian, V.A. Intraneural perineurioma. J. Clin. Neurosci. 2009, 16, 1633–1636. [Google Scholar] [CrossRef]
- Kum, Y.S.; Kim, J.K.; Cho, C.H.; Kim, H.K. Intraneural reticular perineurioma of the hypoglossal nerve. Head Neck 2009, 31, 833–837. [Google Scholar] [CrossRef]
- Carter, J.M.; Wu, Y.; Blessing, M.M.; Folpe, A.L.; Thorland, E.C.; Spinner, R.J.; Jentoft, M.E.; Wang, C.; Baheti, S.; Niu, Z.; et al. Recurrent Genomic Alterations in Soft Tissue Perineuriomas. Am. J. Surg. Pathol. 2018, 42, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.A.; Lemberger, R.J.; Fisher, C.; McCulloch, T.A. Epithelioid perineurioma: An unusual variant. J. Clin. Pathol. 2008, 61, 1130–1132. [Google Scholar] [CrossRef] [Green Version]
- Torres-Mora, J.; Ud Din, N.; Ahrens, W.A.; Folpe, A.L. Pseudolipoblastic perineurioma: An unusual morphological variant of perineurioma that may simulate liposarcoma. Hum. Pathol. 2016, 57, 22–27. [Google Scholar] [CrossRef]
- Rank, J.P.; Rostad, S.W. Perineurioma with ossification: A case report with immunohistochemical and ultrastructural studies. Arch. Pathol. Lab. Med. 1998, 122, 366–370. [Google Scholar]
- Al-Daraji, W.I. Granular perineurioma: The first report of a rare distinctive subtype of perineurioma. Am. J. Dermatopathol. 2008, 30, 163–168. [Google Scholar] [CrossRef]
- Mentzel, T.; Kutzner, H. Reticular and plexiform perineurioma: Clinicopathological and immunohistochemical analysis of two cases and review of perineurial neoplasms of skin and soft tissues. Virchows Arch. 2005, 447, 677–682. [Google Scholar] [CrossRef]
- Zaugg, P.; Maeder, B.; Nobile, A.; Raffoul, W.; Bollmann, C.; di Summa, P.G. Reticular Perineurioma of the Hand: Diagnosis and Treatment of a Rare Case of Hand Mass. J. Hand Surg. Am. 2017, 42, e199–e203. [Google Scholar] [CrossRef]
- Burgues, O.; Monteagudo, C.; Noguera, R.; Revert, A.; Molina, I.; Llombart-Bosch, A. Cutaneous sclerosing Pacinian-like perineurioma. Histopathology 2001, 39, 498–502. [Google Scholar] [CrossRef]
- Colizza, A.; Covello, R.; Greco, A.; Ralli, M.; Coppola, G.; Gilardi, A.; Riminucci, M.; de Vincentiis, M.; Corsi, A. Extraneural Sclerosing Perineurioma of the Tongue. Ear Nose Throat J. 2021, 1455613211020539. [Google Scholar] [CrossRef]
- Giannini, C.; Scheithauer, B.W.; Jenkins, R.B.; Erlandson, R.A.; Perry, A.; Borell, T.J.; Hoda, R.S.; Woodruff, J.M. Soft-tissue perineurioma. Evidence for an abnormality of chromosome 22, criteria for diagnosis, and review of the literature. Am. J. Surg. Pathol. 1997, 21, 164–173. [Google Scholar] [CrossRef]
- Harder, A.; Wesemann, M.; Hagel, C.; Schittenhelm, J.; Fischer, S.; Tatagiba, M.; Nagel, C.; Jeibmann, A.; Bohring, A.; Mautner, V.F.; et al. Hybrid neurofibroma/schwannoma is overrepresented among schwannomatosis and neurofibromatosis patients. Am. J. Surg. Pathol. 2012, 36, 702–709. [Google Scholar] [CrossRef]
- Stahn, V.; Nagel, I.; Fischer-Huchzermeyer, S.; Oyen, F.; Schneppenheim, R.; Gesk, S.; Bohring, A.; Chikobava, L.; Young, P.; Gess, B.; et al. Molecular Analysis of Hybrid Neurofibroma/Schwannoma Identifies Common Monosomy 22 and α-T-Catenin/CTNNA3 as a Novel Candidate Tumor Suppressor. Am. J. Pathol. 2016, 186, 3285–3296. [Google Scholar] [CrossRef] [Green Version]
- Michal, M.; Kazakov, D.V.; Belousova, I.; Bisceglia, M.; Zamecnik, M.; Mukensnabl, P. A benign neoplasm with histopathological features of both schwannoma and retiform perineurioma (benign schwannoma-perineurioma): A report of six cases of a distinctive soft tissue tumor with a predilection for the fingers. Virchows Arch. 2004, 445, 347–353. [Google Scholar] [CrossRef]
