
Dominance of Fructose-Associated Fructobacillus in the Gut Microbiome of Bumblebees (Bombus terrestris) Inhabiting Natural Forest Meadows

 , , , ,
, , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Species, Collection of Specimens, and Bumblebee Habitats
2.2. Gut Dissection and Microbial DNA Extraction
2.3. 16S V3−V4 rRNA Gene Amplification and Illumina MiSeq Sequencing
2.4. 16S Sequence Analyses
2.5. Statistical Analyses
3. Results
3.1. General Profile of the Sequencing Data
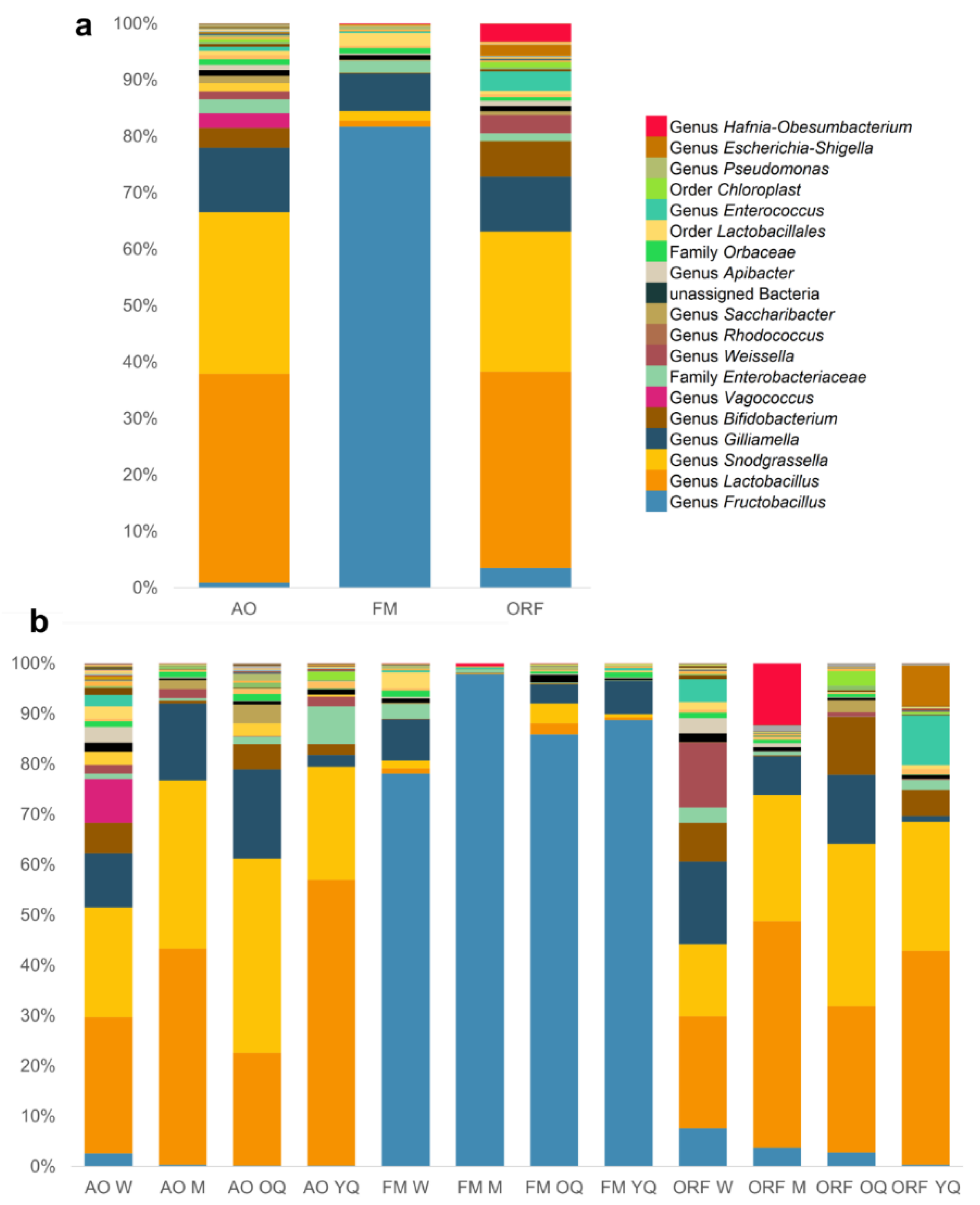
3.2. Bacterial Community Composition
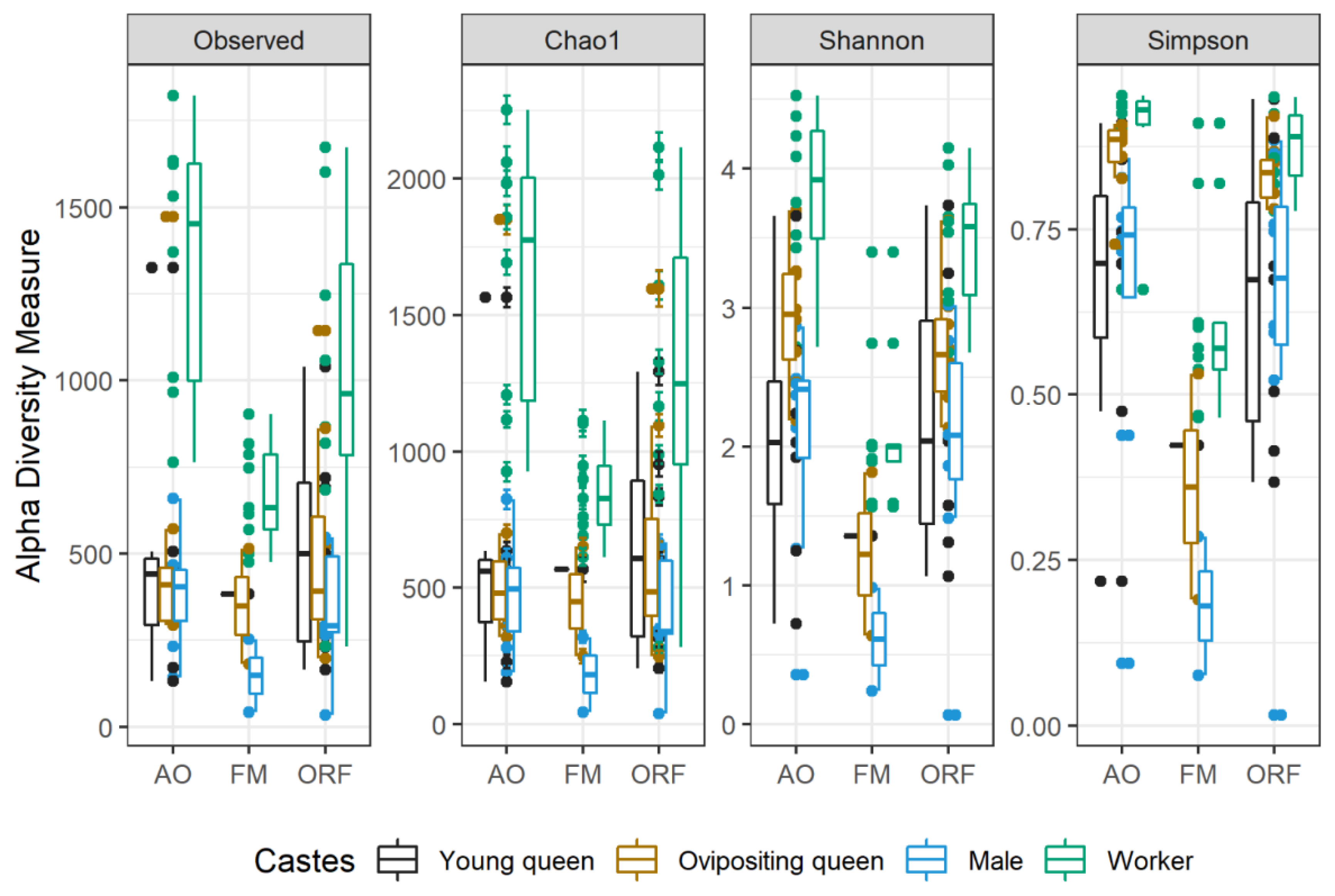
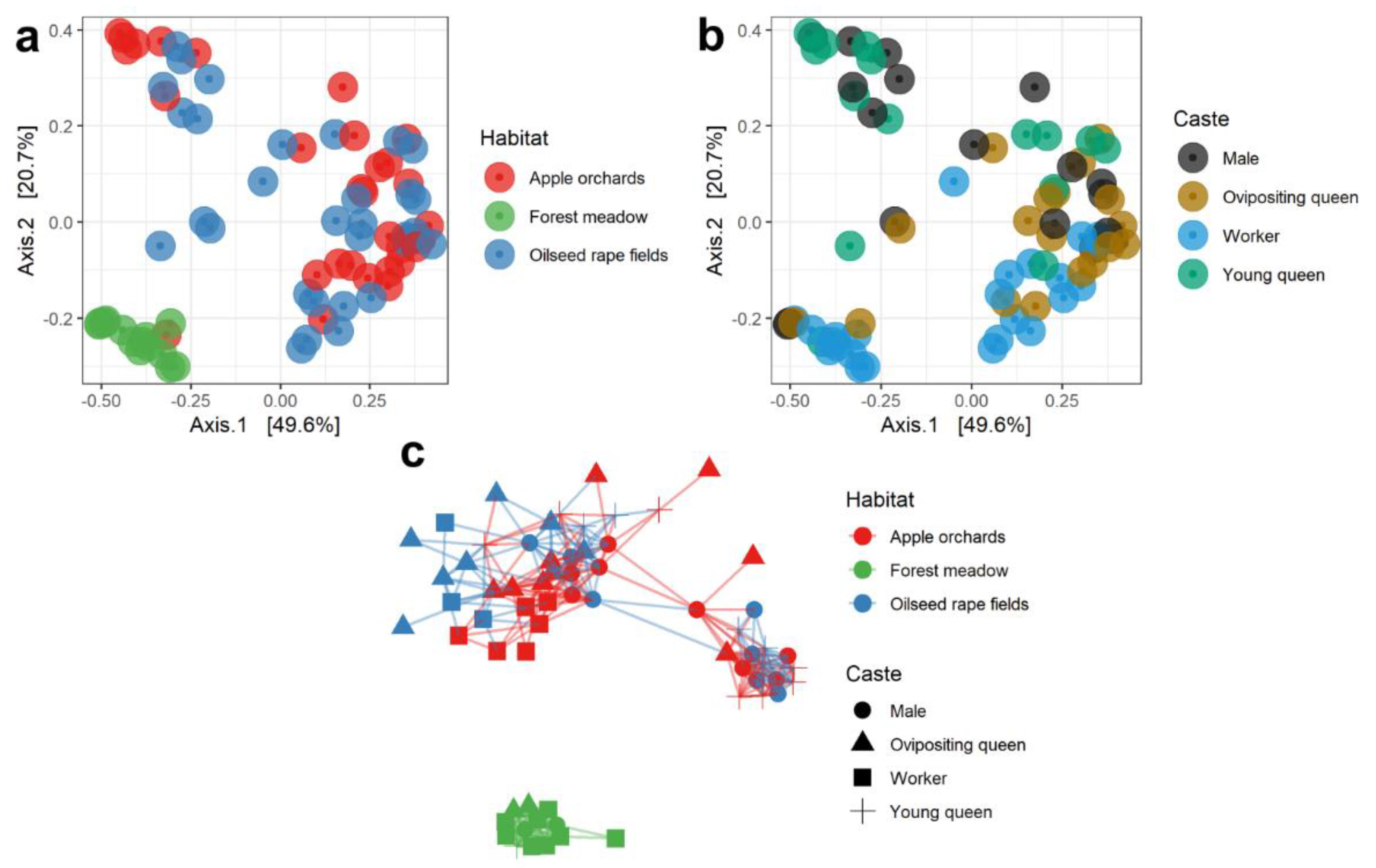
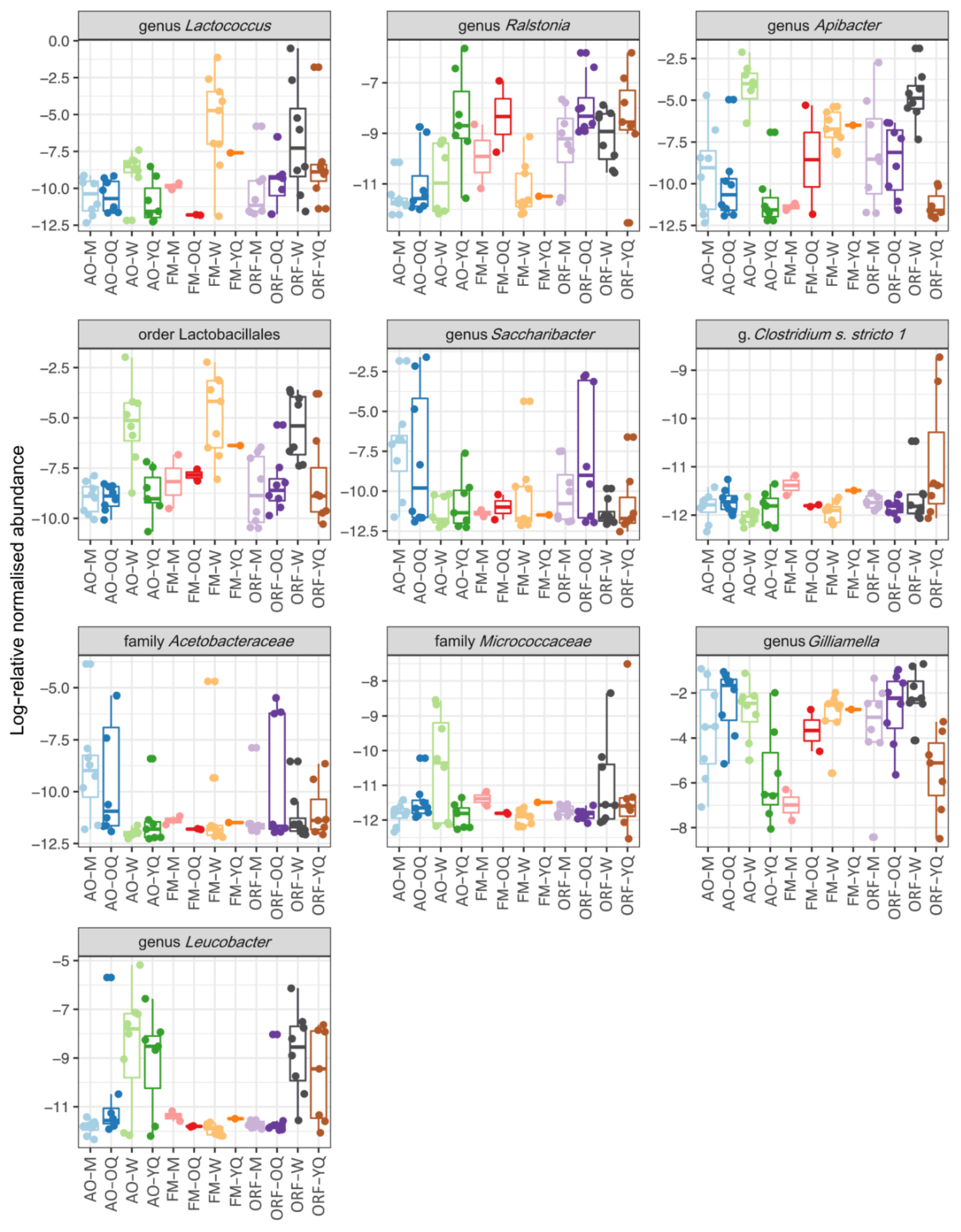
3.3. Comparisons of Microbial Communities from Different Habitats and Castes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meeus, I.; Mommaerts, V.; Billiet, A.; Mosallanejad, H.; Van de Wiele, T.; Wäckers, F.; Smagghe, G. Assessment of Mutualism between Bombus terrestris and Its Microbiota by Use of Microcolonies. Apidologie 2013, 44, 708–719. [Google Scholar] [CrossRef]
- Billiet, A.; Meeus, I.; Cnockaert, M.; Vandamme, P.; Van Oystaeyen, A.; Wäckers, F.; Smagghe, G. Effect of Oral Administration of Lactic Acid Bacteria on Colony Performance and Gut Microbiota in Indoor-Reared Bumblebees (Bombus terrestris). Apidologie 2017, 48, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Bosmans, L.; Pozo, M.I.; Verreth, C.; Crauwels, S.; Wilberts, L.; Sobhy, I.S.; Wäckers, F.; Jacquemyn, H.; Lievens, B. Habitat-Specific Variation in Gut Microbial Communities and Pathogen Prevalence in Bumblebee Queens (Bombus terrestris). PLoS ONE 2018, 13, e0204612. [Google Scholar] [CrossRef] [Green Version]
- Raymann, K.; Moran, N.A. The Role of the Gut Microbiome in Health and Disease of Adult Honey Bee Workers. Curr. Opin. Insect Sci. 2018, 26, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paine, R.T. A Note on Trophic Complexity and Community Stability. Am. Nat. 1969, 103, 91–93. [Google Scholar] [CrossRef]
- Hallmann, C.A.; Sorg, M.; Jongejans, E.; Siepel, H.; Hofland, N.; Schwan, H.; Stenmans, W.; Müller, A.; Sumser, H.; Hörren, T.; et al. More than 75 Percent Decline over 27 Years in Total Flying Insect Biomass in Protected Areas. PLoS ONE 2017, 12, e0185809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollerton, J.; Winfree, R.; Tarrant, S. How Many Flowering Plants Are Pollinated by Animals? Oikos 2011, 120, 321–326. [Google Scholar] [CrossRef]
