Of rAAV and Men: From Genetic Neuromuscular Disorder Efficacy and Toxicity Preclinical Studies to Clinical Trials and Back



Abstract
:1. Introduction
2. Marketed Pharmacological Treatments
3. The Therapeutic Toolbox for Muscle Gene Therapy
3.1. About Wild-Type AAV
3.2. Of the Usage of Recombinant AAV for Central Nervous System (CNS) and Muscle-Specific Targeting
3.3. Restricting Expression by Muscle and CNS-Specific Promoters
4. Translating Preclinical Studies into Clinical Trials
4.1. SMA Trial
4.2. DMD Trials
4.3. XLMTM Trial
5. Improvement of the Therapeutic Toolbox
5.1. Towards Safer Next-Generation Muscle and CNS-Restricted AAVs
5.2. Enhancing the Repertoire of Muscle and CNS-Restricted Promoters
5.3. Detargeting with miRNA-Based Elements
6. From Preclinical Studies to Clinical Trials… and Back: General Point of View
6.1. Defining the Product
6.2. Manufacturing AAV
6.3. Choosing the Best Preclinical Models
6.4. Defining the Dose
6.5. Circumventing Immune Response
6.6. Assessing Long-Term Efficacy
6.7. Assessing Long-Term Toxicity
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sajer, S.; Guardiero, G.S.; Scicchitano, B.M. Myokines in Home-Based Functional Electrical Stimulation-Induced Recovery of Skeletal Muscle in Elderly and Permanent Denervation. Eur. J. Transl. Myol. 2018, 28, 7905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, H.; Carraro, U. Home-Based Functional Electrical Stimulation of Human Permanent Denervated Muscles: A Narrative Review on Diagnostics, Managements, Results and Byproducts Revisited 2020. Diagnostics 2020, 10, 529. [Google Scholar] [CrossRef] [PubMed]
- Carraro, U. Thirty years of translational research in Mobility Medicine: Collection of abstracts of the 2020 Padua Muscle Days. Eur. J. Transl. Myol. 2020, 30, 8826. [Google Scholar] [CrossRef] [PubMed]
- Sarabon, N.; Kozinc, Z.; Lofler, S.; Hofer, C. Resistance Exercise, Electrical Muscle Stimulation, and Whole-Body Vibration in Older Adults: Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Clin. Med. 2020, 9, 2902. [Google Scholar] [CrossRef]
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2020 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. NMD 2019, 29, 980–1018. [Google Scholar] [CrossRef]
- Kraker, J.; Zivkovic, S.A. Autoimmune neuromuscular disorders. Curr. Neuropharmacol. 2011, 9, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Herbelet, S.; Rodenbach, A.; Paepe, B.; De Bleecker, J.L. Anti-Inflammatory and General Glucocorticoid Physiology in Skeletal Muscles Affected by Duchenne Muscular Dystrophy: Exploration of Steroid-Sparing Agents. Int. J. Mol. Sci. 2020, 21, 4596. [Google Scholar] [CrossRef]
- Goto, M.; Komaki, H.; Takeshita, E.; Abe, Y.; Ishiyama, A.; Sugai, K.; Sasaki, M.; Goto, Y.; Nonaka, I. Long-term outcomes of steroid therapy for Duchenne muscular dystrophy in Japan. Brain Dev. 2016, 38, 785–791. [Google Scholar] [CrossRef]
- Biggar, W.D.; Harris, V.A.; Eliasoph, L.; Alman, B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul. Disord. NMD 2006, 16, 249–255. [Google Scholar] [CrossRef]
- Martiniuk, F.; Mehler, M.; Pellicer, A.; Tzall, S.; La Badie, G.; Hobart, C.; Ellenbogen, A.; Hirschhorn, R. Isolation of a cDNA for human acid alpha-glucosidase and detection of genetic heterogeneity for mRNA in three alpha-glucosidase-deficient patients. Proc. Natl. Acad. Sci. USA 1986, 83, 9641–9644. [Google Scholar] [CrossRef] [Green Version]
- FDA. Myozyme Approval Letter. 2006. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/125141s0000_Myozyme_Approv.pdf (accessed on 25 November 2020).
- EMA. Myozyme Alglucosidase Alfa. 2014. Available online: https://www.ema.europa.eu/en/documents/overview/myozyme-epar-summary-public_en.pdf (accessed on 25 November 2020).
- Kishnani, P.S.; Corzo, D.; Nicolino, M.; Byrne, B.; Mandel, H.; Hwu, W.L.; Leslie, N.; Levine, J.; Spencer, C.; McDonald, M.; et al. Recombinant human acid [alpha]-glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology 2007, 68, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Nicolino, M.; Byrne, B.; Wraith, J.E.; Leslie, N.; Mandel, H.; Freyer, D.R.; Arnold, G.L.; Pivnick, E.K.; Ottinger, C.J.; Robinson, P.H.; et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet. Med. Off. J. Am. Coll. Med. Genet. 2009, 11, 210–219. [Google Scholar] [CrossRef] [Green Version]
- van der Ploeg, A.T.; Clemens, P.R.; Corzo, D.; Escolar, D.M.; Florence, J.; Groeneveld, G.J.; Herson, S.; Kishnani, P.S.; Laforet, P.; Lake, S.L.; et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N. Engl. J. Med. 2010, 362, 1396–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, S.H.; Kronn, D.; Leslie, N.D.; Pena, L.D.M.; Tanpaiboon, P.; Gambello, M.J.; Gibson, J.B.; Hillman, R.; Stockton, D.W.; Day, J.W.; et al. Efficacy, safety profile, and immunogenicity of alglucosidase alfa produced at the 4,000-liter scale in US children and adolescents with Pompe disease: ADVANCE, a phase IV, open-label, prospective study. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Grants Accelerated Approval to First Drug for Duchenne Muscular Dystrophy. 2016. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular-dystrophy (accessed on 25 November 2020).
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- England, S.B.; Nicholson, L.V.; Johnson, M.A.; Forrest, S.M.; Love, D.R.; Zubrzycka-Gaarn, E.E.; Bulman, D.E.; Harris, J.B.; Davies, K.E. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 1990, 343, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; Young, G.D.; Milici, A.J.; Voss, J.; DeAlwis, U.; et al. Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology 2018, 90, e2146–e2154. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; Eteplirsen Study, G.; Telethon Foundation, D.M.D.I.N. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Alfano, L.N.; Charleston, J.S.; Connolly, A.M.; Cripe, L.; Donoghue, C.; Dracker, R.; Dworzak, J.; Eliopoulos, H.; Frank, D.E.; Lewis, S.; et al. Long-term treatment with eteplirsen in nonambulatory patients with Duchenne muscular dystrophy. Medicine 2019, 98, e15858. [Google Scholar] [CrossRef] [PubMed]
- Kinane, T.B.; Mayer, O.H.; Duda, P.W.; Lowes, L.P.; Moody, S.L.; Mendell, J.R. Long-Term Pulmonary Function in Duchenne Muscular Dystrophy: Comparison of Eteplirsen-Treated Patients to Natural History. J. Neuromuscul. Dis. 2018, 5, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Eliopoulos, H.; Han, L.; Kinane, T.B.; Lowes, L.P.; Mendell, J.R.; Gordish-Dressman, H.; Henricson, E.K.; McDonald, C.M.; Eteplirsen, I.; et al. Eteplirsen Treatment Attenuates Respiratory Decline in Ambulatory and Non-Ambulatory Patients with Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2019, 6, 213–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F.; Osorio, A.N.; Tulinius, M.; Buccella, F.; Morgenroth, L.P.; Gordish-Dressman, H.; Jiang, J.; Trifillis, P.; Zhu, J.; et al. Safety and effectiveness of ataluren: Comparison of results from the STRIDE Registry and CINRG DMD Natural History Study. J. Comp. Eff. Res. 2020, 9, 341–360. [Google Scholar] [CrossRef] [Green Version]
- FDA. FDA Approves First Drug for Spinal Muscular Atrophy. 2016. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy (accessed on 25 November 2020).
- EMA. First Medicine for Spinal Muscular Atrophy. 2017. Available online: https://www.ema.europa.eu/en/news/first-medicine-spinal-muscular-atrophy (accessed on 25 November 2020).
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. NMD 2019, 29, 842–856. [Google Scholar] [CrossRef] [Green Version]
- FDA. FDA Approves Oral Treatment for Spinal Muscular Atrophy. 2020. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy (accessed on 25 November 2020).
- Poirier, A.; Weetall, M.; Heinig, K.; Bucheli, F.; Schoenlein, K.; Alsenz, J.; Bassett, S.; Ullah, M.; Senn, C.; Ratni, H.; et al. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol. Res. Perspect. 2018, 6, e00447. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [Green Version]
- Sturm, S.; Gunther, A.; Jaber, B.; Jordan, P.; Al Kotbi, N.; Parkar, N.; Cleary, Y.; Frances, N.; Bergauer, T.; Heinig, K.; et al. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br. J. Clin. Pharmacol. 2019, 85, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudet, D.; Methot, J.; Dery, S.; Brisson, D.; Essiembre, C.; Tremblay, G.; Tremblay, K.; de Wal, J.; Twisk, J.; van den Bulk, N.; et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: An open-label trial. Gene Ther. 2013, 20, 361–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yla-Herttuala, S. Endgame: Glybera finally recommended for approval as the first gene therapy drug in the European union. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1831–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- FDA. FDA Approves Novel Gene Therapy to Treat Patients with a Rare Form of Inherited Vision Loss. 2017. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (accessed on 25 November 2020).
- EMA. New Gene Therapy for Rare Inherited Disorder Causing Vision Loss Recommended for Approval. 2018. Available online: https://www.ema.europa.eu/en/documents/press-release/new-gene-therapy-rare-inherited-disorder-causing-vision-loss-recommended-approval_en.pdf (accessed on 25 November 2020).
- FDA. FDA Approves Innovative Gene Therapy to Treat Pediatric Patients with Spinal Muscular Atrophy, a Rare Disease and Leading Genetic Cause of Infant Mortality. 2019. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease (accessed on 25 November 2020).
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- EMA. New Gene Therapy to Treat Spinal Muscular Atrophy (Corrected). 2020. Available online: https://www.ema.europa.eu/en/news/new-gene-therapy-treat-spinal-muscular-atrophy-corrected (accessed on 25 November 2020).
