
Machine Learning-Based Approach Highlights the Use of a Genomic Variant Profile for Precision Medicine in Ovarian Failure

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants and Inclusion Criteria
2.2. Pre-Processing, NGS, and Variant Calling
2.3. Variant Filtering, Processing, and Prioritisation
- A moderate or deleterious effect on protein coding sequence (according to SnpEff annotations). Moderate effect included Missense variants, UTR (5′ + 3′) and splice (acceptor or donor) variants; while deleterious effects included Frameshift, Nosense (stop codon gain/loss) variants, protein to protein contact modifier variants, structural interaction modifier variants and disruptive inframe variants.
- Variants absent from the IGSR database were retained for downstream analysis. Meanwhile, variants present in the IGSR were only kept if their minor allele frequency (MAF) was lower than <0.05, based on the premise that purifying selection decreases allele frequency of variants that confer less fitness.
- Passage of quality criteria for coverage (>100×) as well as several parameters evaluated by GATK: Genotype Quality (GQ), which evaluates the confidence of the genotype attributed to a patient for a certain variant (homozygous for the reference allele, heterozygous or homozygous for the disease-associated allele); Position depth (DP) or total number of reads detected at a given position of the genome; Allele Depth (AD), the number of reads for the variant in that position. Further information is annexed with Supplementary Figure S1.
2.4. Patient Stratification and Ovarian Failure Subtypes Prediction
3. Results
3.1. Clinical Characterisation and Sequencing Quality of the Study Population
3.2. Genomic Variation Hypotheses
3.3. A Genomic Variant Profile Predictive of OF
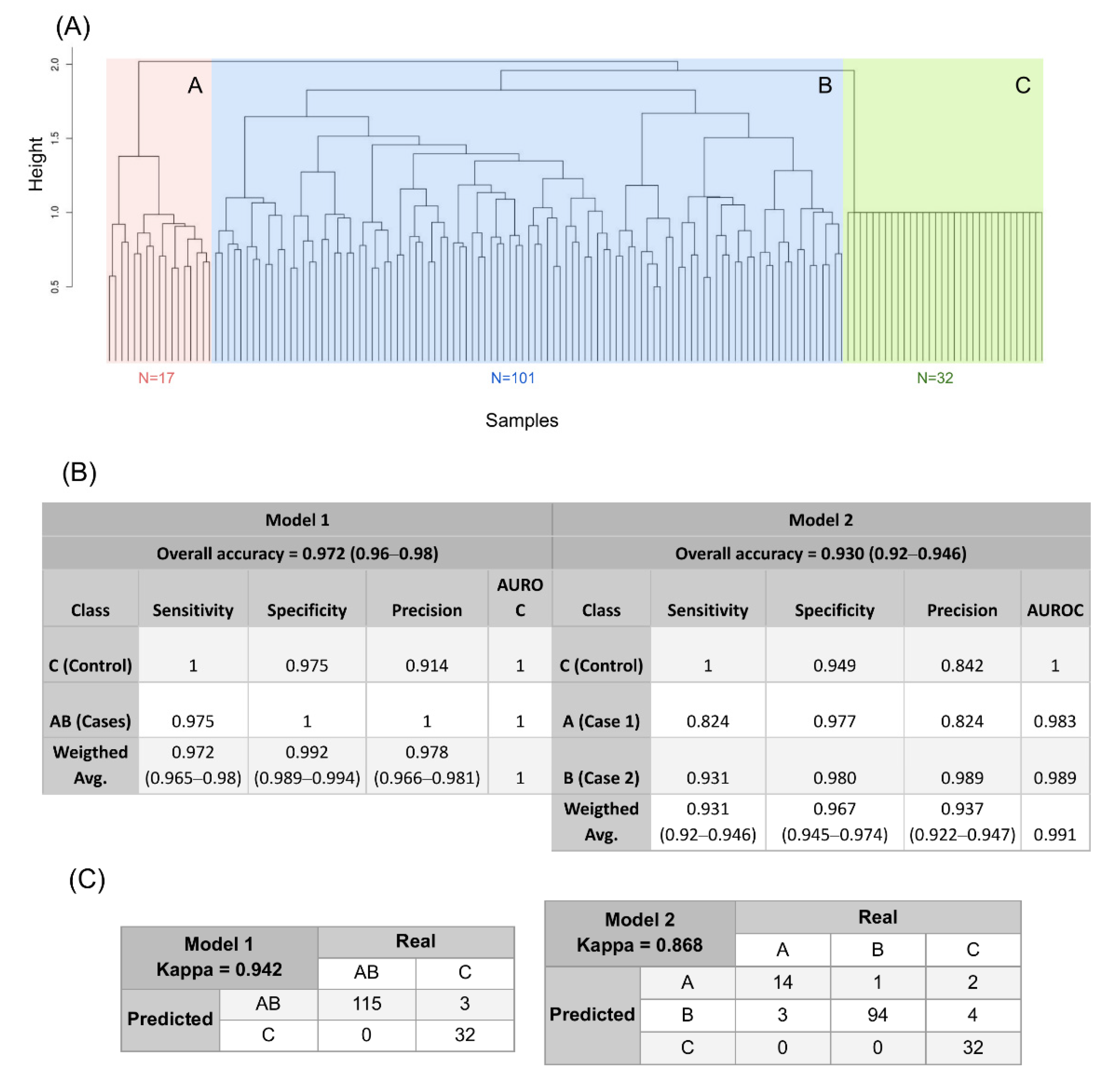
3.4. A New Genomic Taxonomy of OF
3.5. Testing the Genomic Predictive Model in the IGSR Population
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tucker, E.; Grover, S.R.; Bachelot, A.; Touraine, P.; Sinclair, A.H. Premature Ovarian Insufficiency: New Perspectives on Genetic Cause and Phenotypic Spectrum. Endocr. Rev. 2016, 37, 609–635. [Google Scholar] [CrossRef] [PubMed]
- Coulam, C.B.; Adamson, S.C.; Annegers, J.F. Incidence of Premature Ovarian Failure. Obstet. Gynecol. Surv. 1987, 42, 182–183. [Google Scholar] [CrossRef]
- Salvador-Carulla, L.; Bertelli, M.; Martinez-Leal, R. The road to 11th edition of the International Classification of Diseases: Trajectories of scientific consensus and contested science in the classification of intellectual disability/intellectual developmental disorders. Curr. Opin. Psychiatry 2018, 31, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Torrealday, S.; Kodaman, P.; Pal, L. Premature Ovarian Insufficiency—An update on recent advances in understanding and management. F1000Research 2017, 6, 2069. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, E.; Kruszewska, J.; Klicka, K.; Kowalczyk, J.; Grymowicz, M.; Skórska, J.; Pięta, W.; Smolarczyk, R. Premature ovarian insufficiency—Aetiopathology, epidemiology, and diagnostic evaluation. Menopausal Rev. 2018, 17, 105–108. [Google Scholar] [CrossRef]
