Characterization of Two Novel Insect-Specific Viruses Discovered in the Green Leafhopper, Cicadella viridis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and RNA Extraction
2.2. Host Insect Identification
2.3. Transcriptomic and Small RNA (sRNA) Sequencing
2.4. Virus Discovery and Confirmation by Reverse Transcription-PCR (RT-PCR)
2.5. Determination of Viral Genome Termini and Transcript Abundance
2.6. Small RNA Analysis
2.7. Genome Annotation and Phylogenetic Analysis
3. Results and Discussions
3.1. Transcriptome Assembly and Virus Discovery for C. viridis
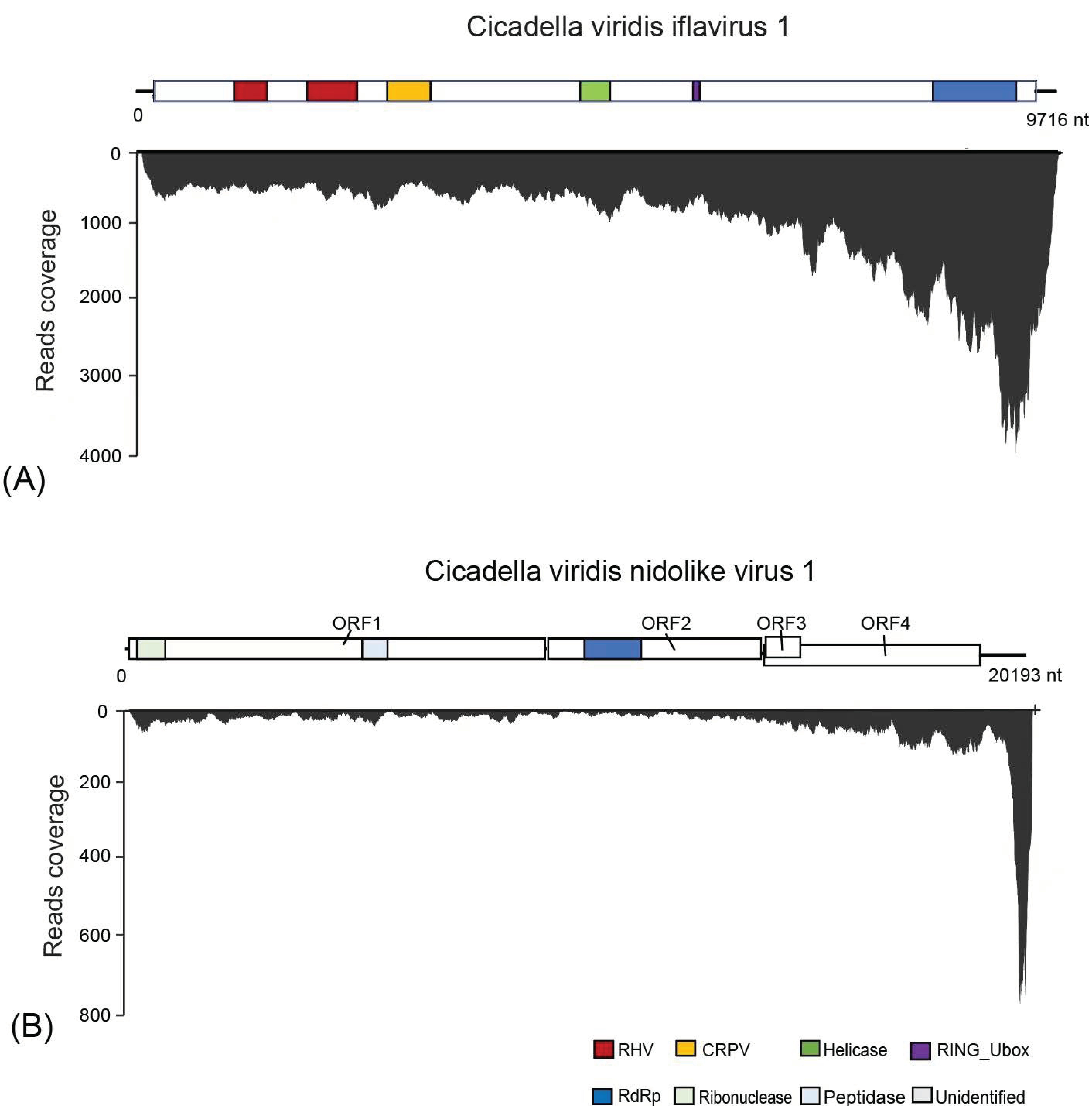
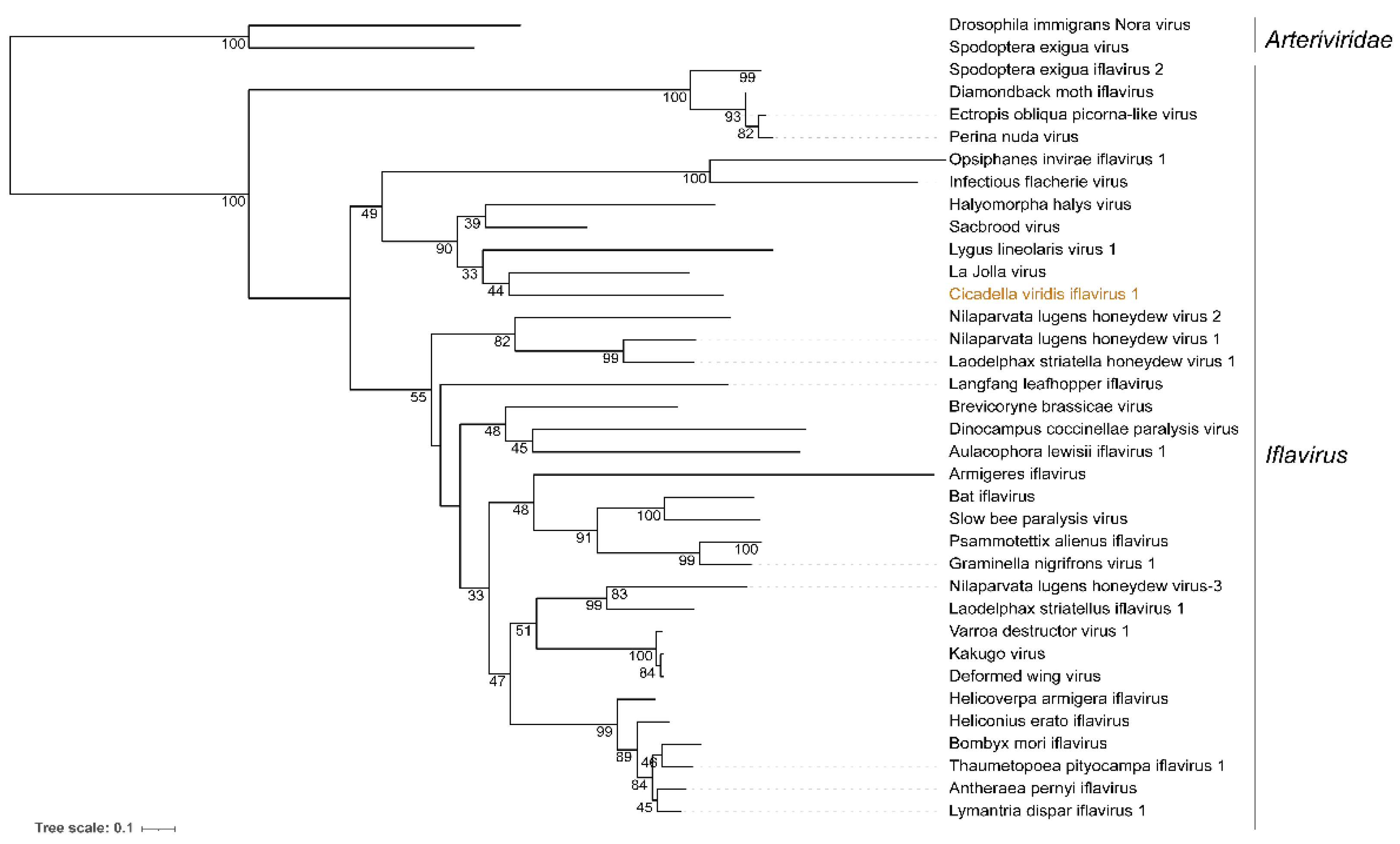
3.2. Cicadella viridis iflavirus 1
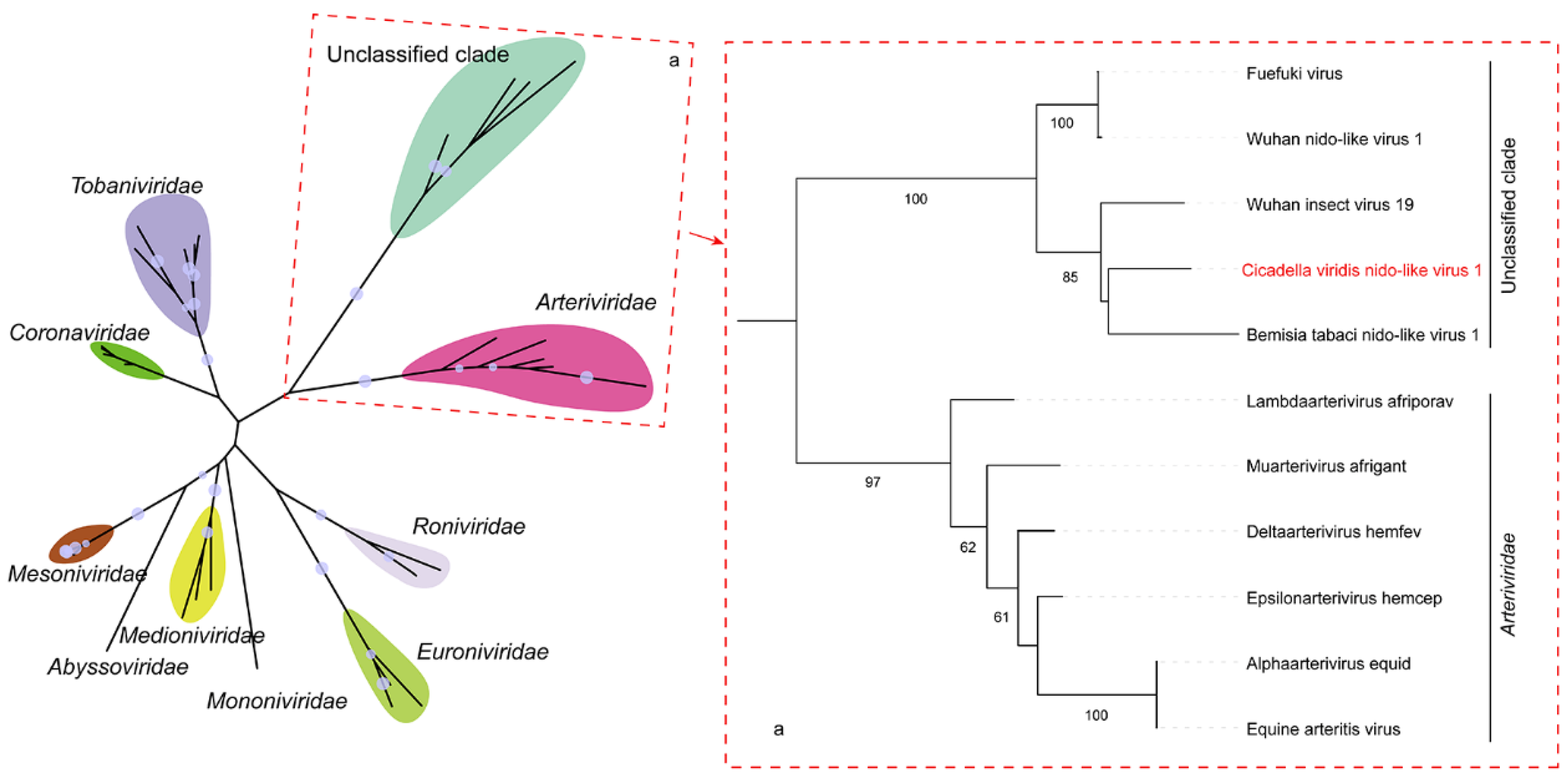
3.3. Cicadella viridis Nido-like Virus 1
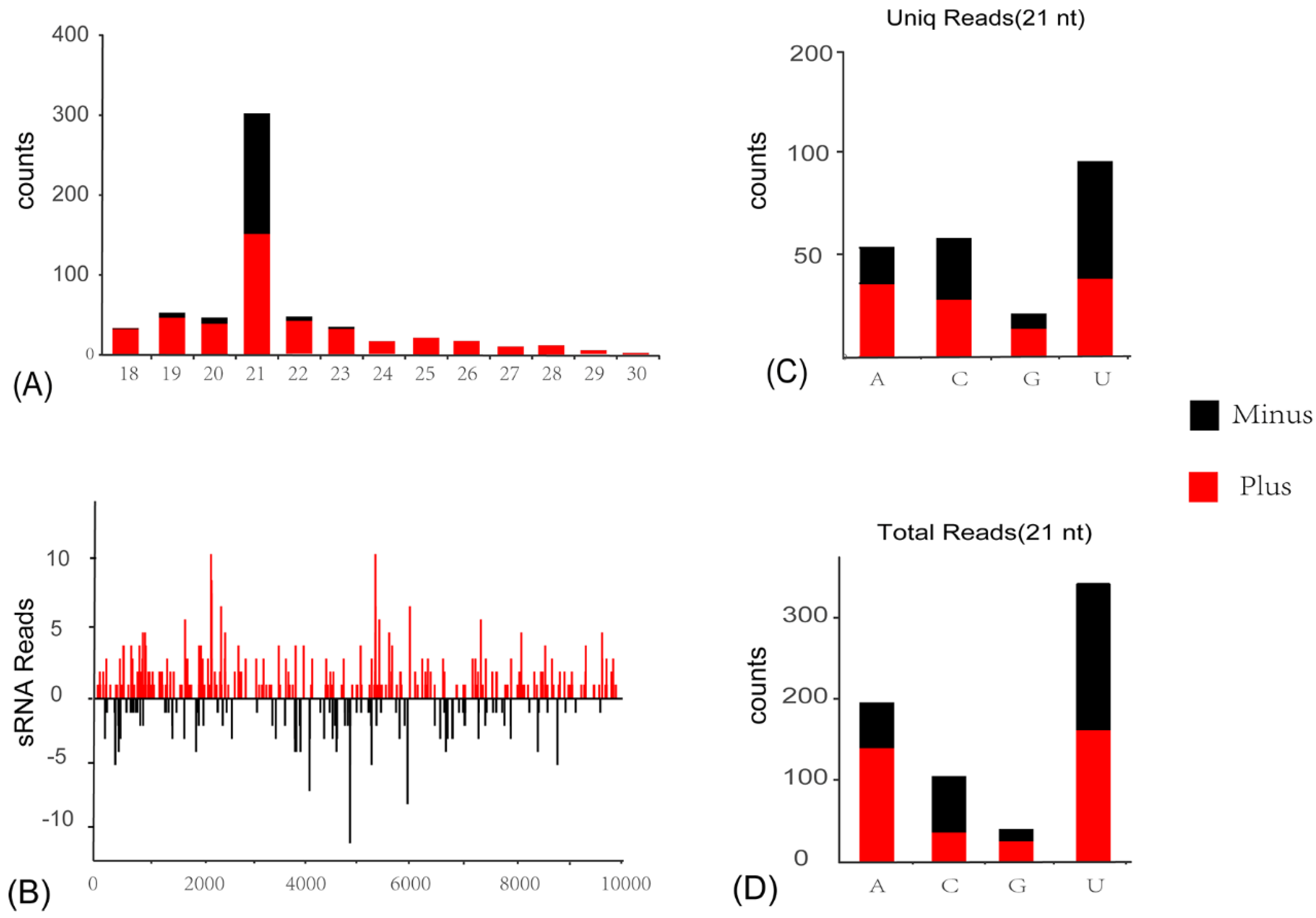
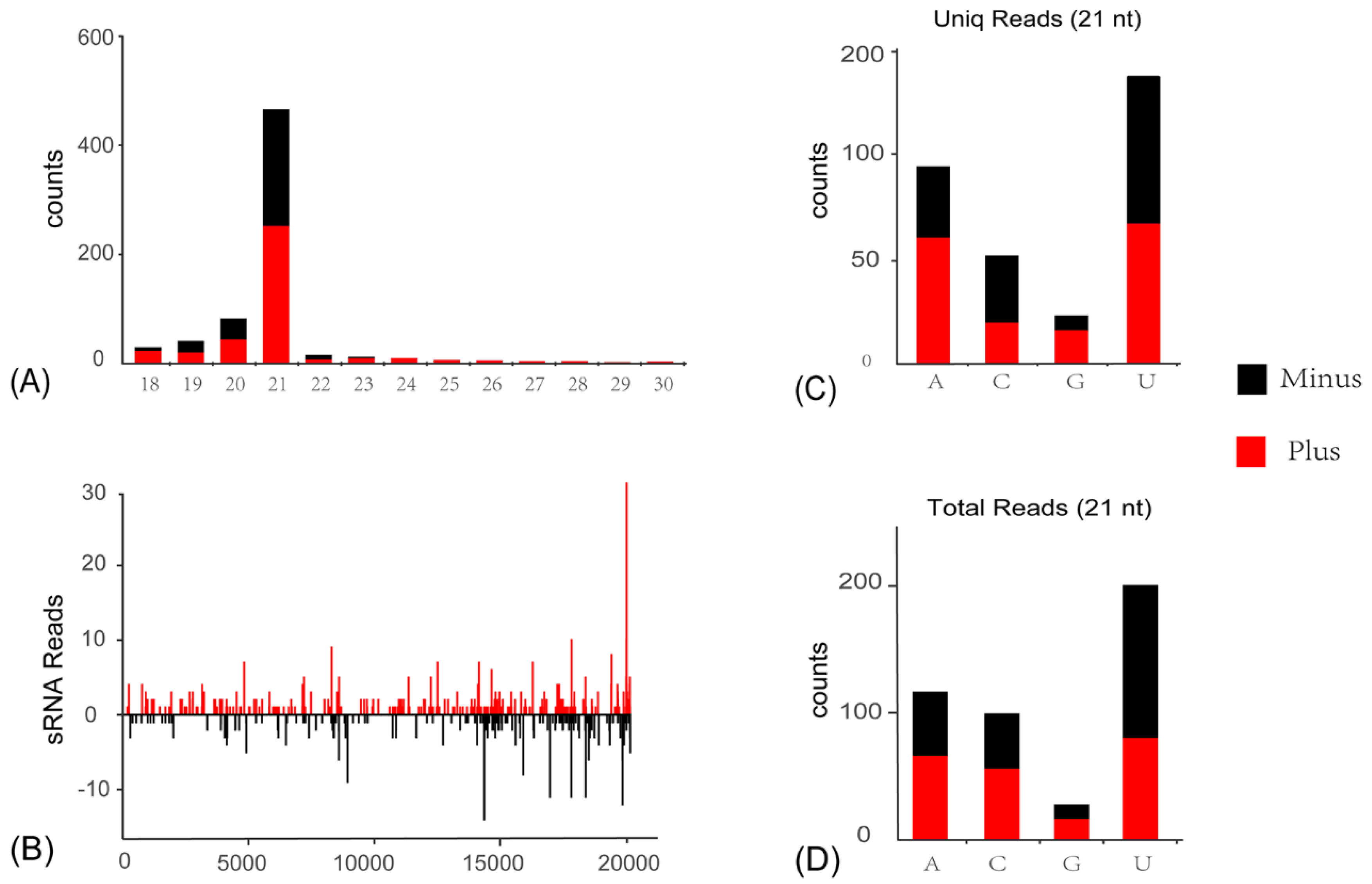
3.4. Activation of Antiviral RNA Interference Pathway in C. viridis Responsive to ISVs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harrison, R.; Hoover, K. Baculoviruses