- Sonntag, F.; Schmidt, K.; Kleinschmidt, J.A. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc. Natl. Acad. Sci. USA 2010, 107, 10220–10225. [Google Scholar] [CrossRef] [Green Version]
- Ogden, P.J.; Kelsic, E.D.; Sinai, S.; Church, G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 2019, 366, 1139–1143. [Google Scholar] [CrossRef]
- Russell, D.W.; Miller, A.D.; Alexander, I.E. Adeno-associated virus vectors preferentially transduce cells in S phase. Proc. Natl. Acad. Sci. USA 1994, 91, 8915–8919. [Google Scholar] [CrossRef] [Green Version]
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–756. [Google Scholar] [CrossRef]
- Hoggan, M.D.; Blacklow, N.R.; Rowe, W.P. Studies of small DNA viruses found in various adenovirus preparations: Physical, biological, and immunological characteristics. Proc. Natl. Acad. Sci. USA 1966, 55, 1467–1474. [Google Scholar] [CrossRef] [Green Version]
- Parks, W.P.; Green, M.; Pina, M.; Melnick, J.L. Physicochemical characterization of adeno-associated satellite virus type 4 and its nucleic acid. J. Virol. 1967, 1, 980–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bantel-Schaal, U.; zur Hausen, H. Characterization of the DNA of a defective human parvovirus isolated from a genital site. Virology 1984, 134, 52–63. [Google Scholar] [CrossRef]
- Rutledge, E.A.; Halbert, C.L.; Russell, D.W. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J. Virol. 1998, 72, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, S.; Wang, L.; Takeuchi, T.; Kanda, T. Two novel adeno-associated viruses from cynomolgus monkey: Pseudotyping characterization of capsid protein. Virology 2004, 330, 375–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Voutetakis, A.; Afione, S.; Zheng, C.; Mandikian, D.; Chiorini, J.A. Adeno-associated virus type 12 (AAV12): A novel AAV serotype with sialic acid- and heparan sulfate proteoglycan-independent transduction activity. J. Virol. 2008, 82, 1399–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Govindasamy, L.; Afione, S.; Kaludov, N.; Agbandje-McKenna, M.; Chiorini, J.A. Molecular characterization of the heparin-dependent transduction domain on the capsid of a novel adeno-associated virus isolate, AAV(VR-942). J. Virol. 2008, 82, 8911–8916. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Vandenberghe, L.H.; Wilson, J.M. New recombinant serotypes of AAV vectors. Curr. Gene Ther. 2005, 5, 285–297. [Google Scholar] [CrossRef]
- Gao, G.; Zhong, L.; Danos, O. Exploiting natural diversity of AAV for the design of vectors with novel properties. Methods Mol. Biol. 2011, 807, 93–118. [Google Scholar] [CrossRef]
- DiMattia, M.A.; Nam, H.J.; Van Vliet, K.; Mitchell, M.; Bennett, A.; Gurda, B.L.; McKenna, R.; Olson, N.H.; Sinkovits, R.S.; Potter, M.; et al. Structural insight into the unique properties of adeno-associated virus serotype 9. J. Virol. 2012, 86, 6947–6958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcedo, R.; Vandenberghe, L.H.; Gao, G.; Lin, J.; Wilson, J.M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 2009, 199, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Calcedo, R.; Wilson, J.M. Humoral Immune Response to AAV. Front. Immunol. 2013, 4, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Summerford, C.; Samulski, R.J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 1998, 72, 1438–1445. [Google Scholar] [CrossRef] [Green Version]
- Mietzsch, M.; Broecker, F.; Reinhardt, A.; Seeberger, P.H.; Heilbronn, R. Differential adeno-associated virus serotype-specific interaction patterns with synthetic heparins and other glycans. J. Virol. 2014, 88, 2991–3003. [Google Scholar] [CrossRef] [Green Version]
- Walters, R.W.; Yi, S.M.; Keshavjee, S.; Brown, K.E.; Welsh, M.J.; Chiorini, J.A.; Zabner, J. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J. Biol. Chem. 2001, 276, 20610–20616. [Google Scholar] [CrossRef] [Green Version]
- Kaludov, N.; Brown, K.E.; Walters, R.W.; Zabner, J.; Chiorini, J.A. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J. Virol. 2001, 75, 6884–6893. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Miller, E.; Agbandje-McKenna, M.; Samulski, R.J. Alpha2,3 and alpha2,6 N-linked sialic acids facilitate efficient binding and transduction by adeno-associated virus types 1 and 6. J. Virol. 2006, 80, 9093–9103. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Bryant, K.D.; Brown, S.M.; Randell, S.H.; Asokan, A. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J. Biol. Chem. 2011, 286, 13532–13540. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.L.; Vandenberghe, L.H.; Bell, P.; Limberis, M.P.; Gao, G.P.; Van Vliet, K.; Agbandje-McKenna, M.; Wilson, J.M. The AAV9 receptor and its modification to improve In Vivo lung gene transfer in mice. J. Clin. Investig. 2011, 121, 2427–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, C.L.; Gurda, B.L.; Van Vliet, K.; Agbandje-McKenna, M.; Wilson, J.M. Identification of the galactose binding domain of the adeno-associated virus serotype 9 capsid. J. Virol. 2012, 86, 7326–7333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pasquale, G.; Davidson, B.L.; Stein, C.S.; Martins, I.; Scudiero, D.; Monks, A.; Chiorini, J.A. Identification of PDGFR as a receptor for AAV-5 transduction. Nat. Med. 2003, 9, 1306–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akache, B.; Grimm, D.; Pandey, K.; Yant, S.R.; Xu, H.; Kay, M.A. The 37/67-kilodalton laminin receptor is a receptor for adeno-associated virus serotypes 8, 2, 3, and 9. J. Virol. 2006, 80, 9831–9836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwakura, Y.; Tamayose, K.; Iwabuchi, K.; Hirai, Y.; Shimada, T.; Matsumoto, K.; Nakamura, T.; Watanabe, M.; Oshimi, K.; Daida, H. Hepatocyte growth factor receptor is a coreceptor for adeno-associated virus type 2 infection. J. Virol. 2005, 79, 609–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summerford, C.; Bartlett, J.S.; Samulski, R.J. AlphaVbeta5 integrin: A co-receptor for adeno-associated virus type 2 infection. Nat. Med. 1999, 5, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Qing, K.; Mah, C.; Hansen, J.; Zhou, S.; Dwarki, V.; Srivastava, A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 1999, 5, 71–77. [Google Scholar] [CrossRef]
- Pillay, S.; Meyer, N.L.; Puschnik, A.S.; Davulcu, O.; Diep, J.; Ishikawa, Y.; Jae, L.T.; Wosen, J.E.; Nagamine, C.M.; Chapman, M.S.; et al. Corrigendum: An essential receptor for adeno-associated virus infection. Nature 2016, 539, 456. [Google Scholar] [CrossRef] [Green Version]
- Pillay, S.; Meyer, N.L.; Puschnik, A.S.; Davulcu, O.; Diep, J.; Ishikawa, Y.; Jae, L.T.; Wosen, J.E.; Nagamine, C.M.; Chapman, M.S.; et al. An essential receptor for adeno-associated virus infection. Nature 2016, 530, 108–112. [Google Scholar] [CrossRef]
- Xiao, P.J.; Samulski, R.J. Cytoplasmic trafficking, endosomal escape, and perinuclear accumulation of adeno-associated virus type 2 particles are facilitated by microtubule network. J. Virol. 2012, 86, 10462–10473. [Google Scholar] [CrossRef] [Green Version]
- Buller, R.M.; Janik, J.E.; Sebring, E.D.; Rose, J.A. Herpes simplex virus types 1 and 2 completely help adenovirus-associated virus replication. J. Virol. 1981, 40, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georg-Fries, B.; Biederlack, S.; Wolf, J.; zur Hausen, H. Analysis of proteins, helper dependence, and seroepidemiology of a new human parvovirus. Virology 1984, 134, 64–71. [Google Scholar] [CrossRef]
- Kotin, R.M.; Menninger, J.C.; Ward, D.C.; Berns, K.I. Mapping and direct visualization of a region-specific viral DNA integration site on chromosome 19q13-qter. Genomics 1991, 10, 831–834. [Google Scholar] [CrossRef]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar] [CrossRef] [Green Version]
- Samulski, R.J.; Zhu, X.; Xiao, X.; Brook, J.D.; Housman, D.E.; Epstein, N.; Hunter, L.A. Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J. 1991, 10, 3941–3950. [Google Scholar] [CrossRef]
- Kotin, R.M.; Linden, R.M.; Berns, K.I. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. 1992, 11, 5071–5078. [Google Scholar] [CrossRef]
- Lamartina, S.; Sporeno, E.; Fattori, E.; Toniatti, C. Characteristics of the adeno-associated virus preintegration site in human chromosome 19: Open chromatin conformation and transcription-competent environment. J. Virol. 2000, 74, 7671–7677. [Google Scholar] [CrossRef] [Green Version]
- Huser, D.; Gogol-Doring, A.; Lutter, T.; Weger, S.; Winter, K.; Hammer, E.M.; Cathomen, T.; Reinert, K.; Heilbronn, R. Integration preferences of wildtype AAV-2 for consensus rep-binding sites at numerous loci in the human genome. PLoS Pathog. 2010, 6, e1000985. [Google Scholar] [CrossRef]
- La Bella, T.; Imbeaud, S.; Peneau, C.; Mami, I.; Datta, S.; Bayard, Q.; Caruso, S.; Hirsch, T.Z.; Calderaro, J.; Morcrette, G.; et al. Adeno-associated virus in the liver: Natural history and consequences in tumour development. Gut 2020, 69, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouze, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef]
- Zhang, H.G.; Wang, Y.M.; Xie, J.F.; Liang, X.; Hsu, H.C.; Zhang, X.; Douglas, J.; Curiel, D.T.; Mountz, J.D. Recombinant adenovirus expressing adeno-associated virus cap and rep proteins supports production of high-titer recombinant adeno-associated virus. Gene Ther. 2001, 8, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.H.; Snyder, R.O.; Schowalter, D.B.; Patijn, G.A.; Donahue, B.; Winther, B.; Kay, M.A. The kinetics of rAAV integration in the liver. Nat. Genet. 1998, 19, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Nakai, H.; Storm, T.A.; Kay, M.A. Recruitment of single-stranded recombinant adeno-associated virus vector genomes and intermolecular recombination are responsible for stable transduction of liver In Vivo. J. Virol. 2000, 74, 9451–9463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, H.; Yant, S.R.; Storm, T.A.; Fuess, S.; Meuse, L.; Kay, M.A. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction In Vivo. J. Virol. 2001, 75, 6969–6976. [Google Scholar] [CrossRef] [Green Version]
- Fisher, K.J.; Jooss, K.; Alston, J.; Yang, Y.; Haecker, S.E.; High, K.; Pathak, R.; Raper, S.E.; Wilson, J.M. Recombinant adeno-associated virus for muscle directed gene therapy. Nat. Med. 1997, 3, 306–312. [Google Scholar] [CrossRef]
- Rutledge, E.A.; Russell, D.W. Adeno-associated virus vector integration junctions. J. Virol. 1997, 71, 8429–8436. [Google Scholar] [CrossRef] [Green Version]
- Nakai, H.; Iwaki, Y.; Kay, M.A.; Couto, L.B. Isolation of recombinant adeno-associated virus vector-cellular DNA junctions from mouse liver. J. Virol. 1999, 73, 5438–5447. [Google Scholar] [CrossRef] [Green Version]
- Nakai, H.; Montini, E.; Fuess, S.; Storm, T.A.; Grompe, M.; Kay, M.A. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat. Genet. 2003, 34, 297–302. [Google Scholar] [CrossRef]
- Miller, D.G.; Trobridge, G.D.; Petek, L.M.; Jacobs, M.A.; Kaul, R.; Russell, D.W. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J. Virol. 2005, 79, 11434–11442. [Google Scholar] [CrossRef] [Green Version]