- Kirshenbaum, M.; Orvieto, R. Premature ovarian insufficiency (POI) and autoimmunity—An update appraisal. J. Assist. Reprod. Genet. 2019, 36, 2207–2215. [Google Scholar] [CrossRef]
- Calik-Ksepka, A.; Grymowicz, M.; Bronkiewicz, W.; Urban, A.; Mierzejewski, K.; Rudnicka, E.; Smolarczyk, R. Spontaneous pregnancy in a patient with premature ovarian insufficiency—Case report. Menopausal Rev. 2018, 17, 139–140. [Google Scholar] [CrossRef]
- Cohen, J.L.; Chabbert-Buffet, N.; Darai, E. Diminished ovarian reserve, premature ovarian failure, poor ovarian responder—A plea for universal definitions. J. Assist. Reprod. Genet. 2015, 32, 1709–1712. [Google Scholar] [CrossRef] [Green Version]
- Pastore, L.M.; Christianson, M.S.; Stelling, J.; Kearns, W.G.; Segars, J.H. Reproductive ovarian testing and the alphabet soup of diagnoses: DOR, POI, POF, POR, and FOR. J. Assist. Reprod. Genet. 2018, 35, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Polson, D.W. Diagnosis and management of premature ovarian failure. Obstet. Gynaecol. 2011, 13, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Vabre, P.; Gatimel, N.; Moreau, J.; Gayrard, V.; Picard-Hagen, N.; Parinaud, J.; Léandri, R. Environmental pollutants, a possible etiology for premature ovarian insufficiency: A narrative review of animal and human data. Environ. Health 2017, 16, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck-Peccoz, P.; Persani, L. Premature ovarian failure. Orphanet J. Rare Dis. 2006, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Iwase, A.; Nakamura, T.; Osuka, S.; Takikawa, S.; Goto, M.; Kikkawa, F. Anti-Müllerian hormone as a marker of ovarian reserve: What have we learned, and what should we know? Reprod. Med. Biol. 2016, 15, 127–136. [Google Scholar] [CrossRef]
- La Marca, A.; Sighinolfi, G.; Papaleo, E.; Cagnacci, A.; Volpe, A.; Faddy, M.J. Prediction of Age at Menopause from Assessment of Ovarian Reserve May Be Improved by Using Body Mass Index and Smoking Status. PLoS ONE 2013, 8, e57005. [Google Scholar] [CrossRef] [Green Version]
- Alipour, F.; Rasekhjahromi, A.; Maalhagh, M.; Sobhanian, S.; Hosseinpoor, M. Comparison of Specificity and Sensitivity of AMH and FSH in Diagnosis of Premature Ovarian Failure. Dis. Markers 2015, 2015, 585604. [Google Scholar] [CrossRef] [Green Version]
- Gleicher, N.; A Kushnir, V.; Barad, D.H. Prospectively assessing risk for premature ovarian senescence in young females: A new paradigm. Reprod. Biol. Endocrinol. 2015, 13, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleicher, N.; Weghofer, A.; Barad, D.H. Defining ovarian reserve to better understand ovarian aging. Reprod. Biol. Endocrinol. 2011, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirnezami, R.; Nicholson, J.; Darzi, A. Preparing for Precision Medicine. N. Engl. J. Med. 2012, 366, 489–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huhtaniemi, I.; Hovatta, O.; La Marca, A.; Livera, G.; Monniaux, D.; Persani, L.; Heddar, A.; Jarzabek, K.; Laisk-Podar, T.; Salumets, A.; et al. Advances in the Molecular Pathophysiology, Genetics, and Treatment of Primary Ovarian Insufficiency. Trends Endocrinol. Metab. 2018, 29, 400–419. [Google Scholar] [CrossRef] [Green Version]