- Hornick, J.L.; Bundock, E.A.; Fletcher, C.D. Hybrid schwannoma/perineurioma: Clinicopathologic analysis of 42 distinctive benign nerve sheath tumors. Am. J. Surg. Pathol. 2009, 33, 1554–1561. [Google Scholar] [CrossRef]
- Dickson, B.C.; Antonescu, C.R.; Demicco, E.G.; Leong, D.I.; Anderson, N.D.; Swanson, D.; Zhang, L.; Fletcher, C.D.M.; Hornick, J.L. Hybrid schwannoma-perineurioma frequently harbors VGLL3 rearrangement. Mod. Pathol. 2021, 34, 1116–1124. [Google Scholar] [CrossRef]
- Nihous, H.; Baud, J.; Azmani, R.; Michot, A.; Perret, R.; Mayeur, L.; de Pinieux, G.; Milin, S.; Angot, E.; Duquenne, S.; et al. Clinicopathologic and Molecular Study of Hybrid Nerve Sheath Tumors Reveals Their Common Association with Fusions Involving VGLL3. Am. J. Surg. Pathol. 2022, 46, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, D.V.; Pitha, J.; Sima, R.; Vanecek, T.; Shelekhova, K.; Mukensnabl, P.; Michal, M. Hybrid peripheral nerve sheath tumors: Schwannoma-perineurioma and neurofibroma-perineurioma. A report of three cases in extradigital locations. Ann. Diagn. Pathol. 2005, 9, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Kacerovska, D.; Michal, M.; Kuroda, N.; Tanaka, A.; Sima, R.; Denisjuk, N.; Kreuzberg, B.; Ricarova, R.; Kazakov, D.V. Hybrid peripheral nerve sheath tumors, including a malignant variant in type 1 neurofibromatosis. Am. J. Dermatopathol. 2013, 35, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Somatilaka, B.N.; Sadek, A.; McKay, R.M.; Le, L.Q. Malignant peripheral nerve sheath tumor: Models, biology, and translation. Oncogene 2022, 41, 2405–2421. [Google Scholar] [CrossRef]
- Knight, S.W.E.; Knight, T.E.; Santiago, T.; Murphy, A.J.; Abdelhafeez, A.H. Malignant Peripheral Nerve Sheath Tumors-A Comprehensive Review of Pathophysiology, Diagnosis, and Multidisciplinary Management. Children 2022, 9, 38. [Google Scholar] [CrossRef]
- Pendleton, C.; Everson, M.C.; Puffer, R.C.; Spinner, R.J. Personal and Familial Malignancy History in Patients with Malignant Peripheral Nerve Sheath Tumors with a Focus on Sporadic Tumors. World Neurosurg. 2020, 141, e778–e782. [Google Scholar] [CrossRef]
- Wakeman, K.M.; Zhang, Q.S.; Bandhlish, A.; Cranmer, L.D.; Ricciotti, R.W.; Mantilla, J.G. Fédération Nationale Des Centres de Lutte Contre Le Cancer (FNCLCC) Grading, Margin Status and Tumor Location Associate With Survival Outcomes in Malignant Peripheral Nerve Sheath Tumors. Am. J. Clin. Oncol. 2022, 45, 28–35. [Google Scholar] [CrossRef]
- Thway, K.; Hamarneh, W.; Miah, A.B.; Fisher, C. Malignant Peripheral Nerve Sheath Tumor with Rhabdomyosarcomatous and Glandular Elements: Rare Epithelial Differentiation in a Triton Tumor. Int. J. Surg. Pathol. 2015, 23, 377–383. [Google Scholar] [CrossRef]
- Meshikhes, A.W.; Duhaileb, M.A.; Amr, S.S. Malignant peripheral nerve sheath tumor with extensive osteosarcomatous and chondrosarcomatous differentiation: A case report. Int. J. Surg. Case Rep. 2016, 25, 188–191. [Google Scholar] [CrossRef] [Green Version]
- Mentzel, T.; Katenkamp, D. Intraneural angiosarcoma and angiosarcoma arising in benign and malignant peripheral nerve sheath tumours: Clinicopathological and immunohistochemical analysis of four cases. Histopathology 1999, 35, 114–120. [Google Scholar] [CrossRef]
- Lu, V.M.; Marek, T.; Gilder, H.E.; Puffer, R.C.; Raghunathan, A.; Spinner, R.J.; Daniels, D.J. H3K27 trimethylation loss in malignant peripheral nerve sheath tumor: A systematic review and meta-analysis with diagnostic implications. J. Neurooncol. 2019, 144, 433–443. [Google Scholar] [CrossRef]