- Nakai, H.; Wu, X.; Fuess, S.; Storm, T.A.; Munroe, D.; Montini, E.; Burgess, S.M.; Grompe, M.; Kay, M.A. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J. Virol. 2005, 79, 3606–3614. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, K.; Lewis, S.M.; Wu, X.; Ma, C.; Munroe, D.J.; Fuess, S.; Storm, T.A.; Kay, M.A.; Nakai, H. DNA palindromes with a modest arm length of greater, similar 20 base pairs are a significant target for recombinant adeno-associated virus vector integration in the liver, muscles, and heart in mice. J. Virol. 2007, 81, 11290–11303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.G.; Rutledge, E.A.; Russell, D.W. Chromosomal effects of adeno-associated virus vector integration. Nat. Genet. 2002, 30, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Xiao, X.; Zhu, X.; Ansardi, D.C.; Epstein, N.D.; Frey, M.R.; Matera, A.G.; Samulski, R.J. Cellular recombination pathways and viral terminal repeat hairpin structures are sufficient for adeno-associated virus integration In Vivo and in vitro. J. Virol. 1997, 71, 9231–9247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Trivedi, N.S.; Carrillo-Carrasco, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.M.; et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 2015, 125, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donsante, A.; Miller, D.G.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Malani, N.; Hamilton, S.R.; Schlachterman, A.; Bussadori, G.; Edmonson, S.E.; Shah, R.; Arruda, V.R.; Mingozzi, F.; Wright, J.F.; et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood 2011, 117, 3311–3319. [Google Scholar] [CrossRef] [PubMed]
- Kaplitt, M.G.; Leone, P.; Samulski, R.J.; Xiao, X.; Pfaff, D.W.; O’Malley, K.L.; During, M.J. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat. Genet. 1994, 8, 148–154. [Google Scholar] [CrossRef]
- Pruchnic, R.; Cao, B.; Peterson, Z.Q.; Xiao, X.; Li, J.; Samulski, R.J.; Epperly, M.; Huard, J. The use of adeno-associated virus to circumvent the maturation-dependent viral transduction of muscle fibers. Hum. Gene Ther. 2000, 11, 521–536. [Google Scholar] [CrossRef]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef]
- Sanftner, L.M.; Sommer, J.M.; Suzuki, B.M.; Smith, P.H.; Vijay, S.; Vargas, J.A.; Forsayeth, J.R.; Cunningham, J.; Bankiewicz, K.S.; Kao, H.; et al. AAV2-mediated gene delivery to monkey putamen: Evaluation of an infusion device and delivery parameters. Exp. Neurol. 2005, 194, 476–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Chirmule, N.; Berta, S.C.; McCullough, B.; Gao, G.; Wilson, J.M. Gene therapy vectors based on adeno-associated virus type 1. J. Virol. 1999, 73, 3994–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louboutin, J.P.; Wang, L.; Wilson, J.M. Gene transfer into skeletal muscle using novel AAV serotypes. J. Gene Med. 2005, 7, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Raz, Y.; Moloney, E.B.; van Putten, M.; Krom, Y.D.; van der Maarel, S.M.; Verhaagen, J.; Raz, V. Differential myofiber-type transduction preference of adeno-associated virus serotypes 6 and 9. Skelet. Muscle 2015, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, S.; Shin, J.H.; Yuasa, K.; Nishiyama, A.; Kira, J.; Okada, T.; Takeda, S. Transduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscle. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 73–80. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, T.; Qiao, C.; Zhou, L.; Wang, B.; Zhang, J.; Chen, C.; Li, J.; Xiao, X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol. 2005, 23, 321–328. [Google Scholar] [CrossRef]
- Nakai, H.; Fuess, S.; Storm, T.A.; Muramatsu, S.; Nara, Y.; Kay, M.A. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J. Virol. 2005, 79, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, K.; Fuess, S.; Storm, T.A.; Gibson, G.A.; McTiernan, C.F.; Kay, M.A.; Nakai, H. Robust systemic transduction with AAV9 vectors in mice: Efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 14, 45–53. [Google Scholar] [CrossRef]
- Pacak, C.A.; Mah, C.S.; Thattaliyath, B.D.; Conlon, T.J.; Lewis, M.A.; Cloutier, D.E.; Zolotukhin, I.; Tarantal, A.F.; Byrne, B.J. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction In Vivo. Circ. Res. 2006, 99, e3–e9. [Google Scholar] [CrossRef] [Green Version]
- Bostick, B.; Ghosh, A.; Yue, Y.; Long, C.; Duan, D. Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Ther. 2007, 14, 1605–1609. [Google Scholar] [CrossRef]
- Pacak, C.A.; Sakai, Y.; Thattaliyath, B.D.; Mah, C.S.; Byrne, B.J. Tissue specific promoters improve specificity of AAV9 mediated transgene expression following intra-vascular gene delivery in neonatal mice. Genet. Vaccines Ther. 2008, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Yue, Y.; Zhang, K.; Lostal, W.; Shin, J.H.; Duan, D. Long-term robust myocardial transduction of the dog heart from a peripheral vein by adeno-associated virus serotype-8. Hum. Gene Ther. 2013, 24, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, R.; Mucci, M.; Addya, S.; Tetreault, R.; Bellinger, D.A.; Nichols, T.C.; Kazazian, H.H., Jr. Long-term efficacy of adeno-associated virus serotypes 8 and 9 in hemophilia a dogs and mice. Hum. Gene Ther. 2006, 17, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Toromanoff, A.; Cherel, Y.; Guilbaud, M.; Penaud-Budloo, M.; Snyder, R.O.; Haskins, M.E.; Deschamps, J.Y.; Guigand, L.; Podevin, G.; Arruda, V.R.; et al. Safety and Efficacy of Regional Intravenous (RI) Versus Intramuscular (IM) Delivery of rAAV1 and rAAV8 to Nonhuman Primate Skeletal Muscle. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1291–1299. [Google Scholar] [CrossRef]
- Gregorevic, P.; Blankinship, M.J.; Allen, J.M.; Crawford, R.W.; Meuse, L.; Miller, D.G.; Russell, D.W.; Chamberlain, J.S. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004, 10, 828–834. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.D. Functional muscle ischemia in Duchenne and Becker muscular dystrophy. Front. Physiol. 2013, 4, 381. [Google Scholar] [CrossRef] [Green Version]
- Katwal, A.B.; Konkalmatt, P.R.; Piras, B.A.; Hazarika, S.; Li, S.S.; John Lye, R.; Sanders, J.M.; Ferrante, E.A.; Yan, Z.; Annex, B.H.; et al. Adeno-associated virus serotype 9 efficiently targets ischemic skeletal muscle following systemic delivery. Gene Ther. 2013, 20, 930–938. [Google Scholar] [CrossRef]
- Arnett, A.L.; Konieczny, P.; Ramos, J.N.; Hall, J.; Odom, G.; Yablonka-Reuveni, Z.; Chamberlain, J.R.; Chamberlain, J.S. Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol. Ther. Methods Clin. Dev. 2014, 1. [Google Scholar] [CrossRef]
- Jiang, H.; Pierce, G.F.; Ozelo, M.C.; de Paula, E.V.; Vargas, J.A.; Smith, P.; Sommer, J.; Luk, A.; Manno, C.S.; High, K.A.; et al. Evidence of multiyear factor IX expression by AAV-mediated gene transfer to skeletal muscle in an individual with severe hemophilia B. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 14, 452–455. [Google Scholar] [CrossRef]
- Bish, L.T.; Morine, K.; Sleeper, M.M.; Sanmiguel, J.; Wu, D.; Gao, G.; Wilson, J.M.; Sweeney, H.L. Adeno-associated virus (AAV) serotype 9 provides global cardiac gene transfer superior to AAV1, AAV6, AAV7, and AAV8 in the mouse and rat. Hum. Gene Ther. 2008, 19, 1359–1368. [Google Scholar] [CrossRef]
- Vandendriessche, T.; Thorrez, L.; Acosta-Sanchez, A.; Petrus, I.; Wang, L.; Ma, L.; De Waele, L.; Iwasaki, Y.; Gillijns, V.; Wilson, J.M.; et al. Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J. Thromb. Haemost. 2007, 5, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Koch, W.J.; Rabinowitz, J.E. Comparative cardiac gene delivery of adeno-associated virus serotypes 1-9 reveals that AAV6 mediates the most efficient transduction in mouse heart. Clin. Transl. Sci. 2010, 3, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Kaspar, B.K. Over the barrier and through the blood: To CNS delivery we go. Cell Cycle 2009, 8, 4017–4018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, S.J.; Matagne, V.; Bachaboina, L.; Yadav, S.; Ojeda, S.R.; Samulski, R.J. Preclinical differences of intravascular AAV9 delivery to neurons and glia: A comparative study of adult mice and nonhuman primates. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Samaranch, L.; Salegio, E.A.; San Sebastian, W.; Kells, A.P.; Foust, K.D.; Bringas, J.R.; Lamarre, C.; Forsayeth, J.; Kaspar, B.K.; Bankiewicz, K.S. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum. Gene Ther. 2012, 23, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duque, S.; Joussemet, B.; Riviere, C.; Marais, T.; Dubreil, L.; Douar, A.M.; Fyfe, J.; Moullier, P.; Colle, M.A.; Barkats, M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 1187–1196. [Google Scholar] [CrossRef]
- Bevan, A.K.; Duque, S.; Foust, K.D.; Morales, P.R.; Braun, L.; Schmelzer, L.; Chan, C.M.; McCrate, M.; Chicoine, L.G.; Coley, B.D.; et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1971–1980. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Pan, X.; Hakim, C.H.; Kodippili, K.; Zhang, K.; Shin, J.H.; Yang, H.T.; McDonald, T.; Duan, D. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum. Mol. Genet. 2015, 24, 5880–5890. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Pan, X.; Hakim, C.H.; Yang, H.T.; Yue, Y.; Zhang, K.; Terjung, R.L.; Duan, D. Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 750–757. [Google Scholar] [CrossRef] [Green Version]
- Kornegay, J.N.; Li, J.; Bogan, J.R.; Bogan, D.J.; Chen, C.; Zheng, H.; Wang, B.; Qiao, C.; Howard, J.F., Jr.; Xiao, X. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Ghosh, A.; Long, C.; Bostick, B.; Smith, B.F.; Kornegay, J.N.; Duan, D. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Malik, V.; Gomez, A.M.; Flanigan, K.M.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Meadows, E.; Lewis, S.; et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Sahenk, Z.; Al-Zaidy, S.; Rodino-Klapac, L.R.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Miller, N.; Yalvac, M.; Dvorchik, I.; et al. Follistatin Gene Therapy for Sporadic Inclusion Body Myositis Improves Functional Outcomes. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 870–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowles, D.E.; McPhee, S.W.; Li, C.; Gray, S.J.; Samulski, J.J.; Camp, A.S.; Li, J.; Wang, B.; Monahan, P.E.; Rabinowitz, J.E.; et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.K.; Collins, S.W.; Conlon, T.J.; Mah, C.S.; Lawson, L.A.; Martin, A.D.; Fuller, D.D.; Cleaver, B.D.; Clement, N.; Phillips, D.; et al. Phase I/II trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: Initial safety and ventilatory outcomes. Hum. Gene Ther. 2013, 24, 630–640. [Google Scholar] [CrossRef] [Green Version]
- Brooks, A.R.; Harkins, R.N.; Wang, P.; Qian, H.S.; Liu, P.; Rubanyi, G.M. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J. Gene Med. 2004, 6, 395–404. [Google Scholar] [CrossRef]
- Qin, L.; Ding, Y.; Pahud, D.R.; Chang, E.; Imperiale, M.J.; Bromberg, J.S. Promoter attenuation in gene therapy: Interferon-gamma and tumor necrosis factor-alpha inhibit transgene expression. Hum. Gene Ther. 1997, 8, 2019–2029. [Google Scholar] [CrossRef] [Green Version]