- Fortuño, C.; Labarta, E. Genetics of primary ovarian insufficiency: A review. J. Assist. Reprod. Genet. 2014, 31, 1573–1585. [Google Scholar] [CrossRef] [Green Version]
- Jiao, X.; Ke, H.; Qin, Y.; Chen, Z.-J. Molecular Genetics of Premature Ovarian Insufficiency. Trends Endocrinol. Metab. 2018, 29, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Patiño, L.C.; Beau, I.; Carlosama, C.; Buitrago, J.C.; González, R.; Suárez, C.F.; Patarroyo, M.A.; Delemer, B.; Young, J.; Binart, N.; et al. New mutations in non-syndromic primary ovarian insufficiency patients identified via whole-exome sequencing. Hum. Reprod. 2017, 32, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Knauff, E.A.H.; Franke, L.; Van Es, M.A.; Van Den Berg, L.H.; Van Der Schouw, Y.T.; Laven, J.S.E.; Lambalk, C.B.; Hoek, A.; Goverde, A.J.; Christin-Maitre, S.; et al. Genome-wide association study in premature ovarian failure patients suggests ADAMTS19 as a possible candidate gene. Hum. Reprod. 2009, 24, 2372–2378. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, B.; Zhang, W.; Chen, B.; Luo, M.; Wang, J.; Wang, X.; Cao, Y.; Kee, K. A homozygous NOBOX truncating variant causes defective transcriptional activation and leads to primary ovarian insufficiency. Hum. Reprod. 2017, 32, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Pasipoularides, A. The new era of whole-exome sequencing in congenital heart disease: Brand-new insights into rare pathogenic variants. J. Thorac. Dis. 2018, 10, S1923–S1929. [Google Scholar] [CrossRef]
- Wang, B.; Li, L.; Zhu, Y.; Zhang, W.; Wang, X.; Chen, B.; Li, T.; Pan, H.; Wang, J.; Kee, K.; et al. Sequence variants of KHDRBS1 as high penetrance susceptibility risks for primary ovarian insufficiency by mis-regulating mRNA alternative splicing. Hum. Reprod. 2017, 32, 2138–2146. [Google Scholar] [CrossRef]
- Tucker, E.J.; Jaillard, S.; Grover, S.R.; van den Bergen, J.; Robevska, G.; Bell, K.M.; Sadedin, S.; Hanna, C.; Dulon, J.; Touraine, P.; et al. TP63-truncating variants cause isolated premature ovarian insufficiency. Hum. Mutat. 2019, 40, 886–892. [Google Scholar] [CrossRef]
- Li, Y.; Vinckenbosch, N.; Tian, G.; Huerta-Sanchez, E.; Jiang, T.; Jiang, H.; Albrechtsen, A.; Andersen, G.; Cao, H.; Korneliussen, T.S.; et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 2010, 42, 969–972. [Google Scholar] [CrossRef]
- Jaillard, S.; Sreenivasan, R.; Beaumont, M.; Robevska, G.; Dubourg, C.; Knarston, I.M.; Akloul, L.; van den Bergen, J.; Odent, S.; Croft, B.; et al. Analysis of NR5A1 in 142 patients with premature ovarian insufficiency, diminished ovarian reserve, or unexplained infertility. Maturitas 2020, 131, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Guo, T.; Gong, Z.; Yu, Y.; Zhang, Y.; Zhao, S.; Qin, Y. Novel FSHR mutations in Han Chinese women with sporadic premature ovarian insufficiency. Mol. Cell. Endocrinol. 2019, 492, 110446. [Google Scholar] [CrossRef]
- Wang, Q.; Li, D.; Cai, B.; Chen, Q.; Li, C.; Wu, Y.; Jin, L.; Wang, X.; Zhang, X.; Zhang, F. Whole-exome sequencing reveals SALL4 variants in premature ovarian insufficiency: An update on genotype-phenotype correlations. Hum. Genet. 2019, 138, 83–92. [Google Scholar] [CrossRef]
