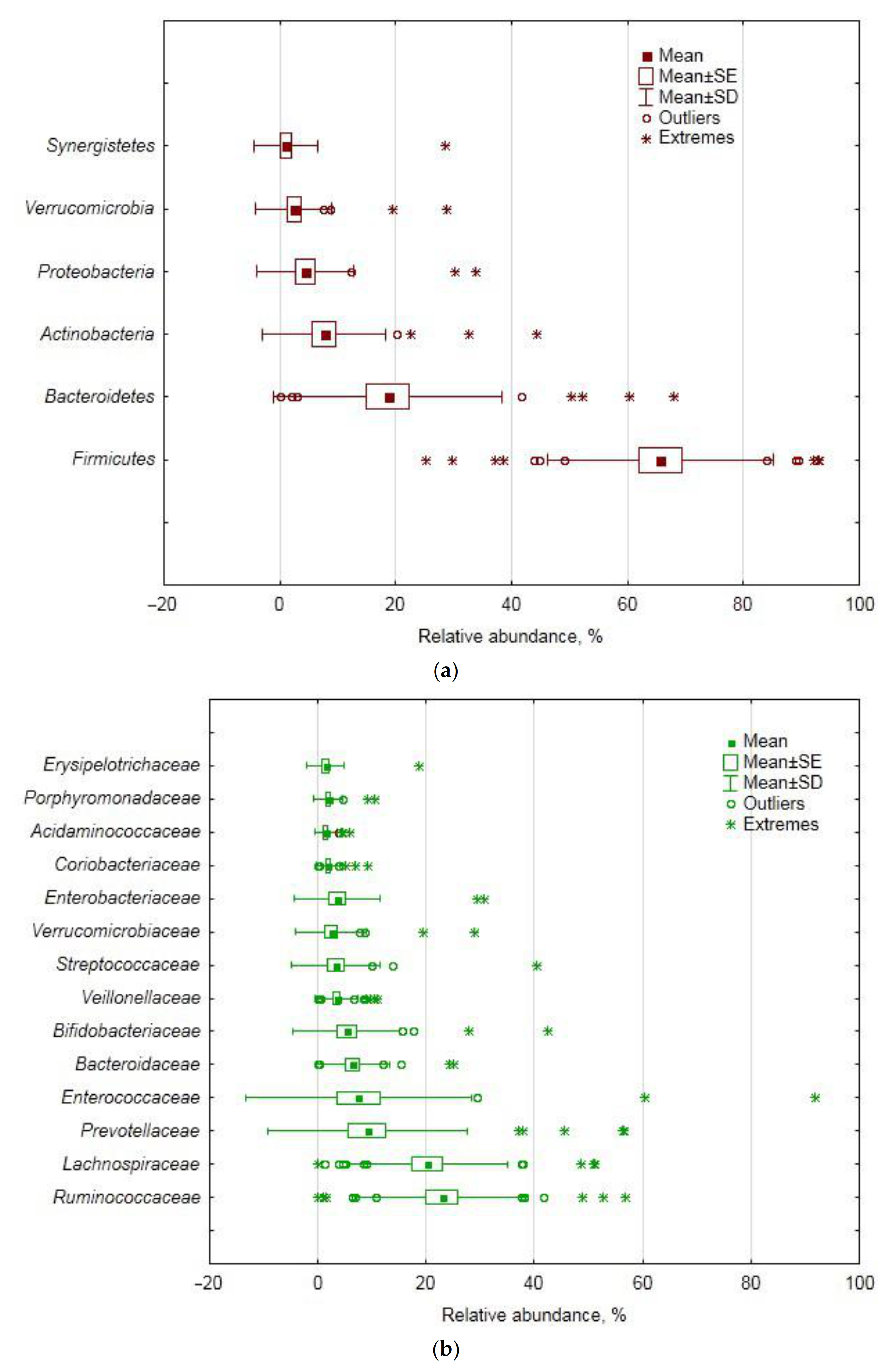
4.1. Fecal Bacteriobiome Composition in GSD Patients before the CCE Surgery
In the studied cohort of GSD-afflicted female patients, the
Firmicutes phylum was the ultimate dominant in the fecal bacteriobiome, with
Bacteroidetes and
Actinobacteria being second and third in the ranking of abundance. In this aspect, our cohort differed from a Chinese one, where
Proteobacteria, instead of
Actinobacteria, were found to be third in abundance, and
Bacteroidetes were almost twice as abundant as in our cohort [
22]. Moreover, in our cohort, the
Firmicutes relative abundance was markedly higher, as compared with the Chinese cohorts [
5,
21]. The apparent discrepancy is most likely due to the fact that one third of the Chinese cohort were males, whereas ours was a purely female one; and to the difference in diet [
23]. As for the biodiversity indices, however, the ones calculated in our study agree well with the indices describing the fecal bacteriobiome of a Chinese cohort of GSD patients [
21].