- Duan, B.; Cheng, L.; Gao, Y.; Yin, F.X.; Su, G.H.; Shen, Q.Y.; Liu, K.; Hu, X.; Liu, X.; Li, G.P. Silencing of fat-1 transgene expression in sheep may result from hypermethylation of its driven cytomegalovirus (CMV) promoter. Theriogenology 2012, 78, 793–802. [Google Scholar] [CrossRef]
- Harms, J.S.; Splitter, G.A. Interferon-gamma inhibits transgene expression driven by SV40 or CMV promoters but augments expression driven by the mammalian MHC I promoter. Hum. Gene Ther. 1995, 6, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.Y.; Zhang, L.; Clift, K.L.; Hulur, I.; Xiang, A.P.; Ren, B.Z.; Lahn, B.T. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS ONE 2010, 5, e10611. [Google Scholar] [CrossRef] [PubMed]
- Al-Zaidy, S.; Pickard, A.S.; Kotha, K.; Alfano, L.N.; Lowes, L.; Paul, G.; Church, K.; Lehman, K.; Sproule, D.M.; Dabbous, O.; et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr. Pulmonol. 2019, 54, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Al-Zaidy, S.A.; Kolb, S.J.; Lowes, L.; Alfano, L.N.; Shell, R.; Church, K.R.; Nagendran, S.; Sproule, D.M.; Feltner, D.E.; Wells, C.; et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: Comparative Study with a Prospective Natural History Cohort. J. Neuromuscul. Dis. 2019, 6, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabbous, O.; Maru, B.; Jansen, J.P.; Lorenzi, M.; Cloutier, M.; Guerin, A.; Pivneva, I.; Wu, E.Q.; Arjunji, R.; Feltner, D.; et al. Survival, Motor Function, and Motor Milestones: Comparison of AVXS-101 Relative to Nusinersen for the Treatment of Infants with Spinal Muscular Atrophy Type 1. Adv. Ther. 2019, 36, 1164–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of Age and Motor Function in a Phase 1/2A Study of Infants with SMA Type 1 Receiving Single-Dose Gene Replacement Therapy. Pediatr. Neurol. 2019, 98, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Hartigan-O’Connor, D.; Kirk, C.J.; Crawford, R.; Mule, J.J.; Chamberlain, J.S. Immune evasion by muscle-specific gene expression in dystrophic muscle. Mol. Ther. J. Am. Soc. Gene Ther. 2001, 4, 525–533. [Google Scholar] [CrossRef]
- Yuasa, K.; Sakamoto, M.; Miyagoe-Suzuki, Y.; Tanouchi, A.; Yamamoto, H.; Li, J.; Chamberlain, J.S.; Xiao, X.; Takeda, S. Adeno-associated virus vector-mediated gene transfer into dystrophin-deficient skeletal muscles evokes enhanced immune response against the transgene product. Gene Ther. 2002, 9, 1576–1588. [Google Scholar] [CrossRef] [Green Version]
- Weeratna, R.D.; Wu, T.; Efler, S.M.; Zhang, L.; Davis, H.L. Designing gene therapy vectors: Avoiding immune responses by using tissue-specific promoters. Gene Ther. 2001, 8, 1872–1878. [Google Scholar] [CrossRef] [Green Version]
- Fabre, E.E.; Bigey, P.; Orsini, C.; Scherman, D. Comparison of promoter region constructs for In Vivo intramuscular expression. J. Gene Med. 2006, 8, 636–645. [Google Scholar] [CrossRef]
- Talbot, G.E.; Waddington, S.N.; Bales, O.; Tchen, R.C.; Antoniou, M.N. Desmin-regulated lentiviral vectors for skeletal muscle gene transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, R.; Huggins, G.S.; Snyder, R.O. Cardiomyocyte-specific gene expression following recombinant adeno-associated viral vector transduction. J. Biol. Chem. 2002, 277, 18979–18985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childers, M.K.; Joubert, R.; Poulard, K.; Moal, C.; Grange, R.W.; Doering, J.A.; Lawlor, M.W.; Rider, B.E.; Jamet, T.; Daniele, N.; et al. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci. Transl. Med. 2014, 6, 220ra210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, P.B.; Kuntz, N.; Smith, B.; Dowling, J.J.; Müller-Felber, W.; Bönnemann, C.G.; Servais, L.; Muntoni, F.; Blaschek, A.; Neuhaus, S.; et al. ASPIRO Gene Therapy Trial In X-Linked Myotubular Myopathy (XLMTM): Update on Preliminary Safety And Efficacy Findings up to 72 Weeks Post-Treatment (1053). Neurology 2020, 94, 1053. [Google Scholar]
- Amacher, S.L.; Buskin, J.N.; Hauschka, S.D. Multiple regulatory elements contribute differentially to muscle creatine kinase enhancer activity in skeletal and cardiac muscle. Mol. Cell. Biol. 1993, 13, 2753–2764. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.G.; Buskin, J.N.; Himeda, C.L.; Fabre-Suver, C.; Hauschka, S.D. Transgenic and tissue culture analyses of the muscle creatine kinase enhancer Trex control element in skeletal and cardiac muscle indicate differences in gene expression between muscle types. Transgenic Res. 2003, 12, 337–349. [Google Scholar] [CrossRef]
- Jaynes, J.B.; Chamberlain, J.S.; Buskin, J.N.; Johnson, J.E.; Hauschka, S.D. Transcriptional regulation of the muscle creatine kinase gene and regulated expression in transfected mouse myoblasts. Mol. Cell. Biol. 1986, 6, 2855–2864. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, F.; Takase, K.; Tozuka, M.; Saha, S.; Okuda, K.; Ishii, N.; Sasaki, S. Muscle creatine kinase/SV40 hybrid promoter for muscle-targeted long-term transgene expression. Int. J. Mol. Med. 2007, 19, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Hauser, M.A.; Robinson, A.; Hartigan-O’Connor, D.; Williams-Gregory, D.A.; Buskin, J.N.; Apone, S.; Kirk, C.J.; Hardy, S.; Hauschka, S.D.; Chamberlain, J.S. Analysis of muscle creatine kinase regulatory elements in recombinant adenoviral vectors. Mol. Ther. J. Am. Soc. Gene Ther. 2000, 2, 16–25. [Google Scholar] [CrossRef]
- Wang, B.; Li, J.; Fu, F.H.; Chen, C.; Zhu, X.; Zhou, L.; Jiang, X.; Xiao, X. Construction and analysis of compact muscle-specific promoters for AAV vectors. Gene Ther. 2008, 15, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Sahenk, Z.; Galloway, G.; Clark, K.R.; Malik, V.; Rodino-Klapac, L.R.; Kaspar, B.K.; Chen, L.; Braganza, C.; Montgomery, C.; Mendell, J.R. AAV1.NT-3 gene therapy for charcot-marie-tooth neuropathy. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Rodino-Klapac, L.R.; Lee, J.S.; Mulligan, R.C.; Clark, K.R.; Mendell, J.R. Lack of toxicity of alpha-sarcoglycan overexpression supports clinical gene transfer trial in LGMD2D. Neurology 2008, 71, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Rosales, X.Q.; Coley, B.D.; Galloway, G.; Lewis, S.; Malik, V.; Shilling, C.; Byrne, B.J.; Conlon, T.; et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann. Neurol. 2010, 68, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Rodino-Klapac, L.R.; Rosales-Quintero, X.; Kota, J.; Coley, B.D.; Galloway, G.; Craenen, J.M.; Lewis, S.; Malik, V.; Shilling, C.; et al. Limb-girdle muscular dystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteins. Ann. Neurol. 2009, 66, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Salva, M.Z.; Himeda, C.L.; Tai, P.W.; Nishiuchi, E.; Gregorevic, P.; Allen, J.M.; Finn, E.E.; Nguyen, Q.G.; Blankinship, M.J.; Meuse, L.; et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Young, S.P.; Li, P.; Di, C.; Brown, T.; Salva, M.Z.; Li, S.; Bird, A.; Yan, Z.; Auten, R.; et al. Correction of multiple striated muscles in murine Pompe disease through adeno-associated virus-mediated gene therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1366–1371. [Google Scholar] [CrossRef]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020. [Google Scholar] [CrossRef]
- Li, X.; Eastman, E.M.; Schwartz, R.J.; Draghia-Akli, R. Synthetic muscle promoters: Activities exceeding naturally occurring regulatory sequences. Nat. Biotechnol. 1999, 17, 241–245. [Google Scholar] [CrossRef]
- Cordier, L.; Gao, G.P.; Hack, A.A.; McNally, E.M.; Wilson, J.M.; Chirmule, N.; Sweeney, H.L. Muscle-specific promoters may be necessary for adeno-associated virus-mediated gene transfer in the treatment of muscular dystrophies. Hum. Gene Ther. 2001, 12, 205–215. [Google Scholar] [CrossRef]
- Fougerousse, F.; Bartoli, M.; Poupiot, J.; Arandel, L.; Durand, M.; Guerchet, N.; Gicquel, E.; Danos, O.; Richard, I. Phenotypic correction of alpha-sarcoglycan deficiency by intra-arterial injection of a muscle-specific serotype 1 rAAV vector. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 53–61. [Google Scholar] [CrossRef]
- Ziegler, R.J.; Lonning, S.M.; Armentano, D.; Li, C.; Souza, D.W.; Cherry, M.; Ford, C.; Barbon, C.M.; Desnick, R.J.; Gao, G.; et al. AAV2 vector harboring a liver-restricted promoter facilitates sustained expression of therapeutic levels of alpha-galactosidase A and the induction of immune tolerance in Fabry mice. Mol. Ther. J. Am. Soc. Gene Ther. 2004, 9, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Poupiot, J.; Costa Verdera, H.; Hardet, R.; Colella, P.; Collaud, F.; Bartolo, L.; Davoust, J.; Sanatine, P.; Mingozzi, F.; Richard, I.; et al. Role of Regulatory T Cell and Effector T Cell Exhaustion in Liver-Mediated Transgene Tolerance in Muscle. Mol. Ther. Methods Clin. Dev. 2019, 15, 83–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolo, L.; Li Chung Tong, S.; Chappert, P.; Urbain, D.; Collaud, F.; Colella, P.; Richard, I.; Ronzitti, G.; Demengeot, J.; Gross, D.A.; et al. Dual muscle-liver transduction imposes immune tolerance for muscle transgene engraftment despite preexisting immunity. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colella, P.; Sellier, P.; Costa Verdera, H.; Puzzo, F.; van Wittenberghe, L.; Guerchet, N.; Daniele, N.; Gjata, B.; Marmier, S.; Charles, S.; et al. AAV Gene Transfer with Tandem Promoter Design Prevents Anti-transgene Immunity and Provides Persistent Efficacy in Neonate Pompe Mice. Mol. Ther. Methods Clin. Dev. 2019, 12, 85–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GlobeNewswire. Solid Biosciences Provides Update Regarding SGT-001 Phase I/II Clinical Hold on IGNITE DMD. 2020. Available online: https://www.globenewswire.com/news-release/2020/05/07/2029328/0/en/Solid-Biosciences-Provides-Update-regarding-SGT-001-Phase-I-II-Clinical-Hold-on-IGNITE-DMD.html (accessed on 25 November 2020).
- GlobeNewswire. Solid Biosciences Announces Clinical Hold On SGT-001 Phase I/II Clinical Trial for Duchenne Muscular Dystrophy. 2018. Available online: https://www.globenewswire.com/news-release/2018/03/14/1422770/0/en/Solid-Biosciences-Announces-Clinical-Hold-On-SGT-001-Phase-I-II-Clinical-Trial-For-Duchenne-Muscular-Dystrophy.html (accessed on 25 November 2020).
- Binks, M. Early, Initial Data from C3391001, a First-In-Human Safety Study of PF-06939926, a Mini-Dystrophin Gene Therapy for the Potential Treatment of DMD; Parent Project Muscular Dystrophy: Hackensack, NJ, USA, 2019. [Google Scholar]
- Novartis. AveXis Presents AVXS-101 IT Data Demonstrating Remarkable Increases in HFMSE Scores and a Consistent Clinically Meaningful Response in Older Patients with SMA Type 2. 2020. Available online: https://www.novartis.com/news/media-releases/avexis-presents-avxs-101-it-data-demonstrating-remarkable-increases-hfmse-scores-and-consistent-clinically-meaningful-response-older-patients-sma-type-2 (accessed on 25 November 2020).
- Pharma, F. Audentes’ Gene Therapy AT132 Hit with FDA Clinical Hold after Second Patient Death. 2020. Available online: https://www.firstwordpharma.com/node/1736153 (accessed on 25 November 2020).