- Laskin, W.B.; Weiss, S.W.; Bratthauer, G.L. Epithelioid variant of malignant peripheral nerve sheath tumor (malignant epithelioid schwannoma). Am. J. Surg. Pathol. 1991, 15, 1136–1145. [Google Scholar] [CrossRef]
- Luzar, B.; Shanesmith, R.; Ramakrishnan, R.; Fisher, C.; Calonje, E. Cutaneous epithelioid malignant peripheral nerve sheath tumour: A clinicopathological analysis of 11 cases. Histopathology 2016, 68, 286–296. [Google Scholar] [CrossRef]
- Dey, B.; Srinivas, B.H.; Badhe, B.; Nachiappa Ganesh, R.; Gochhait, D.; Toi, P.C.; Jinkala, S. Malignant Epithelioid Soft Tissue Tumours- A Pathologist’s Perspective with Review of Literature. Cureus 2020, 12, e12263. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Dong, F.; Garcia, E.P.; Fletcher, C.D.M.; Jo, V.Y. Recurrent SMARCB1 Inactivation in Epithelioid Malignant Peripheral Nerve Sheath Tumors. Am. J. Surg. Pathol. 2019, 43, 835–843. [Google Scholar] [CrossRef]
- Hirose, T.; Scheithauer, B.W.; Sano, T. Perineurial malignant peripheral nerve sheath tumor (MPNST): A clinicopathologic, immunohistochemical, and ultrastructural study of seven cases. Am. J. Surg. Pathol. 1998, 22, 1368–1378. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Langee, C.L.; Stevens, G.L.; Morgan, M.B. Malignant peripheral nerve sheath tumor with perineurial differentiation: “Malignant perineurioma”. J. Cutan. Pathol. 2002, 29, 362–367. [Google Scholar] [CrossRef]
- Mitchell, A.; Scheithauer, B.W.; Doyon, J.; Berthiaume, M.J.; Isler, M. Malignant perineurioma (malignant peripheral nerve sheath tumor with perineural differentiation). Clin. Neuropathol. 2012, 31, 424–429. [Google Scholar] [CrossRef]
- Alexiev, B.A.; Chou, P.M.; Jennings, L.J. Pathology of Melanotic Schwannoma. Arch. Pathol. Lab. Med. 2018, 142, 1517–1523. [Google Scholar] [CrossRef] [Green Version]
- Killeen, R.M.; Davy, C.L.; Bauserman, S.C. Melanocytic schwannoma. Cancer 1988, 62, 174–183. [Google Scholar] [CrossRef]
- Torres-Mora, J.; Dry, S.; Li, X.; Binder, S.; Amin, M.; Folpe, A.L. Malignant melanotic schwannian tumor: A clinicopathologic, immunohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of “melanotic schwannoma”. Am. J. Surg. Pathol. 2014, 38, 94–105. [Google Scholar] [CrossRef]
- Carney, J.A. Psammomatous melanotic schwannoma. A distinctive, heritable tumor with special associations, including cardiac myxoma and the Cushing syndrome. Am. J. Surg. Pathol. 1990, 14, 206–222. [Google Scholar] [CrossRef]
- Wang, L.; Zehir, A.; Sadowska, J.; Zhou, N.; Rosenblum, M.; Busam, K.; Agaram, N.; Travis, W.; Arcila, M.; Dogan, S.; et al. Consistent copy number changes and recurrent PRKAR1A mutations distinguish Melanotic Schwannomas from Melanomas: SNP-array and next generation sequencing analysis. Genes Chromosom. Cancer 2015, 54, 463–471. [Google Scholar] [CrossRef]
- Vallat-Decouvelaere, A.V.; Wassef, M.; Lot, G.; Catala, M.; Moussalam, M.; Caruel, N.; Mikol, J. Spinal melanotic schwannoma: A tumour with poor prognosis. Histopathology 1999, 35, 558–566. [Google Scholar] [CrossRef] [PubMed]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Benign tumors |
|
| Tumors with uncertain malignant potential |
|
| Malignant tumors |
|