- Yang, X.; Touraine, P.; Desai, S.; Humphreys, G.; Jiang, H.; Yatsenko, A.; Rajkovic, A. Gene variants identified by whole-exome sequencing in 33 French women with premature ovarian insufficiency. J. Assist. Reprod. Genet. 2019, 36, 39–45. [Google Scholar] [CrossRef]
- Trakadis, Y.J.; Sardaar, S.; Chen, A.; Fulginiti, V.; Krishnan, A. Machine learning in schizophrenia genomics, a case-control study using 5090 exomes. Am. J. Med. Genet. Neuropsychiatr. Genet. 2019, 180, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.S.W.; Schierding, W.; Wake, M.; Saffery, R.; O’Sullivan, J. Machine Learning SNP Based Prediction for Precision Medicine. Front. Genet. 2019, 10, 267. [Google Scholar] [CrossRef] [Green Version]
- Valdes, G.; Luna, J.; Eaton, E.; Ii, C.B.S.; Ungar, L.H.; Solberg, T.D. MediBoost: A Patient Stratification Tool for Interpretable Decision Making in the Era of Precision Medicine. Sci. Rep. 2016, 6, 37854. [Google Scholar] [CrossRef] [PubMed]
- Fairley, S.; Lowy-Gallego, E.; Perry, E.; Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020, 48, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Sudmant, P.; Rausch, T.; Gardner, E.; Handsaker, R.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.-Y.; et al. An integrated map of structural variation in 2504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Wilcoxon, F. Probability Tables for Individual Comparisons by Ranking Methods. Biometrics 1947, 3, 119–122. [Google Scholar] [CrossRef]
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Fisher, R.A. On the Interpretation of χ2 from Contingency Tables, and the Calculation of P. J. R. Stat. Soc. 1922, 85, 87–94. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2017. [Google Scholar]
- van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009; ISBN 1441412697. [Google Scholar]
- Kosub, S. A note on the triangle inequality for the Jaccard distance. Pattern Recognit. Lett. 2019, 120, 36–38. [Google Scholar] [CrossRef] [Green Version]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Witten, I.H.; Frank, E.; Hall, M.A.; Pal, C.J. Data Mining: Practical Machine Learning Tools and Techniques; Elsevier Inc.: Amsterdam, The Netherlands, 2016; ISBN 9780128042915. [Google Scholar]
- Murtagh, F.; Legendre, P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ishwaran, H. Random forests for genomic data analysis. Genomics 2012, 99, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Guo, X.; Yu, H. Variable selection using Mean Decrease Accuracy and Mean Decrease Gini based on Random Forest. In Proceedings of the 2016 7th IEEE International Conference on Software Engineering and Service Science (ICSESS), Beijing, China, 26–28 August 2016; pp. 219–224. [Google Scholar]
- Xu, K.; Chen, X.; Yang, H.; Xu, Y.; He, Y.; Wang, C.; Huang, H.; Liu, B.; Liu, W.; Li, J.; et al. Maternal Sall4 Is Indispensable for Epigenetic Maturation of Mouse Oocytes. J. Biol. Chem. 2017, 292, 1798–1807. [Google Scholar] [CrossRef] [Green Version]
- Delcour, C.; Amazit, L.; Patino, L.C.; Magnin, F.; Fagart, J.; Delemer, B.; Young, J.; Laissue, P.; Binart, N.; Beau, I. ATG7 and ATG9A loss-of-function variants trigger autophagy impairment and ovarian failure. Genet. Med. 2019, 21, 930–938. [Google Scholar] [CrossRef]