- Van der Sluijs, J.P.; Vaage, N.S. Pollinators and Global Food Security: The Need for Holistic Global Stewardship. Food Ethics 2016, 1, 75–91. [Google Scholar] [CrossRef] [Green Version]
- Mänd, M.; Mänd, R.; Williams, I.H. Bumblebees in the Agricultural Landscape of Estonia. Agric. Ecosyst. Environ. 2002, 89, 69–76. [Google Scholar] [CrossRef]
- Klein, A.-M.; Vaissière, B.E.; Cane, J.H.; Steffan-Dewenter, I.; Cunningham, S.A.; Kremen, C.; Tscharntke, T. Importance of Pollinators in Changing Landscapes for World Crops. Proc. R. Soc. B. 2007, 274, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Happe, A.-K.; Riesch, F.; Rösch, V.; Gallé, R.; Tscharntke, T.; Batáry, P. Small-Scale Agricultural Landscapes and Organic Management Support Wild Bee Communities of Cereal Field Boundaries. Agric. Ecosyst. Environ. 2018, 254, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Koch, H.; Schmid-Hempel, P. Bacterial Communities in Central European Bumblebees: Low Diversity and High Specificity. Microb. Ecol. 2011, 62, 121–133. [Google Scholar] [CrossRef]
- Moran, N.A.; Hansen, A.K.; Powell, J.E.; Sabree, Z.L. Distinctive Gut Microbiota of Honey Bees Assessed Using Deep Sampling from Individual Worker Bees. PLoS ONE 2012, 7, e36393. [Google Scholar] [CrossRef] [Green Version]
- Cariveau, D.P.; Elijah Powell, J.; Koch, H.; Winfree, R.; Moran, N.A. Variation in Gut Microbial Communities and Its Association with Pathogen Infection in Wild Bumble Bees (Bombus). ISME J. 2014, 8, 2369–2379. [Google Scholar] [CrossRef]
- Meeus, I.; Parmentier, L.; Billiet, A.; Maebe, K.; Van Nieuwerburgh, F.; Deforce, D.; Wäckers, F.; Vandamme, P.; Smagghe, G. 16S RRNA Amplicon Sequencing Demonstrates That Indoor-Reared Bumblebees (Bombus terrestris) Harbor a Core Subset of Bacteria Normally Associated with the Wild Host. PLoS ONE 2015, 10, e0125152. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Powell, J.E.; Guo, J.; Evans, J.D.; Wu, J.; Williams, P.; Lin, Q.; Moran, N.A.; Zhang, Z. Two Gut Community Enterotypes Recur in Diverse Bumblebee Species. Curr. Biol. 2015, 25, R652–R653. [Google Scholar] [CrossRef] [Green Version]