- Rodino-klapac, L.R.; Pozsgai, E.R.; Lewis, S.; Griffin, D.A.; Meadows, A.S.; Lehman, K.; Church, K.; Lowes, L.; Mendel, J.R. Systemic Gene Transfer with AAVrh74.MHCK7.SGCB Increased β-sarcoglycan Expression in Patients with Limb Girdle Muscular Dystrophy Type 2E. Neuropediatrics 2019, 50, S1–S55. [Google Scholar]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijngaarde, C.A.; Blank, A.C.; Stam, M.; Wadman, R.I.; van den Berg, L.H.; van der Pol, W.L. Cardiac pathology in spinal muscular atrophy: A systematic review. Orphanet J. Rare Dis. 2017, 12, 67. [Google Scholar] [CrossRef]
- Meyer, K.; Ferraiuolo, L.; Schmelzer, L.; Braun, L.; McGovern, V.; Likhite, S.; Michels, O.; Govoni, A.; Fitzgerald, J.; Morales, P.; et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: A dose-response study in mice and nonhuman primates. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.; et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Bevan, A.K.; Hutchinson, K.R.; Foust, K.D.; Braun, L.; McGovern, V.L.; Schmelzer, L.; Ward, J.G.; Petruska, J.C.; Lucchesi, P.A.; Burghes, A.H.; et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet. 2010, 19, 3895–3905. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, E.; Marais, T.; Chatauret, N.; Benkhelifa-Ziyyat, S.; Duque, S.; Ravassard, P.; Carcenac, R.; Astord, S.; Pereira de Moura, A.; Voit, T.; et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011, 20, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valori, C.F.; Ning, K.; Wyles, M.; Mead, R.J.; Grierson, A.J.; Shaw, P.J.; Azzouz, M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010, 2, 35ra42. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.M. Self-complementary AAV vectors; advances and applications. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Hordeaux, J.; Wang, Q.; Katz, N.; Buza, E.L.; Bell, P.; Wilson, J.M. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 664–668. [Google Scholar] [CrossRef]
- Assinger, A. Platelets and infection—An emerging role of platelets in viral infection. Front. Immunol. 2014, 5, 649. [Google Scholar] [CrossRef] [Green Version]
- Harper, S.Q.; Hauser, M.A.; DelloRusso, C.; Duan, D.; Crawford, R.W.; Phelps, S.F.; Harper, H.A.; Robinson, A.S.; Engelhardt, J.F.; Brooks, S.V.; et al. Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002, 8, 253–261. [Google Scholar] [CrossRef]
- Le Guiner, C.; Montus, M.; Servais, L.; Cherel, Y.; Francois, V.; Thibaud, J.L.; Wary, C.; Matot, B.; Larcher, T.; Guigand, L.; et al. Forelimb treatment in a large cohort of dystrophic dogs supports delivery of a recombinant AAV for exon skipping in Duchenne patients. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1923–1935. [Google Scholar] [CrossRef] [Green Version]
- Goyenvalle, A.; Babbs, A.; Wright, J.; Wilkins, V.; Powell, D.; Garcia, L.; Davies, K.E. Rescue of severely affected dystrophin/utrophin-deficient mice through scAAV-U7snRNA-mediated exon skipping. Hum. Mol. Genet. 2012, 21, 2559–2571. [Google Scholar] [CrossRef] [Green Version]
- Goyenvalle, A.; Vulin, A.; Fougerousse, F.; Leturcq, F.; Kaplan, J.C.; Garcia, L.; Danos, O. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science 2004, 306, 1796–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulin, A.; Barthelemy, I.; Goyenvalle, A.; Thibaud, J.L.; Beley, C.; Griffith, G.; Benchaouir, R.; le Hir, M.; Unterfinger, Y.; Lorain, S.; et al. Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 2120–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bish, L.T.; Sleeper, M.M.; Forbes, S.C.; Wang, B.; Reynolds, C.; Singletary, G.E.; Trafny, D.; Morine, K.J.; Sanmiguel, J.; Cecchini, S.; et al. Long-term restoration of cardiac dystrophin expression in golden retriever muscular dystrophy following rAAV6-mediated exon skipping. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 580–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbash, I.M.; Cecchini, S.; Faranesh, A.Z.; Virag, T.; Li, L.; Yang, Y.; Hoyt, R.F.; Kornegay, J.N.; Bogan, J.R.; Garcia, L.; et al. MRI roadmap-guided transendocardial delivery of exon-skipping recombinant adeno-associated virus restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Gene Ther. 2013, 20, 274–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorevic, P.; Allen, J.M.; Minami, E.; Blankinship, M.J.; Haraguchi, M.; Meuse, L.; Finn, E.; Adams, M.E.; Froehner, S.C.; Murry, C.E.; et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006, 12, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Blankinship, M.J.; Allen, J.M.; Chamberlain, J.S. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, B.D.; Bishaw, Y.; Kagel, D.; Ramos, J.N.; Maricelli, J.W. Micro-dystrophin Gene Therapy Partially Enhances Exercise Capacity in Older Adult mdx Mice. Mol. Ther. Methods Clin. Dev. 2020, 17, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodino-Klapac, L.R.; Janssen, P.M.; Montgomery, C.L.; Coley, B.D.; Chicoine, L.G.; Clark, K.R.; Mendell, J.R. A translational approach for limb vascular delivery of the micro-dystrophin gene without high volume or high pressure for treatment of Duchenne muscular dystrophy. J. Transl. Med. 2007, 5, 45. [Google Scholar] [CrossRef] [Green Version]
- Rodino-Klapac, L.R.; Montgomery, C.L.; Bremer, W.G.; Shontz, K.M.; Malik, V.; Davis, N.; Sprinkle, S.; Campbell, K.J.; Sahenk, Z.; Clark, K.R.; et al. Persistent expression of FLAG-tagged micro dystrophin in nonhuman primates following intramuscular and vascular delivery. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 109–117. [Google Scholar] [CrossRef]
- Foster, H.; Sharp, P.S.; Athanasopoulos, T.; Trollet, C.; Graham, I.R.; Foster, K.; Wells, D.J.; Dickson, G. Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 gene transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Le Guiner, C.; Servais, L.; Montus, M.; Larcher, T.; Fraysse, B.; Moullec, S.; Allais, M.; Francois, V.; Dutilleul, M.; Malerba, A.; et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 16105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostick, B.; Yue, Y.; Lai, Y.; Long, C.; Li, D.; Duan, D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum. Gene Ther. 2008, 19, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Hakim, C.H.; Wasala, N.B.; Pan, X.; Kodippili, K.; Yue, Y.; Zhang, K.; Yao, G.; Haffner, B.; Duan, S.X.; Ramos, J.; et al. A Five-Repeat Micro-Dystrophin Gene Ameliorated Dystrophic Phenotype in the Severe DBA/2J-mdx Model of Duchenne Muscular Dystrophy. Mol. Ther. Methods Clin. Dev. 2017, 6, 216–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostick, B.; Shin, J.H.; Yue, Y.; Duan, D. AAV-microdystrophin therapy improves cardiac performance in aged female mdx mice. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Ohshima-Hosoyama, S.; Kinoshita, K.; Chiyo, T.; Okada, H.; Okada, T.; Takeda, S. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther. 2011, 18, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Hakim, C.H.; Clement, N.; Wasala, L.P.; Yang, H.T.; Yue, Y.; Zhang, K.; Kodippili, K.; Adamson-Small, L.; Pan, X.; Schneider, J.S.; et al. Micro-dystrophin AAV Vectors Made by Transient Transfection and Herpesvirus System Are Equally Potent in Treating mdx Mouse Muscle Disease. Mol. Ther. Methods Clin. Dev. 2020, 18, 664–678. [Google Scholar] [CrossRef]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.T.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L.; et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Investig. 2009, 119, 624–635. [Google Scholar] [CrossRef] [Green Version]
- Griffin, D.A.; Potter, R.A.; Pozsgai, E.R.; Peterson, E.L.; Rodino-Klapac, L.R. Adeno-Associated Virus Serotype rh74 Prevalence in Muscular Dystrophy Population. Mol. Ther. 2019, 27 (Suppl. 1), 342. [Google Scholar]
- Zygmunt, D.A.; Crowe, K.E.; Flanigan, K.M.; Martin, P.T. Comparison of Serum rAAV Serotype-Specific Antibodies in Patients with Duchenne Muscular Dystrophy, Becker Muscular Dystrophy, Inclusion Body Myositis, or GNE Myopathy. Hum. Gene Ther. 2017, 28, 737–746. [Google Scholar] [CrossRef]
- Fu, H.; Meadows, A.S.; Pineda, R.J.; Kunkler, K.L.; Truxal, K.V.; McBride, K.L.; Flanigan, K.M.; McCarty, D.M. Differential Prevalence of Antibodies Against Adeno-Associated Virus in Healthy Children and Patients with Mucopolysaccharidosis III: Perspective for AAV-Mediated Gene Therapy. Hum. Gene Ther. Clin. Dev. 2017, 28, 187–196. [Google Scholar] [CrossRef]
- Potter, R.A.; Griffin, D.A.; Heller, K.N.; Peterson, E.L.; Clark, E.K.; Mendell, J.R.; Rodino-Klapac, L.R. Dose-escalation study of systematically delivered rAAVrh74.MHCK7.micro-dystrophin in the mdx mouse model of Duchenne muscular dystrophy. In Proceedings of the American Society of Gene and Cell Therapy Annual Meeting, Chicago, IL, USA, 16–19 May 2018. [Google Scholar]
- Potter, R.A.; Griffin, D.A.; Heller, K.N.; Peterson, E.L.; Johnson, R.W.; Clark, E.K.; Mendell, J.R.; Rodino-Klapac, L.R. Dose Escalation Study of Systemically Delivered AAVrh74.MHCK7.Micro-Dystrophin in the Mdx Mouse Model of DMD. Mol. Ther. 2018, 26 (Suppl. 1), 5. [Google Scholar]
- Mendell, J.R.; Chicoine, L.G.; Al-Zaidy, S.A.; Sahenk, Z.; Lehman, K.; Lowes, L.; Miller, N.; Alfano, L.; Galliers, B.; Lewis, S.; et al. Gene Delivery for Limb-Girdle Muscular Dystrophy Type 2D by Isolated Limb Infusion. Hum. Gene Ther. 2019, 30, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Asher, D.R.; Thapa, K.; Dharia, S.D.; Khan, N.; Potter, R.A.; Rodino-Klapac, L.R.; Mendell, J.R. Clinical development on the frontier: Gene therapy for duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2020, 20, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.J.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef]
- Solid Biosciences. Letter to the Duchenne Community about the Status of the IGNITE DMD Clinical Trial. 2018. Available online: https://www.solidbio.com/about/media/news/letter-to-the-duchenne-community-about-the-status-of-the-ignite-dmd-clinical-trial (accessed on 25 November 2020).
- Solid Biosciences. Solid Biosciences Announces FDA Lifts Clinical Hold on IGNITE DMD Clinical Trial. 2020. Available online: https://www.solidbio.com/about/media/press-releases/solid-biosciences-announces-fda-lifts-clinical-hold-on-ignite-dmd-clinical-trial (accessed on 25 November 2020).
- Hakim, C.H.; Kodippili, K.; Jenkins, G.; Yang, H.; Pan, X.; Lessa, T.; Leach, S.; Emter, C.; Yue, Y.; Zhang, K.; et al. AAV micro-dystrophin therapy ameliorates muscular dystrophy in young adult Duchenne muscular dystrophy dogs for up to thirty months following injection. Mol. Ther. 2018, 26 (Suppl. 1), 5. [Google Scholar]
- Simon, A.K.; Hollander, G.A.; McMichael, A. Evolution of the immune system in humans from infancy to old age. Proc. Biol. Sci. 2015, 282, 20143085. [Google Scholar] [CrossRef]
- Laporte, J.; Hu, L.J.; Kretz, C.; Mandel, J.L.; Kioschis, P.; Coy, J.F.; Klauck, S.M.; Poustka, A.; Dahl, N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 1996, 13, 175–182. [Google Scholar] [CrossRef]
- Blondeau, F.; Laporte, J.; Bodin, S.; Superti-Furga, G.; Payrastre, B.; Mandel, J.L. Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum. Mol. Genet. 2000, 9, 2223–2229. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.S.; Maehama, T.; Dixon, J.E. Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc. Natl. Acad. Sci. USA 2000, 97, 8910–8915. [Google Scholar] [CrossRef] [Green Version]
- Buj-Bello, A.; Laugel, V.; Messaddeq, N.; Zahreddine, H.; Laporte, J.; Pellissier, J.F.; Mandel, J.L. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 15060–15065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beggs, A.H.; Bohm, J.; Snead, E.; Kozlowski, M.; Maurer, M.; Minor, K.; Childers, M.K.; Taylor, S.M.; Hitte, C.; Mickelson, J.R.; et al. MTM1 mutation associated with X-linked myotubular myopathy in Labrador Retrievers. Proc. Natl. Acad. Sci. USA 2010, 107, 14697–14702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grange, R.W.; Doering, J.; Mitchell, E.; Holder, M.N.; Guan, X.; Goddard, M.; Tegeler, C.; Beggs, A.H.; Childers, M.K. Muscle function in a canine model of X-linked myotubular myopathy. Muscle Nerve 2012, 46, 588–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buj-Bello, A.; Fougerousse, F.; Schwab, Y.; Messaddeq, N.; Spehner, D.; Pierson, C.R.; Durand, M.; Kretz, C.; Danos, O.; Douar, A.M.; et al. AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum. Mol. Genet. 2008, 17, 2132–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elverman, M.; Goddard, M.A.; Mack, D.; Snyder, J.M.; Lawlor, M.W.; Meng, H.; Beggs, A.H.; Buj-Bello, A.; Poulard, K.; Marsh, A.P.; et al. Long-term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve 2017, 56, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Mack, D.L.; Poulard, K.; Goddard, M.A.; Latournerie, V.; Snyder, J.M.; Grange, R.W.; Elverman, M.R.; Denard, J.; Veron, P.; Buscara, L.; et al. Systemic AAV8-Mediated Gene Therapy Drives Whole-Body Correction of Myotubular Myopathy in Dogs. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 839–854. [Google Scholar] [CrossRef] [Green Version]
- Dupont, J.B.; Guo, J.; Renaud-Gabardos, E.; Poulard, K.; Latournerie, V.; Lawlor, M.W.; Grange, R.W.; Gray, J.T.; Buj-Bello, A.; Childers, M.K.; et al. AAV-Mediated Gene Transfer Restores a Normal Muscle Transcriptome in a Canine Model of X-Linked Myotubular Myopathy. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 382–393. [Google Scholar] [CrossRef]