- Patiño, L.C.; Beau, I.; Morel, A.; Delemer, B.; Young, J.; Binart, N.; Laissue, P. Functional evidence implicating NOTCH2 missense mutations in primary ovarian insufficiency etiology. Hum. Mutat. 2019, 40, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Orr, H.A. Fitness and its role in evolutionary genetics. Nat. Rev. Genet. 2009, 10, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Sun, M.; You, L.; Wei, D.; Sun, J.; Liang, X.; Zhang, B.; Jiang, H.; Xu, J.; Chen, Z.-J. ESR1, HK3 and BRSK1 gene variants are associated with both age at natural menopause and premature ovarian failure. Orphanet J. Rare Dis. 2012, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Zhao, H.; Xu, J.; Shi, Y.; Li, Z.; Qiao, J.; Liu, J.; Ren, C.; Chen, S.; Cao, Y.; et al. Association of 8q22.3 locus in Chinese Han with idiopathic premature ovarian failure (POF). Hum. Mol. Genet. 2012, 21, 430–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, J.R.B.; Corre, T.; Esko, T.; Chasman, D.I.; Fischer, K.; Franceschini, N.; He, C.; Kutalik, Z.; Mangino, M.; Rose, L.M.; et al. A genome-wide association study of early menopause and the combined impact of identified variants. Hum. Mol. Genet. 2013, 22, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jia, P.; Wolfinger, R.D.; Chen, X.; Zhao, Z. Gene set analysis of genome-wide association studies: Methodological issues and perspectives. Genomics 2011, 98, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bramble, M.; Goldstein, E.H.; Lipson, A.; Ngun, T.; Eskin, A.; Gosschalk, J.E.; Roach, L.; Vashist, N.; Barseghyan, H.; Lee, E.; et al. A novel follicle-stimulating hormone receptor mutation causing primary ovarian failure: A fertility application of whole exome sequencing. Hum. Reprod. 2016, 31, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Philibert, P.; Paris, F.; Lakhal, B.; Audran, F.; Gaspari, L.; Saâd, A.; Christin-Maitre, S.; Bouchard, P.; Sultan, C. NR5A1 (SF-1) gene variants in a group of 26 young women with XX primary ovarian insufficiency. Fertil. Steril. 2013, 99, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.A.; Balding, D. Effects of population structure on DNA fingerprint analysis in forensic science. Heredity 1991, 66, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipkin, S.M.; Moens, P.B.; Wang, V.; Lenzi, M.; Shanmugarajah, D.; Gilgeous, A.; Thomas, J.; Cheng, J.; Touchman, J.W.; Green, E.D.; et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat. Genet. 2002, 31, 385–390. [Google Scholar] [CrossRef]
- Kumar, T.R.; Wiseman, A.L.; Kala, G.; Kala, S.V.; Matzuk, M.M.; Lieberman, M.W. Reproductive Defects in γ-Glutamyl Transpeptidase-Deficient Mice. Endocrinology 2000, 141, 4270–4277. [Google Scholar] [CrossRef]
- Su, W.; Guan, X.; Zhang, D.; Sun, M.; Yang, L.; Yi, F.; Hao, F.; Feng, X.; Ma, T. Occurrence of multi-oocyte follicles in aquaporin 8-deficient mice. Reprod. Biol. Endocrinol. 2013, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.H.-X.; Yang, Y.; Lam, S.K.; Wong, W.M.; Leung, S.Y.; Yuen, S.T.; Elia, G.; Wright, N.A.; Wong, B.C.-Y. Aberrant epithelial expression of trefoil family factor 2 and mucin 6 in Helicobacter pylori infected gastric antrum, incisura, and body and its association with antralisation. J. Clin. Pathol. 