High inter-individual variation in bacterial sequence reads is quite common in fecal bacteriobiome studies in cohorts of healthy human subjects and of those afflicted by various diseases, including GSD [
21]. Even in cases when the authors [
21] claimed that their results “showed that the individual differences within the group were small”, the huge standard deviations for the OTUs’ relative abundance in their study proved the opposite. Therefore, by structuring down the data variance in our study by principal component analysis, we identified some age- and BMI-related taxa within the studied cohort, as well as some taxa that showed a correlation with blood glucose, bilirubin and transaminase activity.
Some genera of
Lachnospiraceae are known to be important for bile acid metabolism, having 7α-dehydroxylation activity: in the pre-CCE bacteriobiome of the studied cohort, the family accounted for one fifth of the total number of sequence reads, along with
Ruminocccaceae ultimately prevailing at the family level and together accounting for almost half of the abundance; in a Chinese cohort, however, the family with less than 0.3% was not even close to any dominating position [
21]. The latter study also found the relative abundance of
Clostridium to be 0.01%, whereas in our study, the presence of
Clostridium sensu stricto was an order of magnitude more pronounced (
Table 6). The differences are plentiful and most likely attributable to racial, dietary, sex and other characteristics of the cohorts.
Principal component analysis based on covariance allows dealing with the original variance of the data, without standardizing them, easily structuring the variance by extracted principal components, featuring the contribution of the original variables to the new ones (PCs) and reducing the original plethora of variables to fewer ones (PCs), but accounting for most of the original variance in the data. Subjection of the extracted PCs as dependent variables in multiple regression analysis allowed finding links with patients’ demographics and blood biochemical properties and bacterially interpreting them on the basis of a taxon contribution in the respective PC. We believe this approach to be informative for such kind of descriptive study.
The balance between two
Firmicutes classes, namely,
Clostridia and
Bacilli, accounted for almost half of the abundance variance at this taxonomic level, being positively correlated with age at the
p ≤ 0.10 level; the situation was translated in a similar manner at the order level, i.e.,
Clostridiales and
Lactobacillales. Then, at the family level, the balance between
Ruminococcaceae (
Clostridiales) and
Enterococcaceae (
Lactobacillales), accounting for one third of the total data variance (PC1), displayed a statistically significant positive correlation with age. This finding complies with the knowledge that the gut microbiota diversity changes with age [
24,
25], with
Firmicutes taxa increasing. Thus, the increased abundance of the core gut bacterial taxa with age in GSD-compromised subjects seems quite a natural occurrence, not overshadowed by changed bile acid metabolism and other factors. Interestingly, at the genus level, the structure of the data variance shifted, with the
Prevotella (
Bacteroidetes) and
Enterococcus (
Firmicutes) relationship accounting for half of the total data variance at this taxonomic level; the finding underscores the importance of these two core taxa relationships in structuring the gut bacteriome in general and GSD-compromised female patients in particular. As increased plasma levels of bilirubin (secondary to the breakdown of free hemoglobin) were shown to be associated with an increased risk of gallstone disease [
26], the statistically significant positive correlation of the
Prevotella–
Enterococcus balance with blood bilirubin, found in our study, necessitates further investigation of the role of the genera in gallstone formation, both pigmented and cholesterol ones [
27].