and other occluded insect viruses. In Insect Pathology, 2nd ed.; Vega, F.E., Kaya, H.K., Eds.; Academic Press: London, UK, 2012; pp. 73–131. [Google Scholar]
- Chen, Y.P.; Becnel, J.J.; Valles, S.M. RNA Viruses Infecting Pest Insects; Academic Press: San Diego, CA, USA, 2012; pp. 133–170. [Google Scholar]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Holmes, E.C. Predicting virus emergence amid evolutionary noise. Open Biol. 2017, 7, 170189. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Ye, Z.X.; He, Y.J.; Zhang, Y.; Wang, X.; Huang, H.J.; Zhuo, J.C.; Sun, Z.T.; Yan, F.; Chen, J.P.; et al. Discovery of Two Novel Negeviruses in a Dungfly Collected from the Arctic. Viruses 2020, 12, 692. [Google Scholar] [CrossRef]
- Dou, X.; Liu, S.; Soroker, V.; Harari, A.; Jurenka, R. Novel RNA Viruses from the Transcriptome of Pheromone Glands in the Pink Bollworm Moth, Pectinophora gossypiella. Insects 2021, 12, 556. [Google Scholar] [CrossRef] [PubMed]
- Bubici, G.; Prigigallo, M.I.; Garganese, F.; Nugnes, F.; Jansen, M.; Porcelli, F. First Report of Aleurocanthus spiniferus on Ailanthus altissima: Profiling of the Insect Microbiome and MicroRNAs. Insects 2020, 11, 161. [Google Scholar] [CrossRef] [Green Version]
- Nouri, S.; Matsumura, E.E.; Kuo, Y.W.; Falk, B.W. Insect-specific viruses: From discovery to potential translational applications. Curr. Opin. Virol. 2018, 33, 33–41. [Google Scholar] [CrossRef]
- Patterson, E.I.; Villinger, J.; Muthoni, J.N.; Dobel-Ober, L.; Hughes, G.L. Exploiting insect-specific viruses as a novel strategy to control vector-borne disease. Curr. Opin. Insect Sci. 2020, 39, 50–56. [Google Scholar] [CrossRef]
- Huang, H.; Li, J.; Zhang, C.; Chen, J. Hemipteran-Transmitted Plant Viruses: Research Progress and Control Strategies. Front. Agric. Sci. Eng. 2022, 9, 98. [Google Scholar] [CrossRef]