- Tauber, J.P.; McMahon, D.; Ryabov, E.V.; Kunat, M.; Ptaszyńska, A.A.; Evans, J.D. Honeybee Intestines Retain Low Yeast Titers, but No Bacterial Mutualists, at Emergence. Yeast 2021, yea.3665. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, A.; Meeus, I.; Van Nieuwerburgh, F.; Deforce, D.; Vandamme, P.; Smagghe, G. A Different Gut Microbial Community between Larvae and Adults of a Wild Bumblebee Nest (Bombus pascuorum): Gut Bacteria in Larval and Adult Bombus. Insect Sci. 2018, 25, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, J.; Li, K.; Sadd, B.M.; Guo, Y.; Zhuang, D.; Zhang, Z.; Chen, Y.; Evans, J.D.; Guo, J.; et al. Dynamic Changes of Gut Microbial Communities of Bumble Bee Queens through Important Life Stages. mSystems 2019, 4, e00631-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graystock, P.; Goulson, D.; Hughes, W.O.H. Parasites in Bloom: Flowers Aid Dispersal and Transmission of Pollinator Parasites within and between Bee Species. Proc. R. Soc. B. 2015, 282, 20151371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praet, J.; Parmentier, A.; Schmid-Hempel, R.; Meeus, I.; Smagghe, G.; Vandamme, P. Large-Scale Cultivation of the Bumblebee Gut Microbiota Reveals an Underestimated Bacterial Species Diversity Capable of Pathogen Inhibition: Large-Scale Cultivation of the Bumblebee Gut Microbiota. Env. Microbiol. 2018, 20, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Crailsheim, K. The Protein Balance of the Honey Bee Worker. Apidologie 1990, 21, 417–429. [Google Scholar] [CrossRef]
- Moritz, B.; Crailsheim, K. Physiology of Protein Digestion in the Midgut of the Honeybee (Apis mellifera L.). J. Insect Physiol. 1987, 33, 923–931. [Google Scholar] [CrossRef]
- Crailsheim, K.; Schneider, L.H.W.; Hrassnigg, N.; Bühlmann, G.; Brosch, U.; Gmeinbauer, R.; Schöffmann, B. Pollen Consumption and Utilization in Worker Honeybees (Apis mellifera carnica): Dependence on Individual Age and Function. J. Insect Physiol. 1992, 38, 409–419. [Google Scholar] [CrossRef]
- Szolderits, M.J.; Crailsheim, K. A Comparison of Pollen Consumption and Digestion in Honeybee (Apis mellifera carnica) Drones and Workers. J. Insect Physiol. 1993, 39, 877–881. [Google Scholar] [CrossRef]