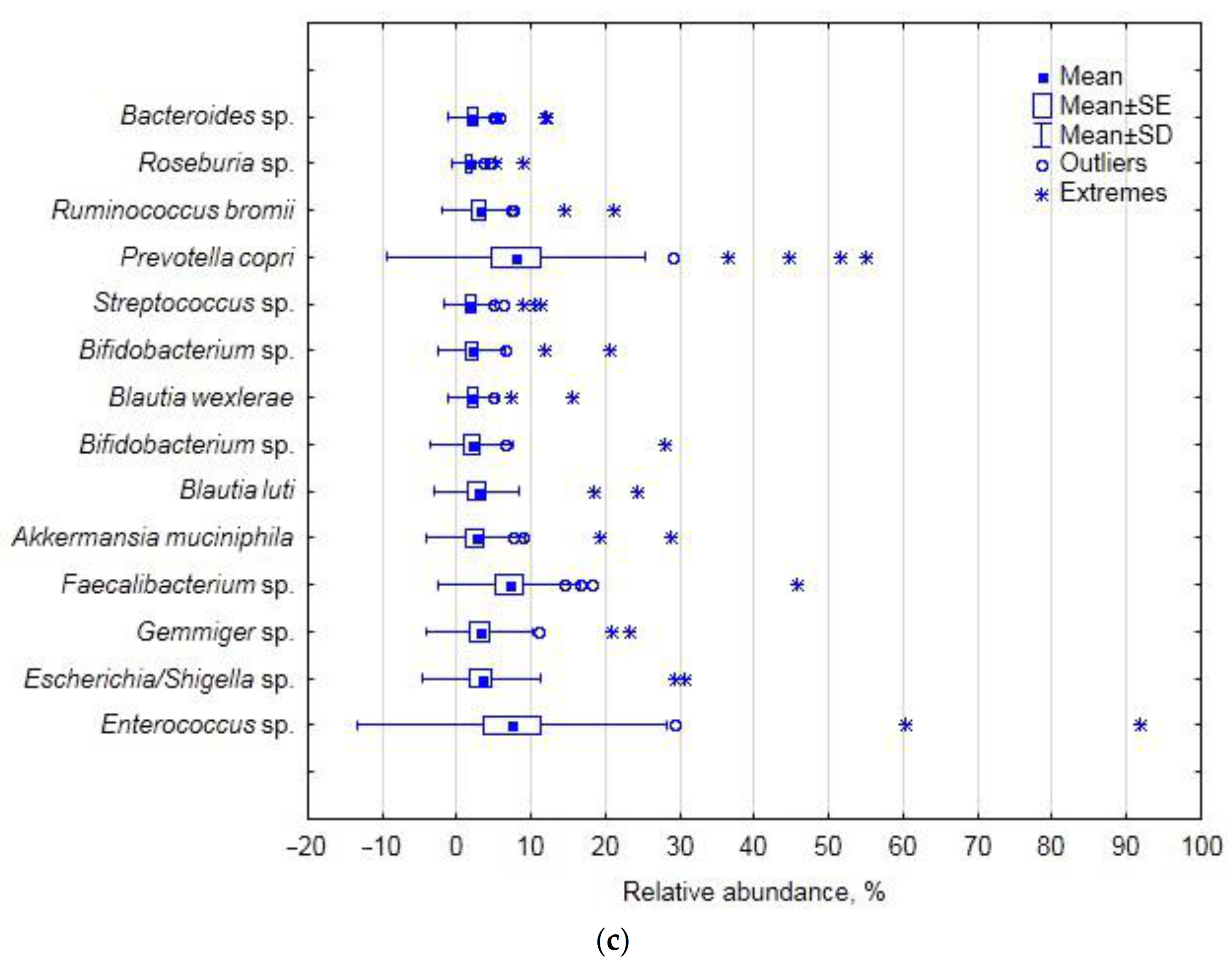
The finding that the balance between
Faecalibacterium, Bifidobacterium and
Gemminer determined 10% of the total data variance and was correlated with blood glucose (
p ≤ 0.05) and BMI (
p ≤ 0.10) may be indicative of the indirect association of the genera with glucose metabolism and insulin sensitivity in overweight patients [
28], but we cannot currently suggest the putative cause–effect mechanism. The joint variation in
Blautia,
Ruminococcus and some unclassified
Ruminococcaceae correlated with blood ALT, AST and bilirubin, and the mechanism of the involvement of these genera has to be comprehensively examined.
