
Reanalysis of Exome Data Identifies Novel SLC25A46 Variants Associated with Leigh Syndrome

 , ,
, ,
Abstract
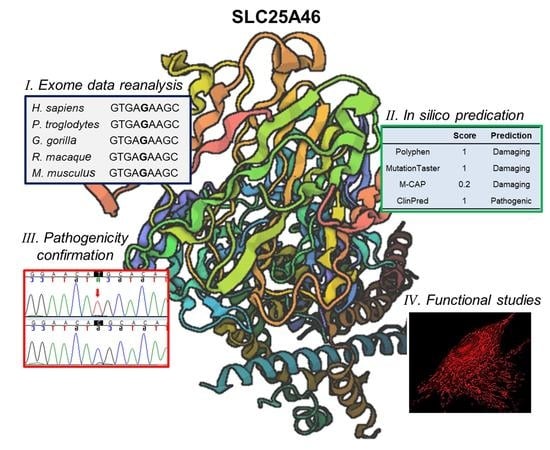
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Proband’s Phenotype
3.2. SLC25A46 Variant Detection and Confirmation
3.3. Protein Structure Analysis Affected by the Missense Variant
3.4. Transcription Analysis of the Donor Splice Region Variant
3.5. Altered Expression of SLC25A46 Mutant Protein
3.6. Increased Mitochondrial Fragmentation in Proband’s Fibroblast Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Asp. Med. 2013, 34, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Aguilar, M.; Baines, C.P. Physiological and pathological roles of mitochondrial SLC25 carriers. Biochem. J. 2013, 454, 371–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases caused by mutations in mitochondrial carrier genes SLC25: A review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janer, A.; Prudent, J.; Paupe, V.; Fahiminiya, S.; Majewski, J.; Sgarioto, N.; Des Rosiers, C.; Forest, A.; Lin, Z.Y.; Gingras, A.C. SLC 25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol. Med. 2016, 8, 1019–1038. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Chao, J.T.; Tavassoli, S.; Wong, A.K.; Choudhary, V.; Young, B.P.; Loewen, C.J.; Prinz, W.A. A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 2014, 12, e1001969. [Google Scholar] [CrossRef] [Green Version]
- Duchesne, A.; Vaiman, A.; Castille, J.; Beauvallet, C.; Gaignard, P.; Floriot, S.; Rodriguez, S.; Vilotte, M.; Boulanger, L.; Passet, B. Bovine and murine models highlight novel roles for SLC25A46 in mitochondrial dynamics and metabolism, with implications for human and animal health. PLoS Genet. 2017, 13, e1006597. [Google Scholar] [CrossRef] [Green Version]
- Steffen, J.; Vashisht, A.A.; Wan, J.; Jen, J.C.; Claypool, S.M.; Wohlschlegel, J.A.; Koehler, C.M. Rapid degradation of mutant SLC25A46 by the ubiquitin-proteasome system results in MFN1/2-mediated hyperfusion of mitochondria. Mol. Biol. Cell 2017, 28, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Abe, K.; Li, Q.; Rosen, S.M.; Nori, N.; Madden, J.A.; Genetti, C.A.; Wojcik, M.H.; Ponnaluri, S.; Gubbels, C.S.; Picker, J.D. Unique bioinformatic approach and comprehensive reanalysis improve diagnostic yield of clinical exomes. Eur. J. Hum. Genet. 2019, 27, 1398–1405. [Google Scholar] [CrossRef]
- Amiott, E.A.; Lott, P.; Soto, J.; Kang, P.B.; McCaffery, J.M.; DiMauro, S.; Abel, E.D.; Flanigan, K.M.; Lawson, V.H.; Shaw, J.M. Mitochondrial fusion and function in Charcot–Marie–Tooth type 2A patient fibroblasts with mitofusin 2 mutations. Exp. Neurol. 2008, 211, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.-M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Suleiman, J.; Almannai, M.; Scaglia, F. Mitochondrial dynamics: Biological roles, molecular machinery, and related diseases. Mol. Genet. Metab. 2018, 125, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Terzenidou, M.E.; Segklia, A.; Kano, T.; Papastefanaki, F.; Karakostas, A.; Charalambous, M.; Ioakeimidis, F.; Papadaki, M.; Kloukina, I.; Chrysanthou-Piterou, M. Novel insights into SLC25A46-related pathologies in a genetic mouse model. PLoS Genet. 2017, 13, e1006656. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Kijima, K.; Numakura, C.; Izumino, H.; Umetsu, K.; Nezu, A.; Shiiki, T.; Ogawa, M.; Ishizaki, Y.; Kitamura, T.; Shozawa, Y. Mitochondrial GTPase mitofusin 2 mutation in Charcot–Marie–Tooth neuropathy type 2A. Hum. Genet. 2005, 116, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Abrams, A.J.; Fontanesi, F.; Tan, N.B.; Buglo, E.; Campeanu, I.J.; Rebelo, A.P.; Kornberg, A.J.; Phelan, D.G.; Stark, Z.; Zuchner, S. Insights into the genotype-phenotype correlation and molecular function of SLC25A46. Hum. Mutat. 2018, 39, 1995–2007. [Google Scholar] [CrossRef]