- Kapheim, K.M.; Rao, V.D.; Yeoman, C.J.; Wilson, B.A.; White, B.A.; Goldenfeld, N.; Robinson, G.E. Caste-Specific Differences in Hindgut Microbial Communities of Honey Bees (Apis mellifera). PLoS ONE 2015, 10, e0123911. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; Vanderplas, J.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kwong, W.K.; Engel, P.; Koch, H.; Moran, N.A. Genomics and Host Specialization of Honey Bee and Bumble Bee Gut Symbionts. Proc. Natl. Acad. Sci. USA 2014, 111, 11509–11514. [Google Scholar] [CrossRef] [Green Version]
- Endo, A.; Maeno, S.; Tanizawa, Y.; Kneifel, W.; Arita, M.; Dicks, L.; Salminen, S. Fructophilic Lactic Acid Bacteria, a Unique Group of Fructose-Fermenting Microbes. Appl. Env. Microbiol. 2018, 84, e01290-18. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.J.; Rusch, D.B.; Stewart, F.J.; Mattila, H.R.; Newton, I.L.G. Saccharide Breakdown and Fermentation by the Honey Bee Gut Microbiome: Fermentation by Honey Bee Gut Microbes. Env. Microbiol. 2015, 17, 796–815. [Google Scholar] [CrossRef]
- McFrederick, Q.S.; Cannone, J.J.; Gutell, R.R.; Kellner, K.; Plowes, R.M.; Mueller, U.G. Specificity between Lactobacilli and Hymenopteran Hosts Is the Exception Rather than the Rule. Appl. Environ. Microbiol. 2013, 79, 1803–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.E.; Sheehan, T.H.; Mott, B.M.; Maes, P.; Snyder, L.; Schwan, M.R.; Walton, A.; Jones, B.M.; Corby-Harris, V. Microbial Ecology of the Hive and Pollination Landscape: Bacterial Associates from Floral Nectar, the Alimentary Tract and Stored Food of Honey Bees (Apis mellifera). PLoS ONE 2013, 8, e83125. [Google Scholar] [CrossRef] [Green Version]
- Rokop, Z.P.; Horton, M.A.; Newton, I.L.G. Interactions between Cooccurring Lactic Acid Bacteria in Honey Bee Hives. Appl. Environ. Microbiol. 2015, 81, 7261–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeno, S.; Tanizawa, Y.; Kanesaki, Y.; Kubota, E.; Kumar, H.; Dicks, L.; Salminen, S.; Nakagawa, J.; Arita, M.; Endo, A. Genomic Characterization of a Fructophilic Bee Symbiont Lactobacillus Kunkeei Reveals Its Niche-Specific Adaptation. Syst. Appl. Microbiol. 2016, 39, 516–526. [Google Scholar] [CrossRef]
- Filannino, P.; Di Cagno, R.; Addante, R.; Pontonio, E.; Gobbetti, M. Metabolism of Fructophilic Lactic Acid Bacteria Isolated from the Apis mellifera L. Bee Gut: Phenolic Acids as External Electron Acceptors. Appl. Environ. Microbiol. 2016, 82, 6899–6911. [Google Scholar] [CrossRef] [Green Version]