Further down the hierarchy at the species level, the
Prevotella–
Enterococcus relationship was manifested by the balance between
Prevotella copri and
Enterococcus sp. (with 40% of the total data variance), which was positively correlated with the cohort’s demographics, i.e., age and BMI (
p ≤ 0.05), and glucose (
p ≤ 0.10) and therefore may be related to glucose metabolism in overweight patients. Interestingly,
Akkermansia muciniphila was found to contribute a small portion of the data variance associated with blood biochemistry (at
p ≤ 0.05) and, hence, generally with the disease-compromised status of the subjects. The increased relative abundance of this mucin-degrading bacterium is often found to be associated with disease [
29,
30]; however, as a propionogenic bacterium,
A. muciniphila is also believed to have several health benefits in humans [
31].
However, as the blood characteristics are far from being specific for gallstone disease diagnostics, it is not possible to implicate the taxa in the changed bile acid metabolism and gallstone formation based on the results of multiple regression analysis, despite the comprehensive outline of the GSD bacteriobiome variation as related to the common blood biochemical properties. The bile acid profiles of the studied patients, if they had been determined, might have been more suggestive in this respect, and we acknowledge this as a drawback in the study.
4.2. Changes in Fecal Bacteriobiome Composition in GSD Patients after the CCE Surgery
In the studied cohort of GSD-afflicted female patients, mainly members of the Firmicutes phylum displayed CCE-related differential abundance.
As for the
Bacteroidetes phylum, its relative abundance did not change after CCE; the result does not agree with the finding of Israeli researchers, for example, when the phylum abundance was shown to be increased in the post-CCE cohort of subjects [
32]. However, the time factor, i.e., the duration of the time span elapsed between CCE and feces sampling, is a critical factor affecting bacterial composition [
33] and may, at least partially, explain the discrepancy between results.
The decreased abundance of
Clostridia (by 7%) and
Clostridiales (by 3–5%), found after CCE in our study, was not reported before. As these taxa are the most species-rich and physiologically diverse components of the fecal bacteriobiome, the CCE-associated shifts in their relative abundance cannot be unequivocally regarded as beneficial or not for human health.
Lachnospiraceae, the most predominant family in the human gut, displayed decreased abundance in the post-CCE bacteriobiome, which is, however, difficult to interpret as (a) most of the OTU-level clusters (69), ascribed to the family in our study (129), could not be taxonomically attributed below the family level, and (b) some genera and species of this family might support/contribute to healthy functions, whereas other genera and species were found to be increased in diseases [
34]. For example,
Blautia and
Roseburia species, often associated with a healthy state, are some of the main short-chain fatty acid producers [
35,
36]; therefore, their post-CCE decreased abundance (
p ≤ 0.10) may hardly be indicative of the better state of the gut bacteriobiome after surgery.
We could not find any information about the effect of CCE on
Actinobacteria/
Coriobacteriales/Coriobacteriaceae representatives, the latter known as pathobionts. As for
Collinsella, the dominant genus in the
Coriobacteriaceae family and the minor dominant in the studied cohort, its increased abundance after CCE (albeit at the
p ≤ 0.10 level) suggests negative implications after such shift [
37,
38,
39,
40]. As for another representative of the family with differential pre- and post-CCE abundance, i.e., the
Gordonibacter genus with just ca. 0.01% of the total number of sequence reads, it is difficult to suggest any ecophysiological significance at such abundance rate, although some genus representatives are known to participate in primary bile acid transformation [
41] or be involved in dietary polyphenol transformations generating more bioactive metabolites [
42].