| Neurofibromatosis type 1 [NF1; MIM # 162200; ORPHA:636] Chromosome 17q11.2 (NF1 gene): heterozygous pathogenic NF1 gene variant in unaffected tissues (e.g., white blood cells) *; Major features: multiple large [>5 mm in pre-pubertal children vs. >1.5 cm in post-pubertal children/adults] and/or small (i.e., the so-called “freckling” in specific places including axillae, groin, peri-oral, sub-mammary) café-au-lait spots * in the skin; two or more iris Lisch nodules (by slit lamp examination) *; two or more eye choroidal abnormalities (i.e., bright, patchy nodules detected by optical coherence tomography, OCT or near-infrared reflectance, NIR) *; one or more neurofibromas (cutaneous, sub-cutaneous/nodular, internal) *; Minor features: macrocephaly/megalencephaly (98th centile); delayed growth (stature 10–25th centile); congenital thoracic deformities; dysmorphic features; Complications: anterolateral long bone dysplasia (bowing) and/or pseudarthrosis *; (kypho) scoliosis; sphenoid wing dysplasia *; osteopenia/porosis; puberty disorders; hypertension; congenital heart defects (CHD); vasculopathy; haemostasis defects; optic pathway glioma (OPG) *; plexiform neurofibroma (PNF) *; learning difficulties; brain high (bright) signal lesions on T2 images (FASI); brainstem gliomas (BSG); increased risk of developing neoplasia [e.g., malignant peripheral nervous sheath tumour (MPNST), breast cancer, phaechromocytoma, neuroblastoma, gastrointestinal neuroendocrine tumour (GIST). NF1 Microdeletion syndrome [ORPHA:97685]: type 1 (1.4 Mb), type 2 (1.2 Mb), type 3 (1.0 Mb), atypical (> 1.4 Mb?): dysmorphisms, congenital heart defects, intellectual disability, neurofibromas (early-onset), sphenoid wing dysplasia, increased frequency of MPNSTs. |
| Spinal neurofibromatosis type 1 [MIM # 162210] Chromosome 17q11.2 (NF1 gene): heterozygous pathogenic NF1 gene variant [missense variants] in unaffected tissues (e.g., white blood cells) or in affected tissues (e.g., neurofibromas); Main features: multiple neurofibromas in all (38) spinal roots in the absence (or with few) classical/typical NF1stigmata (see above) |
| Mosaic (segmental) neurofibromatosis type 1 [MIM # 162210] Chromosome 17q11.2 (NF1 gene): (a) a pathogenic heterozygous NF1 gene variant in a clearly affected tissue with the typical NF1 stigmata [see below] in the absence of a pathogenic heterozygous NF1 gene variant in unaffected tissue (e.g., blood or other body areas); (b) an identical pathogenic heterozygous NF1 gene variant in two anatomically independent affected tissues with the typical NF1 stigmata, in the absence of a pathogenic heterozygous NF1 gene variant in unaffected tissue; or (c) a heterozygous NF1 gene variant with a variant allele fraction of significantly less than 50% in apparently normal tissue (e.g., blood); Main features: NF1 stigmata clearly distributed in a segmental/localised (mosaic) region/pattern: (1) Skin pigmentary manifestations only; (2) Neurofibromas only; (3) Skin pigmentary manifestations and neurofibromas (rare); (4) Plexiform neurofibroma alone; (5) Unilateral iris Lisch nodules; (6) a parent, with a child fulfilling the criteria for NF1, who has only one NF1 diagnostic criterion including small café-au-lait spots (i.e., freckling) in specific places (i.e., axillae, groin, peri-oral, sub-mammary), optic pathway glioma, two or more iris Lisch nodules or two or more choroid abnormalities, distinctive osseous lesion for NF1, two neurofibromas or a plexiform neurofibroma; Complications: decreased frequency of typical NF1 complications (<7%) |
| Watson syndrome [MIM # 193520] Chromosome 17q11.2 (NF1 gene) Main features: pulmonic stenosis, multiple café-au-lait spots, decreased intellectual ability, short stature; this NF1 alternative form is disputed, however, clinicians and pathologists should be aware that individuals harbouring pathogenic NF1 gene variants could manifest these features. |