- Endo, A.; Futagawa-Endo, Y.; Dicks, L.M.T. Isolation and Characterization of Fructophilic Lactic Acid Bacteria from Fructose-Rich Niches. Syst. Appl. Microbiol. 2009, 32, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Makino, T.T.; Ohashi, K.; Sakai, S. How Do Floral Display Size and the Density of Surrounding Flowers Influence the Likelihood of Bumble Bee Revisitation to a Plant? Funct. Ecol. 2007, 21, 87–95. [Google Scholar] [CrossRef]
- Knauer, A.C.; Schiestl, F.P. Bees Use Honest Floral Signals as Indicators of Reward When Visiting Flowers. Ecol Lett 2015, 18, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, J.M.; Cook, S.M.; Wright, G.A.; Osborne, J.L.; Clark, S.J.; Swain, J.L.; Haughton, A.J. Oilseed Rape (Brassica napus) as a Resource for Farmland Insect Pollinators: Quantifying Floral Traits in Conventional Varieties and Breeding Systems. GCB Bioenergy 2017, 9, 1370–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calder, A. Oilseed Rape and Bees; Northern Bee Books: Hebden Bridge, UK, 1986. [Google Scholar]
- Sasaki, H. Changes in Sugar Content during Cold Acclimation and Deacclimation of Cabbage Seedlings. Ann. Bot. 1996, 78, 365–369. [Google Scholar] [CrossRef] [Green Version]
- Akšić, M.F.; Tosti, T.; Nedić, N.; Marković, M.; Ličina, V.; Milojković-Opsenica, D.; Tešić, Ž. Influence of Frost Damage on the Sugars and Sugar Alcohol Composition in Quince (Cydonia oblonga Mill.) Floral Nectar. Acta Physiol. Plant 2015, 37, 1701. [Google Scholar] [CrossRef]
- Lin, I.W.; Sosso, D.; Chen, L.-Q.; Gase, K.; Kim, S.-G.; Kessler, D.; Klinkenberg, P.M.; Gorder, M.K.; Hou, B.-H.; Qu, X.-Q.; et al. Nectar Secretion Requires Sucrose Phosphate Synthases and the Sugar Transporter SWEET9. Nature 2014, 508, 546–549. [Google Scholar] [CrossRef]
- Borghi, M.; Perez de Souza, L.; Yoshida, T.; Fernie, A.R. Flowers and Climate Change: A Metabolic Perspective. New Phytol. 2019, 224, 1425–1441. [Google Scholar] [CrossRef] [Green Version]
- Kehrberger, S.; Holzschuh, A. How Does Timing of Flowering Affect Competition for Pollinators, Flower Visitation and Seed Set in an Early Spring Grassland Plant? Sci. Rep. 2019, 9, 15593. [Google Scholar] [CrossRef]