- Phillips, A.; Belle, A.; Guo, J.; Ton, J.; Stinchcombe, T.; Buj-Bello, A.; Gray, J.T. Nonhuman Primate Safety and Potency of an AAV Vector for XLMTM Produced by Transient Transfection at 500L. Mol. Ther. 2017, 25 (Suppl. 1), 102–103. [Google Scholar]
- Kuntz, N.; Shieh, P.B.; Smith, B.; Bonnemann, C.G.; Dowling, J.J.; Lawlor, M.W.; Müller-Felber, W.; Noursalehi, M.; Rico, S.; Servais, L.; et al. ASPIRO phase 1/2 gene therapy trial In X-linked myotubular myopathy: Preliminary safety and efficacy findings. Neuromuscul. Disord. 2018, 28 (Suppl. 2), S91. [Google Scholar] [CrossRef]
- Kuntz, N.; Shieh, P.B.; Smith, B.; Bonnemann, C.G.; Dowling, J.J.; Lawlor, M.W.; Müller-Felber, W.; Noursalehi, M.; Rico, S.; Servais, L.; et al. ASPIRO phase 1/2 gene therapy trail in X-linked myotubular myopathy (XLMTM): Preliminary safety and efficacy findings. Mol. Ther. 2018, 26 (Suppl. 1), 4. [Google Scholar]
- Kuntz, N.; Shie, P.B.; Smith, B.; Bonnemann, C.G.; Dowling, J.J.; Lawlor, M.W.; Mavilio, F.; Muller-Felber, W.; Noursalehi, M.; Rico, S.; et al. Gene therapy for X-linked myotubular myopathy with AT132 (rAAV8-Des-hMTM1): Preliminary results from the ASPIRO phase-1/2 study. Hum. Gene Ther. 2018, 27, A2–A169. [Google Scholar]
- Somanathan, S.; Calcedo, R.; Wilson, J.M. Adenovirus-Antibody Complexes Contributed to Lethal Systemic Inflammation in a Gene Therapy Trial. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Flotte, T.R. Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum. Gene Ther. 2020, 31, 695–696. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Ortiz, J.; Ojala, D.S.; Westesson, O.; Weinstein, J.R.; Wong, S.Y.; Steinsapir, A.; Kumar, S.; Holmes, I.; Schaffer, D.V. AAV ancestral reconstruction library enables selection of broadly infectious viral variants. Gene Ther. 2015, 22, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Asokan, A.; Conway, J.C.; Phillips, J.L.; Li, C.; Hegge, J.; Sinnott, R.; Yadav, S.; DiPrimio, N.; Nam, H.J.; Agbandje-McKenna, M.; et al. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat. Biotechnol. 2010, 28, 79–82. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.Y.; Yuan, Z.; Cao, Z.; Wang, B.; Qiao, C.; Li, J.; Xiao, X. A muscle-targeting peptide displayed on AAV2 improves muscle tropism on systemic delivery. Gene Ther. 2009, 16, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Ying, Y.; Muller, O.J.; Goehringer, C.; Leuchs, B.; Trepel, M.; Katus, H.A.; Kleinschmidt, J.A. Heart-targeted adeno-associated viral vectors selected by In Vivo biopanning of a random viral display peptide library. Gene Ther. 2010, 17, 980–990. [Google Scholar] [CrossRef]
- Shen, S.; Horowitz, E.D.; Troupes, A.N.; Brown, S.M.; Pulicherla, N.; Samulski, R.J.; Agbandje-McKenna, M.; Asokan, A. Engraftment of a galactose receptor footprint onto adeno-associated viral capsids improves transduction efficiency. J. Biol. Chem. 2013, 288, 28814–28823. [Google Scholar] [CrossRef] [Green Version]
- Tarantal, A.F.; Lee, C.C.I.; Martinez, M.L.; Asokan, A.; Samulski, R.J. Systemic and Persistent Muscle Gene Expression in Rhesus Monkeys with a Liver De-Targeted Adeno-Associated Virus Vector. Hum. Gene Ther. 2017, 28, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Pulicherla, N.; Shen, S.; Yadav, S.; Debbink, K.; Govindasamy, L.; Agbandje-McKenna, M.; Asokan, A. Engineering liver-detargeted AAV9 vectors for cardiac and musculoskeletal gene transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1070–1078. [Google Scholar] [CrossRef] [Green Version]
- Tulalamba, W.; Weinmann, J.; Pham, Q.H.; El Andari, J.; VandenDriessche, T.; Chuah, M.K.; Grimm, D. Distinct transduction of muscle tissue in mice after systemic delivery of AAVpo1 vectors. Gene Ther. 2020, 27, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Fitzpatrick, Z.; Harris, A.F.; Maitland, S.A.; Ferreira, J.S.; Zhang, Y.; Ma, S.; Sharma, R.B.; Gray-Edwards, H.L.; Johnson, J.A.; et al. In Vivo Selection Yields AAV-B1 Capsid for Central Nervous System and Muscle Gene Therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1247–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakim, C.H.; Yue, Y.; Shin, J.H.; Williams, R.R.; Zhang, K.; Smith, B.F.; Duan, D. Systemic gene transfer reveals distinctive muscle transduction profile of tyrosine mutant AAV-1, -6, and -9 in neonatal dogs. Mol. Ther. Methods Clin. Dev. 2014, 1, 14002. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, J.; Xiao, X. Directed evolution of adeno-associated virus (AAV) as vector for muscle gene therapy. Methods Mol. Biol. 2011, 709, 127–139. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, J.; Drouin, L.M.; Agbandje-McKenna, M.; Chen, C.; Qiao, C.; Pu, D.; Hu, X.; Wang, D.Z.; Li, J.; et al. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and In Vivo selection. Proc. Natl. Acad. Sci. USA 2009, 106, 3946–3951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, C.; Zhang, W.; Yuan, Z.; Shin, J.H.; Li, J.; Jayandharan, G.R.; Zhong, L.; Srivastava, A.; Xiao, X.; Duan, D. Adeno-associated virus serotype 6 capsid tyrosine-to-phenylalanine mutations improve gene transfer to skeletal muscle. Hum. Gene Ther. 2010, 21, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Liu, Y.; Shi, Z.; Liu, H.; Wei, Y.; Yang, L. Bat adeno-associated viruses as gene therapy vectors with the potential to evade human neutralizing antibodies. Gene Ther. 2019, 26, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Maheshri, N.; Koerber, J.T.; Kaspar, B.K.; Schaffer, D.V. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat. Biotechnol. 2006, 24, 198–204. [Google Scholar] [CrossRef]
- Tse, L.V.; Klinc, K.A.; Madigan, V.J.; Castellanos Rivera, R.M.; Wells, L.F.; Havlik, L.P.; Smith, J.K.; Agbandje-McKenna, M.; Asokan, A. Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc. Natl. Acad. Sci. USA 2017, 114, E4812–E4821. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wu, S.; Albright, B.; Hirsch, M.; Li, W.; Tseng, Y.S.; Agbandje-McKenna, M.; McPhee, S.; Asokan, A.; Samulski, R.J. Development of Patient-specific AAV Vectors After Neutralizing Antibody Selection for Enhanced Muscle Gene Transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Sarcar, S.; Tulalamba, W.; Rincon, M.Y.; Tipanee, J.; Pham, H.Q.; Evens, H.; Boon, D.; Samara-Kuko, E.; Keyaerts, M.; Loperfido, M.; et al. Next-generation muscle-directed gene therapy by in silico vector design. Nat. Commun. 2019, 10, 492. [Google Scholar] [CrossRef] [PubMed]
- Piekarowicz, K.; Bertrand, A.T.; Azibani, F.; Beuvin, M.; Julien, L.; Machowska, M.; Bonne, G.; Rzepecki, R. A Muscle Hybrid Promoter as a Novel Tool for Gene Therapy. Mol. Ther. Methods Clin. Dev. 2019, 15, 157–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porro, F.; Bortolussi, G.; Barzel, A.; De Caneva, A.; Iaconcig, A.; Vodret, S.; Zentilin, L.; Kay, M.A.; Muro, A.F. Promoterless gene targeting without nucleases rescues lethality of a Crigler-Najjar syndrome mouse model. EMBO Mol. Med. 2017, 9, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Barzel, A.; Paulk, N.K.; Shi, Y.; Huang, Y.; Chu, K.; Zhang, F.; Valdmanis, P.N.; Spector, L.P.; Porteus, M.H.; Gaensler, K.M.; et al. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature 2015, 517, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Powell, S.K.; Rivera-Soto, R.; Gray, S.J. Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov. Med. 2015, 19, 49–57. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Lee, E.J.; Baek, M.; Gusev, Y.; Brackett, D.J.; Nuovo, G.J.; Schmittgen, T.D. Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. RNA 2008, 14, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Vechetti, I.J., Jr.; Wen, Y.; Chaillou, T.; Murach, K.A.; Alimov, A.P.; Figueiredo, V.C.; Dal-Pai-Silva, M.; McCarthy, J.J. Life-long reduction in myomiR expression does not adversely affect skeletal muscle morphology. Sci. Rep. 2019, 9, 5483. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.D.; Gentner, B.; Cantore, A.; Colleoni, S.; Amendola, M.; Zingale, A.; Baccarini, A.; Lazzari, G.; Galli, C.; Naldini, L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 2007, 25, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Roudaut, C.; Le Roy, F.; Suel, L.; Poupiot, J.; Charton, K.; Bartoli, M.; Richard, I. Restriction of calpain3 expression to the skeletal muscle prevents cardiac toxicity and corrects pathology in a murine model of limb-girdle muscular dystrophy. Circulation 2013, 128, 1094–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, A.; Jungmann, A.; Kurreck, J.; Poller, W.; Katus, H.A.; Vetter, R.; Fechner, H.; Muller, O.J. microRNA122-regulated transgene expression increases specificity of cardiac gene transfer upon intravenous delivery of AAV9 vectors. Gene Ther. 2011, 18, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Yuan, Z.; Li, J.; He, B.; Zheng, H.; Mayer, C.; Li, J.; Xiao, X. Liver-specific microRNA-122 target sequences incorporated in AAV vectors efficiently inhibits transgene expression in the liver. Gene Ther. 2011, 18, 403–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, B.D.; Venneri, M.A.; Zingale, A.; Sergi Sergi, L.; Naldini, L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat. Med. 2006, 12, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Cantore, A.; Annoni, A.; Sergi, L.S.; Lombardo, A.; Della Valle, P.; D’Angelo, A.; Naldini, L. A microRNA-regulated lentiviral vector mediates stable correction of hemophilia B mice. Blood 2007, 110, 4144–4152. [Google Scholar] [CrossRef] [Green Version]
- Majowicz, A.; Maczuga, P.; Kwikkers, K.L.; van der Marel, S.; van Logtenstein, R.; Petry, H.; van Deventer, S.J.; Konstantinova, P.; Ferreira, V. Mir-142-3p target sequences reduce transgene-directed immunogenicity following intramuscular adeno-associated virus 1 vector-mediated gene delivery. J. Gene Med. 2013, 15, 219–232. [Google Scholar] [CrossRef]
- Carpentier, M.; Lorain, S.; Chappert, P.; Lalfer, M.; Hardet, R.; Urbain, D.; Peccate, C.; Adriouch, S.; Garcia, L.; Davoust, J.; et al. Intrinsic Transgene Immunogenicity Gears CD8(+) T-cell Priming After rAAV-Mediated Muscle Gene Transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Boisgerault, F.; Gross, D.A.; Ferrand, M.; Poupiot, J.; Darocha, S.; Richard, I.; Galy, A. Prolonged gene expression in muscle is achieved without active immune tolerance using microrRNA 142.3p-regulated rAAV gene transfer. Hum. Gene Ther. 2013, 24, 393–405. [Google Scholar] [CrossRef]
- Belbellaa, B.; Reutenauer, L.; Messaddeq, N.; Monassier, L.; Puccio, H. High levels of frataxin overexpression leads to mitochondrial and cardiac toxicity in mouse model. Mol. Ther. Methods Clin. Dev. 2020, 19, 120–138. [Google Scholar] [CrossRef]
- Kraszewska, I.; Tomczyk, M.; Andrysiak, K.; Biniecka, M.; Geisler, A.; Fechner, H.; Zembala, M.; Stepniewski, J.; Dulak, J.; Jazwa-Kusior, A. Variability in Cardiac miRNA-122 Level Determines Therapeutic Potential of miRNA-Regulated AAV Vectors. Mol. Ther. Methods Clin. Dev. 2020, 17, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhang, Q.; Ma, X.; Wang, J.; Liang, T. miRNA and mRNA expression analysis reveals potential sex-biased miRNA expression. Sci. Rep. 2017, 7, 39812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; He, H.W.; Wang, Z.M.; Zhao, H.; Lian, X.Q.; Wang, Y.S.; Zhu, J.; Yan, J.J.; Zhang, D.G.; Yang, Z.J.; et al. Plasma levels of lipometabolism-related miR-122 and miR-370 are increased in patients with hyperlipidemia and associated with coronary artery disease. Lipids Health Dis. 2012, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puzzo, F.; Colella, P.; Biferi, M.G.; Bali, D.; Paulk, N.K.; Vidal, P.; Collaud, F.; Simon-Sola, M.; Charles, S.; Hardet, R.; et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid alpha-glucosidase. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterna, J.C.; Moccetti, T.; Mura, A.; Feldon, J.; Bueler, H. Influence of promoter and WHV post-transcriptional regulatory element on AAV-mediated transgene expression in the rat brain. Gene Ther. 2000, 7, 1304–1311. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.D.; Naldini, L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat. Rev. Genet. 2009, 10, 578–585. [Google Scholar] [CrossRef]
- Dressman, D.; Araishi, K.; Imamura, M.; Sasaoka, T.; Liu, L.A.; Engvall, E.; Hoffman, E.P. Delivery of alpha- and beta-sarcoglycan by recombinant adeno-associated virus: Efficient rescue of muscle, but differential toxicity. Hum. Gene Ther. 2002, 13, 1631–1646. [Google Scholar] [CrossRef]
- Ayuso, E.; Blouin, V.; Lock, M.; McGorray, S.; Leon, X.; Alvira, M.R.; Auricchio, A.; Bucher, S.; Chtarto, A.; Clark, K.R.; et al. Manufacturing and characterization of a recombinant adeno-associated virus type 8 reference standard material. Hum. Gene Ther. 2014, 25, 977–987. [Google Scholar] [CrossRef] [Green Version]
- Prior, H.; Baldrick, P.; Beken, S.; Booler, H.; Bower, N.; Brooker, P.; Brown, P.; Burlinson, B.; Burns-Naas, L.A.; Casey, W.; et al. Opportunities for use of one species for longer-term toxicology testing during drug development: A cross-industry evaluation. Regul. Toxicol. Pharmacol. RTP 2020, 113, 104624. [Google Scholar] [CrossRef]
- EMA. Guideline on Quality, Non-Clinical and Clinical Requirements for Investigational Advanced Therapy Medicinal Products in Clinical Trials. 2019. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-non-clinical-clinical-requirements-investigational-advanced-therapy_en.pdf (accessed on 25 November 2020).