| Neurofibromatosis/Noona syndrome [MIM # 601321; ORPHA:638] Chromosome 17q11.2 (NF1 gene) Chromosome 12q24.13 (PTPN11 gene) Main features: typical NF1 features + Noonan syndrome features [i.e., facial dysmorphic features (large forehead, hypertelorism, ptosis, down-slanting ocular rims, low-set/dysmorphic ears); webbed neck; congenital heart defects; thoracic deformities; learning difficulties; this NF1 alternative form is disputed, however, clinicians and pathologists should be aware that individuals harbouring pathogenic NF1/PTPN11 gene variants could manifest these features. |
| Constitutional mismatch repair cancer syndrome [MMRCS; MIM # 276300] Including: Turcot and Lynch [HNPCC1] syndromes or hereditary non-poliposis colo-rectal cancer (HNPCC) syndrome. Chromosome 2p21p16 (MSH2 gene); 2p16.3 (MSH6 gene); 3p22.2 (MLH1 gene); 7p22.1: biallelic DNA mismatch repair pathogenic variants. Main features: NF1-like macules and/or peripheral nerve tumours and malignant gliomas. |
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Treatment/Prognosis |
|
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Treatment/Prognosis |
|
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Treatment/Prognosis |
|
| Definition |
|
| Histological diagnosis (presence of at least two of the following features) |
|
| NF2-related schwannomatosis [NF2/MERLIN-schwannoma predisposing syndrome (NF2/MERLIN-SPS)] (Previously, neurofibromatosis type 2 or NF2) [MIM # 101000; ORPHA:637] Chromosome 22q12.2 (NF2/MERLIN moesin-ezrin-radixin-like gene): identical NF2 gene pathogenic variant in at least two anatomically distinctNF2related tumours (e.g., schwannoma, meningioma and/or ependymoma); NF2 gene variants in unaffected tissues (e.g., blood) and major/minor criteria (see below); Main features: (1) Gardner type (adulthood): Bilateral (or, sometimes, unilateral) VIII cranial nerve (vestibular) schwannoma(s) *; schwannomas of cranial nerves *; multiple meningiomas, ependymomas, schwannomas *; early-onset (posterior subcapsular or cortical) cataracts *; skin schwannomas (NF2 plaques) *; nodular schwannomas *; (2) Wishart (severe) type (childhood): prior to appearance of VIII nerve schwannomas * and/or nervous system tumours (meningiomas, ependymomas) *, non-VIII-cranial nerve schwannomas (e.g., mixed nerves, V, VII) *; early-onset (posterior subcapsular or cortical) cataract *; epiretinal membranes/hamartomas *; skin schwannomas (NF2 plaques) * diffused over body; brain cortical dysplasia; bone dysplasia; (3) Congenital type (neonatal/< 1 year): small bilateral VIII nerve schwannomas * stable for decade(s); optic nerve sheath meningioma(s) *; epiretinal membranes/hamartomas *; early-onset (posterior subcapsular or cortical) cataract *; skin schwannomas (NF2 plaques) * in atypical places (face, arms, legs) later disappearing; ependymomas *; spinal cord schwannomas and meningiomas *; brain cortical dysplasia; |
| Mosaic (segmental)NF2-related schwannomatosis [Mosaic NF2/MERLIN-schwannoma predisposing syndrome] (Mosaic NF2/MERLIN-SPS) (Previously, mosaic neurofibromatosis type 2 or mosaic NF2) [MIM # 101000; ORPHA:637] Chromosome 22q12.2 (NF2/MERLIN moesin-ezrin-radixin-like gene): NF2 gene pathogenic variant (variant allele fraction) in unaffected tissue (e.g., blood) < 50% Main features: NF2 stigmata distributed in a segmental/localised (mosaic) distribution(e.g., unilateral VIII nerve schwannoma, ipsilateral meningiomas, schwannomas); Diagnosis ➔ NF2 gene pathogenic variant in unaffected tissue (e.g., blood) < 50% |