- Acosta, A.L.; Giannini, T.C.; Imperatriz-Fonseca, V.L.; Saraiva, A.M. Worldwide Alien Invasion: A Methodological Approach to Forecast the Potential Spread of a Highly Invasive Pollinator. PLoS ONE 2016, 11, e0148295. [Google Scholar] [CrossRef]
- Vásquez, A.; Forsgren, E.; Fries, I.; Paxton, R.J.; Flaberg, E.; Szekely, L.; Olofsson, T.C. Symbionts as Major Modulators of Insect Health: Lactic Acid Bacteria and Honeybees. PLoS ONE 2012, 7, e33188. [Google Scholar] [CrossRef]
- Powell, J.E.; Martinson, V.G.; Urban-Mead, K.; Moran, N.A. Routes of Acquisition of the Gut Microbiota of the Honey Bee Apis mellifera. Appl. Environ. Microbiol. 2014, 80, 7378–7387. [Google Scholar] [CrossRef] [Green Version]
- Filannino, P.; Di Cagno, R.; Tlais, A.Z.A.; Cantatore, V.; Gobbetti, M. Fructose-Rich Niches Traced the Evolution of Lactic Acid Bacteria toward Fructophilic Species. Crit. Rev. Microbiol. 2019, 45, 65–81. [Google Scholar] [CrossRef]
- Endo, A.; Salminen, S. Honeybees and Beehives Are Rich Sources for Fructophilic Lactic Acid Bacteria. Syst. Appl. Microbiol. 2013, 36, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, D.; Castelli, L.; Porrini, M.P.; Garrido, P.M.; Eguaras, M.J.; Zunino, P.; Antúnez, K. Lactobacillus Kunkeei Strains Decreased the Infection by Honey Bee Pathogens Paenibacillus Larvae and Nosema ceranae. Benef. Microbes 2018, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Berríos, P.; Fuentes, J.A.; Salas, D.; Carreño, A.; Aldea, P.; Fernández, F.; Trombert, A.N. Inhibitory Effect of Biofilm-Forming Lactobacillus Kunkeei Strains against Virulent Pseudomonas aeruginosa in Vitro and in Honeycomb Moth (Galleria mellonella) Infection Model. Benef. Microbes 2018, 9, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Ptaszyńska, A.A.; Latoch, P.; Hurd, P.J.; Polaszek, A.; Michalska-Madej, J.; Grochowalski, Ł.; Strapagiel, D.; Gnat, S.; Załuski, D.; Gancarz, M.; et al. Amplicon Sequencing of Variable 16S RRNA from Bacteria and ITS2 Regions from Fungi and Plants, Reveals Honeybee Susceptibility to Diseases Results from Their Forage Availability under Anthropogenic Landscapes. Pathogens 2021, 10, 381. [Google Scholar] [CrossRef]
- Pachla, A.; Ptaszyńska, A.A.; Wicha, M.; Kunat, M.; Wydrych, J.; Oleńska, E.; Małek, W. Insight into Probiotic Properties of Lactic Acid Bacterial Endosymbionts of Apis mellifera L. Derived from the Polish Apiary. Saudi Journal of Biological Sciences 2021, 28, 1890–1899. [Google Scholar] [CrossRef]