- Eldomery, M.K.; Coban-Akdemir, Z.; Harel, T.; Rosenfeld, J.A.; Gambin, T.; Stray-Pedersen, A.; Küry, S.; Mercier, S.; Lessel, D.; Denecke, J. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Shashi, V.; Schoch, K.; Spillmann, R.; Cope, H.; Tan, Q.K.-G.; Walley, N.; Pena, L.; McConkie-Rosell, A.; Jiang, Y.-H.; Stong, N. A comprehensive iterative approach is highly effective in diagnosing individuals who are exome negative. Genet. Med. 2019, 21, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Meng, L.; Normand, E.A.; Xia, F.; Song, X.; Ghazi, A.; Rosenfeld, J.; Magoulas, P.L.; Braxton, A.; Ward, P. Reanalysis of clinical exome sequencing data. N. Engl. J. Med. 2019, 380, 2478–2480. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Boesten, I.; Hellebrekers, D.; Mulder-den Hartog, N.; de Coo, I.; Smeets, H.; Gerards, M. Novel pathogenic SLC25A46 splice-site mutation causes an optic atrophy spectrum disorder. Clin. Genet. 2017, 91, 121–125. [Google Scholar] [CrossRef]
- Braunisch, M.; Gallwitz, H.; Abicht, A.; Diebold, I.; Holinski-Feder, E.; Van Maldergem, L.; Lammens, M.; Kovács-Nagy, R.; Alhaddad, B.; Strom, T. Extension of the phenotype of biallelic loss-of-function mutations in SLC25A46 to the severe form of pontocerebellar hypoplasia type I. Clin. Genet. 2018, 93, 255–265. [Google Scholar] [CrossRef]
- Krawczak, M.; Thomas, N.S.; Hundrieser, B.; Mort, M.; Wittig, M.; Hampe, J.; Cooper, D.N. Single base-pair substitutions in exon–intron junctions of human genes: Nature, distribution, and consequences for mRNA splicing. Hum. Mutat. 2007, 28, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Rossing, M.; Albrechtsen, A.; Skytte, A.-B.; Jensen, U.B.; Ousager, L.B.; Gerdes, A.-M.; Nielsen, F.C.; vO Hansen, T. Genetic screening of the FLCN gene identify six novel variants and a Danish founder mutation. J. Hum. Genet. 2017, 62, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Kobayashi, H.; Baba, M.; Ito, T.; Tanaka, R.; Nakatani, Y. Splice-site mutation causing partial retention of intron in the FLCN gene in Birt-Hogg-Dubé syndrome: A case report. BMC Med. Genom. 2018, 11, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Peng, Y.; Hufnagel, R.B.; Hu, Y.-C.; Zhao, C.; Queme, L.F.; Khuchua, Z.; Driver, A.M.; Dong, F.; Lu, Q.R. Loss of SLC25A46 causes neurodegeneration by affecting mitochondrial dynamics and energy production in mice. Hum. Mol. Genet. 2017, 26, 3776–3791. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| This Study | Nguyen et al. [22] | Braunisch et al. [23] | |
|---|---|---|---|
| SLC25A46 mutations | c.283+5G>A; c.1039C>T | c.283+3G>T (homozygous) | c.42C>G; c.462+1G>A |
| SLC25A46 proteins | Splicing defect; p.[Arg347Cys] | Splicing defect | p.[Tyr14Ter]; Splicing defect |
| Age of onset | <1 month | Birth | Birth |
| Age of death | 7 months | 7 days | 1 day/18 days |
| Cause of death | Respiratory insufficiency | Respiratory insufficiency | Respiratory insufficiency |
| Optic atrophy | + | + | Unknown |
| Cerebellar or brainstem atrophy | + | + | + |
| Hypotonia | + | + | + |
| Other features | Global developmental delay, decreased NAA to choline ratio and decreased NAA to creatine ratio, with no lactate peak detected | Increase lactic acid level and lactate to pyruvate ratio, and decrease in cytochrome c oxidase activity | Pontocerebellar hypoplasia, respiratory defect, neurogenic lesion; loss of spinal motor neurons |
| Mitochondrial defects | Mitochondrial fragmentation | Mitochondrial fragmentation | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Madden, J.A.; Lin, J.; Shi, J.; Rosen, S.M.; Schmitz-Abe, K.; Agrawal, P.B. Reanalysis of Exome Data Identifies Novel SLC25A46 Variants Associated with Leigh Syndrome. J. Pers. Med. 2021, 11, 1277. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11121277
Li Q, Madden JA, Lin J, Shi J, Rosen SM, Schmitz-Abe K, Agrawal PB. Reanalysis of Exome Data Identifies Novel SLC25A46 Variants Associated with Leigh Syndrome. Journal of Personalized Medicine. 2021; 11(12):1277. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11121277
Chicago/Turabian StyleLi, Qifei, Jill A. Madden, Jasmine Lin, Jiahai Shi, Samantha M. Rosen, Klaus Schmitz-Abe, and Pankaj B. Agrawal. 2021. "Reanalysis of Exome Data Identifies Novel SLC25A46 Variants Associated with Leigh Syndrome" Journal of Personalized Medicine 11, no. 12: 1277. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11121277