Interestingly, although specific bacterial species such as
Helicobacter and
Salmonella were shown to be involved in the pathogenesis of cholesterol gallstones [
43], we did not find
Helicobacter at all, and found only one
Salmonella OTU with 0.3 and 0.6% abundance in pre- and post-CCE subcohorts, respectively; the finding infers potentially different bacterial involvement in GSD etiology in cohorts of different sex and ethnicity.
The actual number of OTUs per sample observed in our study was practically the same as the number obtained by the same methodology for post-CCE patients in Korea [
44]. However, as the latter study did not report whether the control group, i.e., non-CCE control patients, was also diagnosed with GSD, it is not possible to compare our results about the CCE effect on the fecal bacteriobiome with those results in their entirety, only for the post-CCE subcohort. For instance, in our study, CCE did not affect the gut bacteriobiome species richness, whereas compared with the independent control group, CCE decreased it [
43]. Notably, the α-biodiversity index (Shannon) reported in the aforementioned study was much higher than the one reported here (4.9 vs. 2.8): in our view, the discrepancy may be attributed to both the sex composition of the Korean cohort and the dietary habits, etc. At the same time, for the Israeli cohort of GDS patients, the pre- and post-CCE values of the Shannon index did not differ [
31], being close to, but slightly lower than, those in our study (2.1 vs. 2.8, respectively). We cannot help but emphasize that often the studies claiming to reveal the effect of CCE on the gut microbiota performed comparisons between the post-CCE patients with GSD and the healthy subjects without a GSD history [
33,
43,
44]. We believe such approach does not seem to be adequate for aiming to examine the effect of CCE per se, as only a direct comparison between pre- and post-CCE conditions of one and the same cohort of GSD-affected subjects is pertinent for the goal of revealing microbiome shifts associated with the surgery and potential biomarkers of the latter.
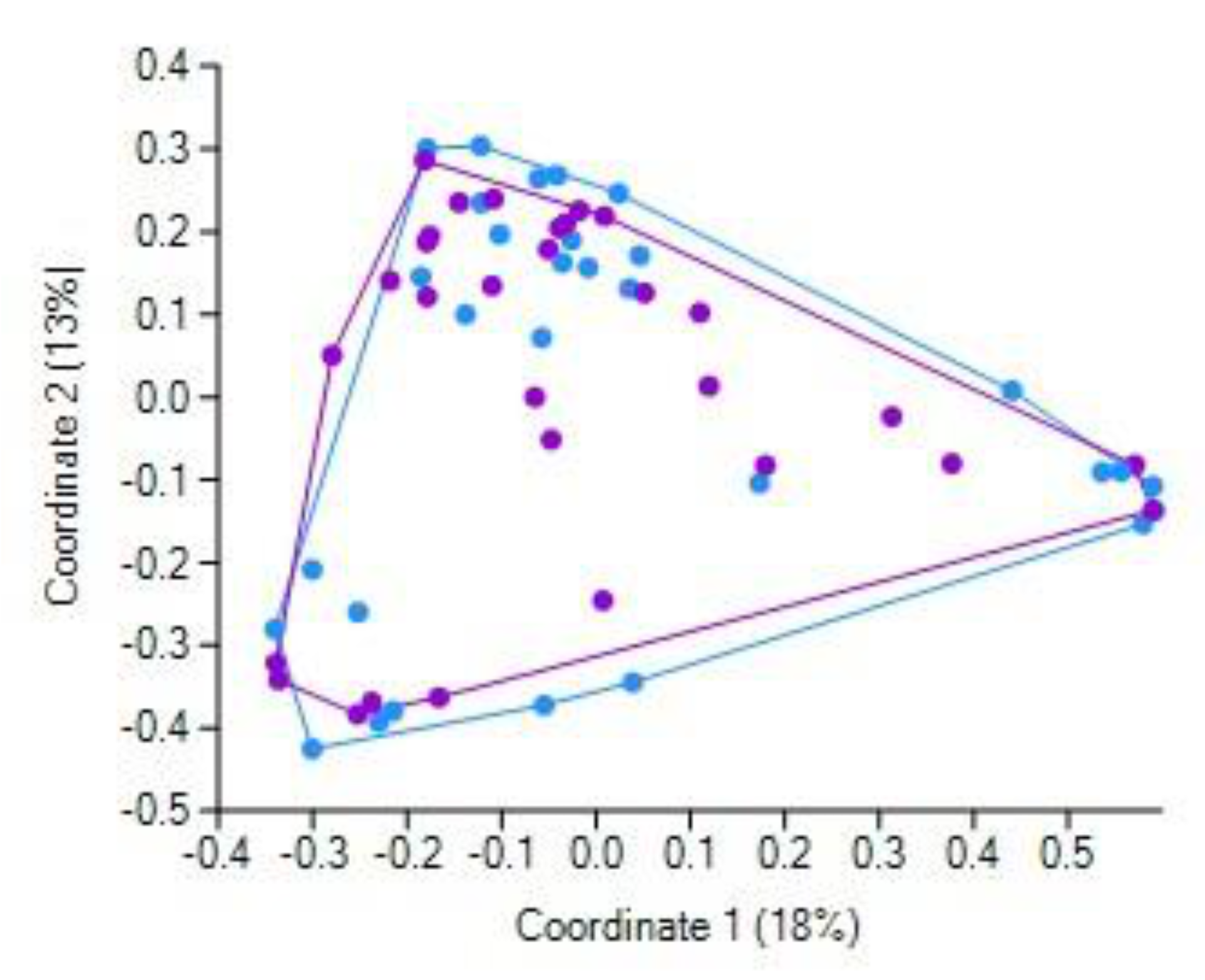
It was shown that CCE did not markedly affect the bile acid profile in the GSD patients [