- Sõber, V.; Leps, M.; Kaasik, A.; Mänd, M.; Teder, T. Forest Proximity Supports Bumblebee Species Richness and Abundance in Hemi-Boreal Agricultural Landscape. Agric. Ecosyst. Environ. 2020, 298, 106961. [Google Scholar] [CrossRef]
- Gancarz, M.; Hurd, P.J.; Latoch, P.; Polaszek, A.; Michalska-Madej, J.; Grochowalski, Ł.; Strapagiel, D.; Gnat, S.; Załuski, D.; Rusinek, R.; et al. Dataset of the Next-Generation Sequencing of Variable 16S RRNA from Bacteria and ITS2 Regions from Fungi and Plants Derived from Honeybees Kept under Anthropogenic Landscapes. Data in Brief 2021, 36, 107019. [Google Scholar] [CrossRef] [PubMed]
- Vásquez, A.; Olofsson, T.C. The Lactic Acid Bacteria Involved in the Production of Bee Pollen and Bee Bread. J. Apic. Res. 2009, 48, 189–195. [Google Scholar] [CrossRef]
- Koch, H.; Schmid-Hempel, P. Socially Transmitted Gut Microbiota Protect Bumble Bees against an Intestinal Parasite. Proc. Natl. Acad. Sci. USA 2011, 108, 19288–19292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Huang, M.-F.; Qiu, L.-F.; Song, R.-H.; Zhang, Z.-H.; Ding, Y.-W.; Zhou, X.; Zhang, H.; Zheng, H. Diversity and Functional Analysis of Chinese Bumblebee Gut Microbiota Reveal the Metabolic Niche and Antibiotic Resistance Variation of Gilliamella. Insect Sci. 2021, 28, 302–314. [Google Scholar] [CrossRef]
- Bakker, E.S.; Olff, H.; Vandenberghe, C.; De Maeyer, K.; Smit, R.; Gleichman, J.M.; Vera, F.W.M. Ecological Anachronisms in the Recruitment of Temperate Light-Demanding Tree Species in Wooded Pastures. J Appl Ecology 2004, 41, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Kull, K.; Zobel, M. High Species Richness in an Estonian Wooded Meadow. Journal of Vegetation Science 1991, 2, 715–718. [Google Scholar] [CrossRef]
- Motta, E.V.S.; Raymann, K.; Moran, N.A. Glyphosate Perturbs the Gut Microbiota of Honey Bees. Proc Natl Acad Sci USA 2018, 115, 10305–10310. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Habitat Type | Caste | Number of Insects Used (Number of Samples) | Observed OTU Richness | Chao1 | Coverage (%) | Shannon | Simpson |
|---|---|---|---|---|---|---|---|
| Apple orchards | ovipositing queen | 24 (24) | 356.5 ± 239.48 | 468.83 ± 352.81 | 74.2 ± 4.91 | 2.92 ± 0.48 | 0.86 ± 0.06 |