- Bolling, B.G.; Olea-Popelka, F.J.; Eisen, L.; Moore, C.G.; Blair, C.D. Transmission dynamics of an insect-specific flavivirus in a naturally infected Culex pipiens laboratory colony and effects of co-infection on vector competence for West Nile virus. Virology 2012, 427, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Marklewitz, M.; Zirkel, F.; Kurth, A.; Drosten, C.; Junglen, S. Evolutionary and phenotypic analysis of live virus isolates suggests arthropod origin of a pathogenic RNA virus family. Proc. Natl. Acad. Sci. USA 2015, 112, 7536–7541. [Google Scholar] [CrossRef] [Green Version]
- Asgari, S.; Johnson, K.N. Hytrosaviruses: Structure and Genomic Properties. In Insect Virolog; Asgari, S., Johnson, K.N., Eds.; Caister Academic Press: Norfolk, UK, 2010; pp. 103–121. [Google Scholar]
- Jankielsohn, A. The Importance of Insects in Agricultural Ecosystems. Adv. Entomol. 2018, 6, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, C. Keys to the families of Cicadomorpha and subfamilies and tribes of Cicadellidae (Hemiptera: Auchenorrhyncha). Fla. Entomol. 2005, 88, 502–517. [Google Scholar] [CrossRef]