2004, 57, 861–866. [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, K.; Yasuda, M.; Kajiwara, H.; Itoh, J.; Miyazawa, M.; Hirasawa, T.; Muramatsu, T.; Murakami, M.; Mikami, M.; Osamura, R.Y. Alterations in Mucin Expression in Ovarian Mucinous Tumors: Immunohistochemical Analysis of MUC2, MUC5AC, MUC6, and CD10 Expression. Acta Histochem. Cytochem. 2008, 41, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Jiao, X.; Simpson, J.L.; Chen, Z.-J. Genetics of primary ovarian insufficiency: New developments and opportunities. Hum. Reprod. Update 2015, 21, 787–808. [Google Scholar] [CrossRef]
- Feng, Y.; Vlassis, A.; Roques, C.; LaLonde, M.; González-Aguilera, C.; Lambert, J.; Lee, S.; Zhao, X.; Alabert, C.; Johansen, J.V.; et al. BRPF 3-HBO 1 regulates replication origin activation and histone H3K14 acetylation. EMBO J. 2016, 35, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Swiech, L.; Kisiel, K.; Czolowska, R.; Zientarski, M.; Borsuk, E. Accumulation and dynamics of proteins of the MCM family during mouse oogenesis and the first embryonic cell cycle. Int. J. Dev. Biol. 2007, 51, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Won, M.; Luo, Y.; Lee, D.-H.; Shin, E.; Suh, D.-S.; Kim, T.-H.; Jin, H.; Bae, J. BAX is an essential key mediator of AP5M1-induced apoptosis in cervical carcinoma cells. Biochem. Biophys. Res. Commun. 2019, 518, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Lussier, J.G.; Diouf, M.N.; Lévesque, V.; Sirois, J.; Ndiaye, K. Gene expression profiling of upregulated mRNAs in granulosa cells of bovine ovulatory follicles following stimulation with hCG. Reprod. Biol. Endocrinol. 2017, 15, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navani, S. The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 6 May 2020).
- Desai, S.; Wood-Trageser, M.; Matic, J.; Chipkin, J.; Jiang, H.; Bachelot, A.; Dulon, J.; Sala, C.; Barbieri, C.; Cocca, M.; et al. MCM8 and MCM9 Nucleotide Variants in Women with Primary Ovarian Insufficiency. J. Clin. Endocrinol. Metab. 2017, 102, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Dondik, Y.; Lei, Z.; Gaskins, J.; Pagidas, K. Minichromosome maintenance complex component 8 and 9 gene expression in the menstrual cycle and unexplained primary ovarian insufficiency. J. Assist. Reprod. Genet. 2019, 36, 57–64. [Google Scholar] [CrossRef]
- Lee, K.Y.; Im, J.-S.; Shibata, E.; Park, J.; Handa, N.; Kowalczykowski, S.C.; Dutta, A. MCM8-9 complex promotes resection of double-strand break ends by MRE11-RAD50-NBS1 complex. Nat. Commun. 2015, 6, 7744. [Google Scholar] [CrossRef]
- Zhang, X.; Abreu, J.G.; Yokota, C.; MacDonald, B.T.; Singh, S.; Coburn, K.L.A.; Cheong, S.-M.; Zhang, M.M.; Ye, Q.-Z.; Hang, H.C.; et al. Tiki1 Is Required for Head Formation via Wnt Cleavage-Oxidation and Inactivation. Cell 2012, 149, 1565–1577. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Kawamura, K.; Takae, S.; Deguchi, M.; Yang, Q.; Kuo, C.; Hsueh, A.J.W. Oocyte-derived R-spondin2 promotes ovarian follicle development. FASEB J. 2013, 27, 2175–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedini, A.; Zamberlam, G.; Lapointe, E.; Tourigny, C.; Boyer, A.; Paquet, M.; Hayashi, K.; Honda, H.; Kikuchi, A.; Price, C.; et al. WNT5a is required for normal ovarian follicle development and antagonizes gonadotropin responsiveness in granulosa cells by suppressing canonical WNT signaling. FASEB J. 2016, 30, 1534–1547. [Google Scholar] [CrossRef] [Green Version]