- EMA. ICH Guideline S6 (R1)–Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-s6r1-preclinical-safety-evaluation-biotechnology-derived-pharmaceuticals-step-5_en.pdf (accessed on 25 November 2020).
- Gao, K.; Li, M.; Zhong, L.; Su, Q.; Li, J.; Li, S.; He, R.; Zhang, Y.; Hendricks, G.; Wang, J.; et al. Empty Virions In AAV8 Vector Preparations Reduce Transduction Efficiency And May Cause Total Viral Particle Dose-Limiting Side-Effects. Mol. Ther. Methods Clin. Dev. 2014, 1, 20139. [Google Scholar] [CrossRef]
- Deland, F.H.; North, W.A. Relationship between liver size and body size. Radiology 1968, 91, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Davidoff, A.M.; Ng, C.Y.; Zhou, J.; Spence, Y.; Nathwani, A.C. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood 2003, 102, 480–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronzitti, G.; Gross, D.A.; Mingozzi, F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front. Immunol. 2020, 11, 670. [Google Scholar] [CrossRef] [PubMed]
- Chicoine, L.G.; Montgomery, C.L.; Bremer, W.G.; Shontz, K.M.; Griffin, D.A.; Heller, K.N.; Lewis, S.; Malik, V.; Grose, W.E.; Shilling, C.J.; et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 338–347. [Google Scholar] [CrossRef] [Green Version]
- Mingozzi, F.; High, K.A. Overcoming the Host Immune Response to Adeno-Associated Virus Gene Delivery Vectors: The Race between Clearance, Tolerance, Neutralization, and Escape. Annu. Rev. Virol. 2017, 4, 511–534. [Google Scholar] [CrossRef]
- Meliani, A.; Boisgerault, F.; Hardet, R.; Marmier, S.; Collaud, F.; Ronzitti, G.; Leborgne, C.; Costa Verdera, H.; Simon Sola, M.; Charles, S.; et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018, 9, 4098. [Google Scholar] [CrossRef] [Green Version]
- Mingozzi, F.; Anguela, X.M.; Pavani, G.; Chen, Y.; Davidson, R.J.; Hui, D.J.; Yazicioglu, M.; Elkouby, L.; Hinderer, C.J.; Faella, A.; et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci. Transl. Med. 2013, 5, 194ra192. [Google Scholar] [CrossRef] [Green Version]
- Leborgne, C.; Barbon, E.; Alexander, J.M.; Hanby, H.; Delignat, S.; Cohen, D.M.; Collaud, F.; Muraleetharan, S.; Lupo, D.; Silverberg, J.; et al. IgG-cleaving endopeptidase enables In Vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020, 26, 1096–1101. [Google Scholar] [CrossRef]
- Jordan, S.C.; Lorant, T.; Choi, J.; Kjellman, C.; Winstedt, L.; Bengtsson, M.; Zhang, X.; Eich, T.; Toyoda, M.; Eriksson, B.M.; et al. IgG Endopeptidase in Highly Sensitized Patients Undergoing Transplantation. N. Engl. J. Med. 2017, 377, 442–453. [Google Scholar] [CrossRef]
- Elmore, Z.C.; Oh, D.K.; Simon, K.E.; Fanous, M.M.; Asokan, A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Jonuschies, J.; Antoniou, M.; Waddington, S.; Boldrin, L.; Muntoni, F.; Thrasher, A.; Morgan, J. The human desmin promoter drives robust gene expression for skeletal muscle stem cell-mediated gene therapy. Curr. Gene Ther. 2014, 14, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.; Lau, A.; Wang, Z.; Zhang, Y.; Zhang, F.; Grompe, M.; Kay, M.A. Ribosomal DNA integrating rAAV-rDNA vectors allow for stable transgene expression. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1912–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas, L.E.; Grieves, J.L.; Zaraspe, K.; La Perle, K.M.; Fu, H.; McCarty, D.M. Patterns of scAAV vector insertion associated with oncogenic events in a mouse model for genotoxicity. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 2098–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GlobeNewswire. Sarepta Therapeutics and Genethon Announce a Gene Therapy Research Collaboration for the Treatment of Duchenne Muscular Dystrophy. 2017. Available online: https://www.globenewswire.com/news-release/2017/06/21/1027114/0/en/Sarepta-Therapeutics-and-Genethon-Announce-a-Gene-Therapy-Research-Collaboration-for-the-Treatment-of-Duchenne-Muscular-Dystrophy.html (accessed on 25 November 2020).


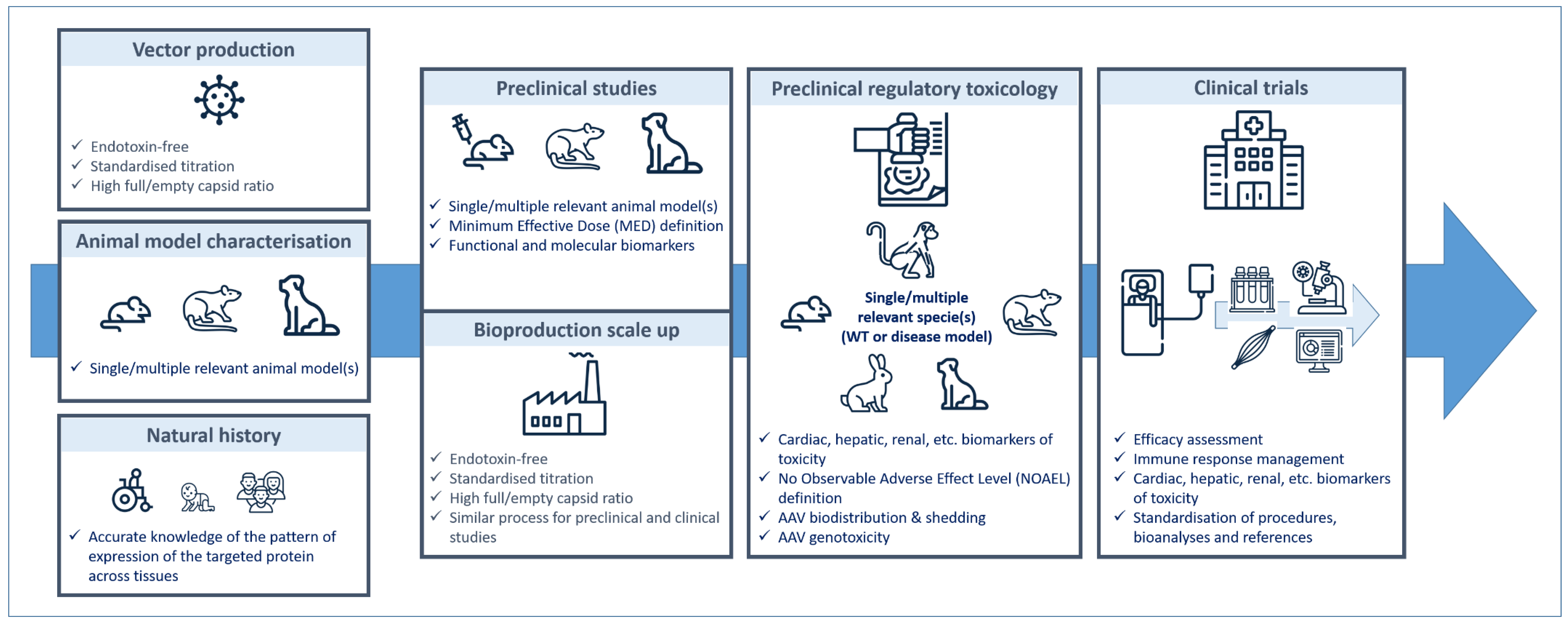{kind=link}
| PRODUCT/ADMINISTRATION | CLINICAL DESIGN | SERIOUS ADVERSE EVENTS | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DISEASE | AAV Serotype | Promoter | Transgene | Name | Administration/Dose | Clinical Trial ID (Study Name) Sponsor/Collaborator | Study Phase/Status | Study Timelines (Clinical Follow-Up) | Age, Gender, Actual or Estimated/ Planned Number of Participants Enrolled | |
| DMD | AAV9 | CK8 | Micro-dystrophin | SGT-001 | Intravenous 2 doses | NCT03368742 (IGNITE DMD) Solid Biosciences, LLC | Phase ½ active, not recruiting | 2017–2024 (2 years) | 4 to 17 years, males, n = 16/same as current | Complement activation kidney failure, platelet count drop (n = 1 at 5 × 1013 vg/kg) + cardiopulmonary insufficiency (n = 1 at 2 × 1014 vg/kg) [185,186] |
| DMD | AAVrh74 | MHCK7 | Micro-dystrophin | SRP-9001 | Intravenous 2 × 1014 vg/kg | NCT03375164 Sarepta Therapeutics, Inc. | Phase ½ active, not recruiting | 2018–2021 (3 years) | 3 months to 7 years, males, n = 4/12 | No serious adverse events [177] |
| DMD | AAVrh74 | MHCK7 | Micro-dystrophin | SRP-9001 | Intravenous 1 dose | NCT03769116 Sarepta Therapeutics, Inc. | Phase 2 active, not recruiting | 2018–2026 (5 years) | 4 years to 7 years, males, n = 41/24 | - |
| DMD | AAV9 | Human muscle-specific | Mini-dystrophin | PF-06939926 | Intravenous 1 × 1014 vg/kg 3 × 1014 vk/kg | NCT03362502 Pfizer | Phase 1B active, not recruiting | 2018–2026 (5 years) | 4 years and older, males, n = 30/12 | Antibody response, complement activation, acute kidney injury, haemolysis, thrombocytopenia (n = 1 at 3 × 1014 vg/kg) [187] |
| SMA | AAV9 | Hybrid CMV enhancer/chicken β-actin promoter | Human SMN | AVXS-101 | Intravenous 6.7 × 1013 vg/kg 2 × 1014 vg/kg | NCT02122952 AveXis, Inc. | Phase 1 completed | 2014–2017 (2 years) | Up to 6 months of age, males and female, n = 15/9 | Elevated serum aminotransferase levels (˃10× normal level) [44] |
| SMA | AAV9 | Hybrid CMV enhancer/chicken β-actin promoter | Human SMN | AVXS-101 | Intravenous therapeutic dose | NCT03306277 (STR1VE) AveXis, Inc. | Phase 3 completed | 2017–2019 (18 months of age) | Up to 6 months of age, males and females, n = 22/15 | - |
| SMA | AAV9 | Hybrid CMV enhancer/chicken β-actin promoter | Human SMN | AVXS-101 | Intrathecal 6 × 1013 vg 1.2 × 1014 vg 2.4 × 1014 vg | NCT03381729 (STRONG) AveXis, Inc. | Phase 1 suspended (on clinical hold pending further discussions regarding pre-clinical findings) | 2017–2021 (15 months) | 6 to 60 months of age, males and females, n = 51/27 | SAE mainly related to the disease itself (n = 7). Transaminitis events probably related to treatment (n = 2). [188] |
| SMA | AAV9 | Hybrid CMV enhancer/chicken β-actin promoter | Human SMN | AVXS-101 | Intravenous | NCT03461289 (STRIVE-EU) AveXis, Inc. | Phase 3 completed | 2018–2020 (18 months of age) | Up to 6 months of age, males and females, n = 33/30 | - |
| SMA | AAV9 | Hybrid CMV enhancer/chicken β-actin promoter | Human SMN | AVXS-101 | Intravenous 1.1 × 1014 vg/kg | NCT03505099 (SPR1NT) AveXis, Inc./PRA Health Sciences | Phase 3 active, not recruiting | 2018–2021 (18 and 24 months of age) | Up to 42 days, males and females, n = 30/44 | - |
| SMA | AAV9 | Hybrid CMV enhancer/chicken β-actin promoter | Human SMN | AVXS-101 | Intravenous single dose | NCT03837184 AveXis, Inc./PRA Health Sciences | Phase 3 active, not recruiting | 2019–2021 (18 months of age) | Up to 6 months of age, males and females, n = 2/6 | - |
| XLMTM | AAV8 | Des | Human MTM1 | AT132 | Intravenous 1 × 1014 vg/kg 3 × 1014 vg/kg | NCT03199469 (ASPIRO) Audentes Therapeutics | Phase ½ active, not recruiting (FDA placed on clinical hold since June 2020) | 2017–2024 (5 years) | Up to 5 years, males, n = 24/12 | Progressive liver dysfunction, hyperbilirubinemia, death from sepsis or gastrointestinal bleeding (n = 3/17 at 3 × 1014 vg/kg) [189] |
| Pompe | AAV2/8 | Liver-specific promoter | hGAA | ACTUS-101 | Intravenous 2 doses | NCT03533673 Asklepios Biopharmaceuticals, INC./Duke University and National Institute of Arthritis and Musculoskelatal and Skin Diseases (NIAMS) | Phase ½ recruiting | 2018–2022 (52 weeks) | 18 years and older, males and females, n = 8/6 | - |
| Pompe | AAV | Liver-specific promoter | hGAA | SPK-3006 | Intravenous dose escalation | NCT04093349 (RESOLUTE) Spark Therapeutics | Phase ½ Recruiting | 2020–2023 (52 weeks) | 18 years and older, males and females, n = 20/same as current | - |