| SMARCB1-related schwannomatosis (SWNTS 1) LZTR1-related schwannomatosis (SWNTS2) [SMARCB1/LZTR1-schwannoma predisposing syndrome (SMARCB1/LZTR1-SPS)] [SWNTS1, MIM # 162091; SWNTS2, MIM # 615670] Chromosome 22q11.23 (SMARCB1 gene); chromosome 22q11.21 (LZTR1 gene): (a) at least one pathologically confirmed schwannoma or hybrid nerve sheath tumour and SMARCB1 or LZTR1 pathogenic variant in an unaffected tissue (e.g., blood); (b) a common SMARCB1 or LZTR1 variant in two anatomically distinct tumours; Main features: multiple non-VIII cranial nerve, non-intradermal, cranial, spinal and peripheral schwannomas (in the absence of NF2 stigmata); SWNTS1 = additional extra-axial, extra-medullary meningiomas and occasionally unilateral VIII nerve schwannomas; SWNTS2 = later onset of disease (up to 60 years), schwannomas affecting various body regions (extremities, spinal cord, chest wall, subcutaneous); |
| MosaicSMARCB1-related schwannomatosis (SWNTS 1) MosaicLZTR1-related schwannomatosis (SWNTS2) [Mosaic SMARCB1/LZTR1-schwannoma predisposing syndrome] (Mosaic SMARCB1/LZTR1-SPS) [Mosaic SWNTS1, MIM # 162091; Mosaic SWNTS2, MIM # 615670] Chromosome 22q11.23 (SMARCB1 gene); chromosome 22q11.21 (LZTR1 gene): (a) at least one pathologically confirmed and SMARCB1 or LZTR1 pathogenic variant in an unaffected tissue (e.g., blood) in < 50% cells analysed; (b) a common SMARCB1 or LZTR1 variant in two anatomically distinct tumours Main features: see the above features of above SWNTS1 and SWNTS2 |
| 22q-related-scwhanommatosis [22q-Schwannoma predisposing syndrome (22q-SPS)] Chromosome 22q12.2 (NF2/MERLIN moesin-ezrin-radixin-like gene): Main features: patients who do NOT meet criteria for NF2/MERLIN-related-schwannomatosis, SMARCB1-related-schwanomatosis (SWNTS1) or LZTR1-related-scwhannomatosis (SWNTS2) but have both: (1) LOH of the same chromosome 22q markers in two anatomically distinct tumours (e.g., schwannoma or hybrid nerve sheath tumour); (2) A different NF2 pathogenic variant in each tumour but not in the unaffected tissue. |
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Treatment/Prognosis |
|
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Treatment/Prognosis |
|
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Treatment/Prognosis |
|
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Treatment/Prognosis |
|
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Treatment/Prognosis |
|
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Molecular Diagnostic Features |
|
| Treatment/Prognosis |
|
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Molecular Diagnostic Features |
|
| Treatment/Prognosis |
|
|
| Definition |
|
| Clinical Features |
|
| Gross Pathology |
|
| Histopathology |
|
| Immunohistochemistry |
|
| Treatment/Prognosis |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magro, G.; Broggi, G.; Angelico, G.; Puzzo, L.; Vecchio, G.M.; Virzì, V.; Salvatorelli, L.; Ruggieri, M. Practical Approach to Histological Diagnosis of Peripheral Nerve Sheath Tumors: An Update. Diagnostics 2022, 12, 1463. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12061463
Magro G, Broggi G, Angelico G, Puzzo L, Vecchio GM, Virzì V, Salvatorelli L, Ruggieri M. Practical Approach to Histological Diagnosis of Peripheral Nerve Sheath Tumors: An Update. Diagnostics. 2022; 12(6):1463. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12061463
Chicago/Turabian StyleMagro, Gaetano, Giuseppe Broggi, Giuseppe Angelico, Lidia Puzzo, Giada Maria Vecchio, Valentina Virzì, Lucia Salvatorelli, and Martino Ruggieri. 2022. "Practical Approach to Histological Diagnosis of Peripheral Nerve Sheath Tumors: An Update" Diagnostics 12, no. 6: 1463. https://0-doi-org.brum.beds.ac.uk/10.3390/diagnostics12061463