31], leading the authors to conclude that the modified fecal bile acid composition results from inherently aberrant bile acid metabolism, leading, in turn, to gallstone formation. In general, our finding that the pre- and post-CCE fecal bacteriobiome profiles were not overall differentially distinct (as revealed by principal coordinate analysis) apparently complies with this conclusion. It should be emphasized that the repeated collection of feces samples was performed 1–3 days after the surgery, i.e., quite soon. Therefore, we are inclined to believe that it was a very short time to interpret the observed differential abundance of some bacterial taxa as solely resultant from the changed inflow of bile acids; the overall post-surgery condition most likely contributed significantly, if not primarily and predominantly, to the observed short-term CCE-related shifts in fecal bacterial assemblages. It should be noted that the overall post-surgery condition included ceftriaxone treatment of all patients. This beta-lactam antibiotic is able to kill a broad spectrum of bacteria [
45], thus potentially shaping the gut bacteriobiome [
46], especially when administered orally. Therefore, despite the very short time between the surgery and stool collection in this study, and hence the short time of antibiotic treatment, the revealed changes in the fecal bacteriobiome might have resulted, in part, due to the antibiotic per se. However, we should also emphasize that our study did not aim at discriminating between the effects of post-CCE altered bile profiles and antibiotic therapy; we aimed at profiling the gut bacteriobiome, referring to post-CCE as a single factor, as such embracing many factors, aspects, nuances, etc. We described the fecal bacteriobiome just at the starting point of patients embarking on the rest of life without a gallbladder. Whether the longer-term shifts in the gut microbiota after CCE will occur and to what degree and at what rate remain to be determined in future studies, which, hopefully, will also elucidate if gut microbes can act as the main character in the broad scenery of liver diseases [
47].


 ,
, 



{kind=link}
{kind=link}
{kind=link}