| Pompe | AAV8 | Hybrid liver/desmin promoter | hGAA | AT845 | Intravenous 2 doses | NCT04174105 (FORTIS) Audentes Therapeutics | Phase ½ Recruiting | 2020–2027 (5 years) | 18 to 80 years, males and females, n = 8/same as current | - |
| Danon | AAV9 | CAG | hLAMP2B | RP-A501 | Intravenous 2 doses | NCT03882437 Rocket Pharmaceuticals Inc. | Phase 1 recruiting | 2019–2023 (3 years) | 8 years to 14 years and 15 years and older, males, n = 24/same as current | - |
| LGMD2E | scAAV rh74 | MHCK7 | SGCB | SRP-9003 | Intravenous 5 × 1013 vg/kg | NCT03652259 Sarepta Therapeutics, Inc. | Phase ½ active, not recruiting | 2018–2020 (3 years) | 4 to 15 years, males and females, n = 6/9 | Elevated liver enzymes associated with transient increase in bilirubin (n = 1) [190] |
| Batten disease | AAV2 | CU | hCLN2 | - | CNS administration 3 × 1012 vg | NCT00151216 Weill Medical College of Cornell University/Nathan’s Battle Foundation | Phase 1 completed | 2004–2019 (18 months) | 3 to 18 years, males and females, n = 10/11 | - |
| Batten disease | AAVrh.10 | CU | hCLN2 | - | Direct CNS administration 9 × 1011 vg/2.85 × 1011 vg | NCT01414985 Weill Medical College of Cornell University | Phase ½ completed | 2010–2017 (18 months) | 3 to 18 years, males and females, n = 8/16 | - |
| Batten disease | AAVrh.10 | CU | hCLN2 | - | Direct CNS administration 9 × 1011 vg 2.85 × 1011 vg | NCT01161576 Weill Medical College of Cornell University/National Institute of Health | Phase 1 active, not recruiting | 2010–2032 (18 months) | 2 to 18 years, males and females, n = 25/16 | - |
| Batten disease | scAAV9 | CB | CLN6 | AT-GTX-501 | Intrathecal | NCT02725580 Amicus Therapeutics | Phase 1/2A active, not recruiting | 2016–2021 (24 months) | 1 year and older, males and females, n = 13/6 | - |
| Batten disease | scAAV9 | P546 | CLN3 | AT-GTX-502 | Intrathecal 2 doses | NCT03770572 Amicus Therapeutics | Phase 1/2A active, not recruiting | 2018–2023 (36 months) | 3 to 10 years, males and females, n = 7/same as current | - |
| GSD1a | AAV8 | Native promoter | G6Pase | DTX401 | Intravenous 3 doses | NCT03517085 Ultragenyx Pharmaceutical INC | Phase 1/2 recruiting | 2018–2020 (52 weeks) | 18 years and older, males and females, n = 18/9 | No treatment-related serious adverse events reported to date |
| Reference. | Promoter | Codon Optimisation | Dose vg/per Mouse | Dose vg/kg of Body Weight | Expression in CNS | Mean Survival (Days) | Adverse Events |
|---|---|---|---|---|---|---|---|
| [196] | PGK | Yes | 4.5 × 1010 | 3 × 1013 | SC: 80–140% of WT levels Brain: low | 160 d (in 100% mice) |
|
| [194] | CBA | No | 5 × 1011 | 3.3 × 1014 | SC: 42% of WT levels | >250 d (n = 4, 1 death at d 97) |
|
| [197] | CMV | Yes | 1 × 1011 | 6.7 × 1013 | Lumbar SC: 66.5% MN Thoracic SC: 45% MN Cervical SC: 55% MN | 69 d (in 80% of mice) |
|
| Trial Promoter/Product Name/Reference | Vector | Promoter | Micro-Dystrophin Domains | Dose vg/kg of Body Weight | Expression in SM | NSAA | Serious Adverse Events |
|---|---|---|---|---|---|---|---|
| Sarepta SRP-9001-101 [177] | AAVrh74 | MHCK7 (SM and cardiac) | coΔR4-R23/ΔCT | 2 × 1014 | 95.8% of normal | 5.5 points increase after 1 year | |
| Pfizer PF-06939926 | AAV9 | Human muscle specific | - | 1 × 1014 3 × 1014 | 23.6% of normal 29.5% of normal | 2 points increase after 1 year | In 1 patient at 3 × 1014 vg/kg: complement activation, acute kidney failure, thrombocytopenia |
| Solid SGT-001 | AAV9 | CK8 | ΔR2-R15/ΔR18-R22/ΔCT | 2 × 1014 2 doses | Complement activation, acute kidney failure, thrombocytopenia (2 SAEs in 6th patient) |
| Reference | AAV Name | Parental AAV | Method | Compared with | Receptor | Muscle Transduction | Heart Transduction | Liver Transduction | Immune Response |
|---|---|---|---|---|---|---|---|---|---|
| [253] | AAV2i8 | AAV2 | Rational design: replacement of receptor-binding hexapeptide with corresponding residue in AAV8 | AAV2/AAV8 | Not HS | =AAV8 >AAV2 | =AAV8 >AAV2 | <AAV2 and AAV8 (40-fold lower) | Lower cross reactivity to AAV2 antibody |
| [257] | AAV9 (rhesus monkey) | <AAV9 (122-fold lower) | <AAV9 (46-fold lower) | <AAV9 (11-fold lower) | ND | ||||
| [256] | AAV2i8G9 | AAV2i8 | Rational design: graft galactose-binding footprint of AAV9 in VP3 AAV2i8 | AAV2i8/AAV9 | Not HS Glycan | >AAV2i8>AAV9 | AAV2i8 < AAV2i8G9 < AAV9 | AAV2i8 < AAV2i8G9 < AAV9 (5-fold lower) | ND |
| [146] | AAV2.5 | AAV2 | Rational design: AAV2 capsid with 5 mutations from AAV1 | AAV2 | HS | >AAV2 (2- to 5-fold) | NA | NA | -No cellular immune response to capsid. -Lower cross-reaction to AAV2 NAb |
| [254] | AAV2 587 MTP | AAV2 | Rational design: insertion of muscle-targeting peptide in AAV2 capsid | AAV2 | Not heparin | ≥AAV2 (2-fold) | >AAV2 (7-fold) | <AAV2 (2.5-fold) | ND |
| [258] | AAV9.45 | AAV9 | Directed evolution: random mutagenesis of surface-exposed regions of AAV9 | AAV9 | ND | ≈AAV9 | ≈AAV9 | <AAV9 (10- to 25-fold lower) | ND |
| [259] | AAV po1 | NA | Natural pig isolate | AAV9 | ND | <AAV9 (≈2–4-fold) | <AAV9 (≈3-fold) | <AAV9 (≈140-fold) | ND |
| AAV5 | ND | >AAV5 (1.5-fold) | <AAV5 (30-fold) | <AAV5 (≈125-fold) | No pre-existing immunity No cross-neutralisation by antisera against all common AAVs | ||||
| [261] | AAV9-Y731F AAV1-Y445F/Y731F | AAV9 AAV1 | Rational design: tyrosine mutations | Other mutants (no AAV of reference) | ND | Skeletal muscle > heart ≈ liver liver (3–10-fold lower) < skeletal muscle (3–10-fold lower) < heart | ND | ||
| [255] | AAV2-VNSTRLP | AAV2 | Directed evolution: from AAV2 display peptide library with in vitro selection for heart tropism | AAV2 AAV9 | ND | ≈AAV2 <AAV9 | >AAV2 (>10-fold) <AAV9 | <AAV2 (≈10-fold) <AAV9 | ND |
| [263] | AAVM41 | AAV1/6/7/8 | Directed evolution by shuffling the capsids of AAV1 to AAV9 and in vivo selection on skeletal muscle | AAV9 AAV6 | ND | <AAV9 >AAV6 | ≈AAV9 >AAV6 (up to 13-fold) | <AAV9 <AAV6 | Lower cross reactivity |
| [260] | AAVB1 | AAV8 AAVrh43 (mostly) | Directed evolution of DNA shuffled library and selection on brain tissues | AAV9 | Not SA Not Galactose | >AAV9 (10- to 26-fold higher) | >AAV9 (14-fold higher) | <AAV9 (3.6-fold lower) | Modestly more resistant to neutralisation than AAV9 |
| [252] | AAVC4 AAVC7 AAVG4 | NA | Ancestral reconstruction from NHP and human AAV by combinatorial variation of 32 amino acids and selection on muscle cells | AAV1 | Not SA Not galactose Not HS | >AAV1 (10–31-fold higher) | NA | NA | Not resistant to neutralisation with IVIG |
| [265] | AAV10HB | NA | Isolation from bat faecal and intestinal tissues | AAV2 AAV8 | ND | Ratio muscle/liver = 8.8 Ratio muscle/liver < 1 for AAV2 and AAV8 | Reduced neutralisation with IVIG ≈AAV2 | ||
| [266] | Several variants: r2.4/r2.15 | AAV2 | Directed evolution: random mutagenesis and selection of heparin binding or neutralising serum binding | AAV2 | Not heparin | ND | ND | ND | Reduced neutralisation with serum/AAV2 |
| [268] | Mus12 | Capsid shuffled library | Directed evolution: shuffled library selected on patients’ sera and amplified in vivo in mouse muscle | AAV1/AAV2/AAV2.5/AAV6/AAV8/AAV9 | ND | IM ≈ AAV6 ≈ AAV9 IV < AAV9 | ND | ND | Immune escape |
| [267] | CAM130 | AAV1 | Directed evolution: rational mutagenesis on AAV1 capsid residues in contact with antibodies, library generation and evolution on vascular endothelial cells | AAV1 | ND | ND | >AAV1 (2-fold) | =AAV1 | Neutralisation escape to murine, NHP and human sera |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buscara, L.; Gross, D.-A.; Daniele, N. Of rAAV and Men: From Genetic Neuromuscular Disorder Efficacy and Toxicity Preclinical Studies to Clinical Trials and Back. J. Pers. Med. 2020, 10, 258. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm10040258
Buscara L, Gross D-A, Daniele N. Of rAAV and Men: From Genetic Neuromuscular Disorder Efficacy and Toxicity Preclinical Studies to Clinical Trials and Back. Journal of Personalized Medicine. 2020; 10(4):258. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm10040258
Chicago/Turabian StyleBuscara, Laurine, David-Alexandre Gross, and Nathalie Daniele. 2020. "Of rAAV and Men: From Genetic Neuromuscular Disorder Efficacy and Toxicity Preclinical Studies to Clinical Trials and Back" Journal of Personalized Medicine 10, no. 4: 258. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm10040258