| young queen | 63 (21) | 311 ± 227.07 | 428.3 ± 323.22 | 73.37 ± 7.81 | 2.07 ± 0.96 | 0.66 ± 0.24 | |
| worker | 72 (24) | 841.75 ± 253.51 | 1156.98 ± 352.39 | 72.9 ± 3.31 | 3.82 ± 0.59 | 0.89 ± 0.09 | |
| male | 72 (24) | 259.13 ± 90.78 | 345.63 ± 137.85 | 75.89 ± 6.07 | 2.05 ± 0.83 | 0.65 ± 0.26 | |
| Oilseed rape fields | ovipositing queen | 24 (24) | 336.13 ± 191.18 | 450.09 ± 269.67 | 76.22 ± 4.28 | 2.71 ± 0.46 | 0.83 ± 0.05 |
| young queen | 63 (21) | 338.57 ± 177.04 | 455.59 ± 231.6 | 74.25 ± 5.69 | 2.21 ± 1.01 | 0.64 ± 0.23 | |
| worker | 72 (24) | 659 ± 264.68 | 895.48 ± 376.07 | 74.58 ± 5.03 | 3.46 ± 0.49 | 0.88 ± 0.06 | |
| male | 72 (24) | 234.88 ± 114.47 | 308.42 ± 146.96 | 76.17 ± 3.01 | 1.98 ± 0.92 | 0.62 ± 0.28 | |
| Forest meadows | ovipositing queen | 6 (6) | 232.5 ± 196.66 | 288.1 ± 196.66 | 81.06 ±1.1 | 1.22 ± 0.82 | 0.36 ± 0.24 |
| young queen | 18 (6) | 264 | 372.78 | 70.82 | 1.36 | 0.429 | |
| worker | 18 (6) | 415.89 ± 93.36 | 583.84 ± 131.11 | 71.32 ± 2.2 | 2.12 ± 0.59 | 0.62 ± 0.15 | |
| male | 36 (6) | 116.5 ± 120.92 | 173.5 ± 188.79 | 71.65 ± 8.28 | 0.61 ± 0.52 | 0.18 ± 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krams, R.; Gudra, D.; Popovs, S.; Willow, J.; Krama, T.; Munkevics, M.; Megnis, K.; Jõers, P.; Fridmanis, D.; Contreras Garduño, J.; et al. Dominance of Fructose-Associated Fructobacillus in the Gut Microbiome of Bumblebees (Bombus terrestris) Inhabiting Natural Forest Meadows. Insects 2022, 13, 98. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13010098
Krams R, Gudra D, Popovs S, Willow J, Krama T, Munkevics M, Megnis K, Jõers P, Fridmanis D, Contreras Garduño J, et al. Dominance of Fructose-Associated Fructobacillus in the Gut Microbiome of Bumblebees (Bombus terrestris) Inhabiting Natural Forest Meadows. Insects. 2022; 13(1):98. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13010098
Chicago/Turabian StyleKrams, Ronalds, Dita Gudra, Sergejs Popovs, Jonathan Willow, Tatjana Krama, Maris Munkevics, Kaspars Megnis, Priit Jõers, Davids Fridmanis, Jorge Contreras Garduño, and et al. 2022. "Dominance of Fructose-Associated Fructobacillus in the Gut Microbiome of Bumblebees (Bombus terrestris) Inhabiting Natural Forest Meadows" Insects 13, no. 1: 98. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13010098