- Chawengsaksophak, K.; Svingen, T.; Ng, E.T.; Epp, T.; Spiller, C.; Clark, C.; Cooper, H.; Koopman, P. Loss of Wnt5a Disrupts Primordial Germ Cell Migration and Male Sexual Development in Mice1. Biol. Reprod. 2012, 86, 1–12. [Google Scholar] [CrossRef]
- Ibañez-Tallon, I.; Gorokhova, S.; Heintz, N. Loss of function of axonemal dynein Mdnah5 causes primary ciliary dyskinesia and hydrocephalus. Hum. Mol. Genet. 2002, 11, 715–721. [Google Scholar] [CrossRef]
- Hu, J.; Lessard, C.; Longstaff, C.; O’Brien, M.; Palmer, K.; Reinholdt, L.; Eppig, J.; Schimenti, J.; Handel, M.A. ENU-induced mutant allele of Dnah1, ferf1, causes abnormal sperm behavior and fertilization failure in mice. Mol. Reprod. Dev. 2019, 86, 416–425. [Google Scholar] [CrossRef]
- Areal, L.; Pereira, L.P.; Ribeiro, F.; Olmo, I.G.; Muniz, M.R.; do Carmo Rodrigues, M.; Costa, P.F.; Martins-Silva, C.; Ferguson, S.S.G.; Guimarães, D.A.M.; et al. Role of Dynein Axonemal Heavy Chain 6 Gene Expression as a Possible Biomarker for Huntington’s Disease: A Translational Study. J. Mol. Neurosci. 2017, 63, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yagi, H.; Onuoha, E.O.; Damerla, R.R.; Francis, R.; Furutani, Y.; Tariq, M.; King, S.M.; Hendricks, G.; Cui, C.; et al. DNAH6 and Its Interactions with PCD Genes in Heterotaxy and Primary Ciliary Dyskinesia. PLoS Genet. 2016, 12, e1005821. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Chromosome and Position | Change at Sequence and Aa Level | Rs | Type of Change | Amino Acid Class, Polarity, and Charge Change | Gene | Function | N Cases Affected | Coverage | Accession Number |
|---|---|---|---|---|---|---|---|---|---|
| 2; 84897501 2 | c.6356A > G, p.Tyr2119Cys | rs17025409 | Missense variant | Aromatic polar neutral > sulfuric nonpolar neutral | DNAH6 | Microtubule activity | 17 | 146 (18.08) | NM_001370.1 |
| 2; 84932720 2 | c.8576A > G, p.Lys2859Arg | rs61750773 | Missense variant | Basic polar positive > basic polar positive | DNAH6 | Microtubule activity | 19 | 146 (20.02) | NM_001370.1 |
| 2; 85059227 2 | c.1034C > T, p.Arg345His | rs61744273 | Missense variant | Basic polar positive > basic aromatic polar positive-neutral | TRABD2A | Negative regulation of WNT signalling pathway | 18 | 287 (42.23) | NM_001277053.1 |
| 5; 79950724 3 | c.181G > C, p.Ala60Pro | rs2001675 | Missense variant | Aliphatic nonpolar neutral > cyclic nonpolar neutral | MSH3 | DNA repair | 15 | 145 (57.73) | NM_002439.4 |
| 6; 36168628 1 | c.529A > G, p.Ser177Gly | rs45504893 | Missense variant | Hydroxylic polar neutral > aliphatic nonpolar neutral | BRPF3 | Chromatin organisation | 22 | 320 (44.1) | NM_015695.2 |
| 9; 69391207 4 | c.715G > A, p.Ala239Thr | Missense variant | Aliphatic nonpolar neutral > hydroxylic polar neutral | ANKRD20A4 | Unknown | 13 | 125 (8.03) | NM_001098805.1 | |
| 11; 1017471 2 | c.5330G > A, p.Gly1777Asp | Missense variant | Aliphatic nonpolar neutral > acid acidic polar negative | MUC6 | Cytoprotection of epithelial surfaces | 13 | 448 (194.38) | NM_005961.2 | |