- Mazzoni, V.; Lucchi, F.C.A.; Santini, L. Leafhoppers and planthoppers vectors in Ligurian and Tuscan vineyards. IOBC/WPRS Bull. 2001, 24, 263–266. [Google Scholar]
- Arzone, A.; Vidano, C.; Arnò, C. Predators and parasitoids of Empoasca vitis and Zygina rhamni (Rhynchota Auchenorrhyncha). In Proceedings of the 6th Auchenorrhyncha Meeting, Turin, Italy, 7–11 September 1987. [Google Scholar]
- Michalik, A.; Jankowska, W.; Kot, M.; Golas, A.; Szklarzewicz, T. Symbiosis in the green leafhopper, Cicadella viridis (Hemiptera, Cicadellidae). Association in statu nascendi? Arthropod Struct. Dev. 2014, 43, 579–587. [Google Scholar] [CrossRef]
- Khine, M.O.; Wang, H.; Raza, A.; Liu, Y.; Wang, X. The complete genomic sequence of a new iflavirus from the leafhopper Psammotettix alienus. Arch. Virol. 2020, 165, 1883–1886. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Liu, W.; Cao, M.; Wang, X. Sequence analysis and genomic organization of a novel chuvirus, Taiyuan leafhopper virus. Arch. Virol. 2019, 164, 617–620. [Google Scholar] [CrossRef]