| 11; 1017504 1,2 | c.5297C > T, p.Thr1766Ile | Missense variant | Hydroxylic polar neutral > aliphatic nonpolar neutral | MUC6 | Cytoprotection of epithelial surfaces | 31 | 448 (147.15) | NM_005961.2 | |
| 14; 57755564 1 | c.1435G > A, p.Ala479Thr | rs35759976 | Missense variant | Aliphatic nonpolar neutral > hydroxylic polar neutral | AP5M1 | Apoptosis | 22 | 179 (14.19) | NM_018229.3 |
| 16; 25239809 2,3 | c.782G > A, p.Arg261Gln | rs111840156 | Missense variant | Basic polar positive > amide polar neutral | AQP8 | Cellular response to cAMP | 13 | 265 (24.69) | NM_001169.2 |
| 16; 84902483 1 | c.880A > T, p.Met294Leu | rs72799568 | Missense variant | Sulfuric nonpolar neutral > aliphatic nonpolar neutral | CRISPLD2 | Extracellular matrix assembly | 21 | 247 (145.71) | NM_031476.3 |
| 16; 88902199 1 | c.692C > G, p.Ala237Gly | rs34745339 | Structural interaction variant, missense variant | Aliphatic nonpolar neutral > aliphatic nonpolar neutral | GALNS | Degradation of glycosaminoglycans | 20 | 214 (33.02) | NM_000512.4 |
| 17; 71232990 2 | c.1369C > A, p.Arg457Ser | rs61729639 | Missense variant | Basic polar positive > hydroxylic polar neutral | SPEP1 | Unknown | 14 | 180 (38.16) | NM_001288771.1 |
| 22; 17450929 2 | c.841G > A, p.Ala281Thr | rs61741409 | Missense variant | Aliphatic nonpolar neutral > hydroxylic polar neutral | GAB4 | Unknown | 14 | 236 (36.89) | NM_001037814.1 |
| 22; 17450952 2 | c.818T > C, p.Leu273Pro | rs11703655 | Missense variant | Aliphatic nonpolar neutral > cyclic nonpolar neutral | GAB4 | Unknown | 14 | 236 (43.66) | NM_001037814.1 |
| 22; 25024326 3 | c.1534G > A, p.Val512Ile | Structural interaction variant, missense variant | Aliphatic nonpolar neutral > aliphatic nonpolar neutral | GGT1 | Proteolysis | 14 | 112 (17.48 | NM_013430.2 | |
| 22; 35802661 1 | c.539C > G, p.Thr180Ser | rs2307340 | Missense variant | Hydroxylic polar neutral > hydroxylic polar neutral | MCM5 | DNA replication initiation | 20 | 309 (92.27) | NM_006739.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henarejos-Castillo, I.; Aleman, A.; Martinez-Montoro, B.; Gracia-Aznárez, F.J.; Sebastian-Leon, P.; Romeu, M.; Remohi, J.; Patiño-Garcia, A.; Royo, P.; Alkorta-Aranburu, G.; et al. Machine Learning-Based Approach Highlights the Use of a Genomic Variant Profile for Precision Medicine in Ovarian Failure. J. Pers. Med. 2021, 11, 609. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11070609
Henarejos-Castillo I, Aleman A, Martinez-Montoro B, Gracia-Aznárez FJ, Sebastian-Leon P, Romeu M, Remohi J, Patiño-Garcia A, Royo P, Alkorta-Aranburu G, et al. Machine Learning-Based Approach Highlights the Use of a Genomic Variant Profile for Precision Medicine in Ovarian Failure. Journal of Personalized Medicine. 2021; 11(7):609. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11070609
Chicago/Turabian StyleHenarejos-Castillo, Ismael, Alejandro Aleman, Begoña Martinez-Montoro, Francisco Javier Gracia-Aznárez, Patricia Sebastian-Leon, Monica Romeu, Jose Remohi, Ana Patiño-Garcia, Pedro Royo, Gorka Alkorta-Aranburu, and et al. 2021. "Machine Learning-Based Approach Highlights the Use of a Genomic Variant Profile for Precision Medicine in Ovarian Failure" Journal of Personalized Medicine 11, no. 7: 609. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11070609