- Fu, Y.; Cao, M.; Wang, H.; Du, Z.; Liu, Y.; Wang, X. Discovery and characterization of a novel insect-specific reovirus isolated from Psammotettix alienus. J. Gen. Virol. 2020, 101, 884–892. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Huang, H.J.; Ye, Z.X.; Wang, X.; Yan, X.T.; Zhang, Y.; He, Y.J.; Qi, Y.H.; Zhang, X.D.; Zhuo, J.C.; Lu, G.; et al. Diversity and infectivity of the RNA virome among different cryptic species of an agriculturally important insect vector: Whitefly Bemisia tabaci. NPJ Biofilms Microbiomes 2021, 7, 43. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [Green Version]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [Green Version]
- Enjuanes, L.; Gorbalenya, A.; De Groot, R.; Cowley, J.; Ziebuhr, J.; Snijder, E. Nidovirales. Encycl. Virol. 2008, 419–430. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pmc/articles/PMC7150171/ (accessed on 7 March 2022).
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar]
- Gulyaeva, A.A.; Gorbalenya, A.E. A nidovirus perspective on SARS-CoV-2. Biochem. Biophys. Res. Commun. 2021, 538, 24–34. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.-L.; Ye, Z.-X.; Chen, J.-P.; Zhang, C.-X.; Huang, H.-J.; Li, J.-M. Characterization of Two Novel Insect-Specific Viruses Discovered in the Green Leafhopper, Cicadella viridis. Insects 2022, 13, 378. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13040378
Li L-L, Ye Z-X, Chen J-P, Zhang C-X, Huang H-J, Li J-M. Characterization of Two Novel Insect-Specific Viruses Discovered in the Green Leafhopper, Cicadella viridis. Insects. 2022; 13(4):378. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13040378
Chicago/Turabian StyleLi, Li-Li, Zhuang-Xin Ye, Jian-Ping Chen, Chuan-Xi Zhang, Hai-Jian Huang, and Jun-Min Li. 2022. "Characterization of Two Novel Insect-Specific Viruses Discovered in the Green Leafhopper, Cicadella viridis" Insects 13, no. 4: 378. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13040378