Genetic Variation in Insect Vectors: Death of Typology?

Yale University, 21 Sachem Street, New Haven, CT 06520-8105, USA
Insects 2018, 9(4), 139; https://0-doi-org.brum.beds.ac.uk/10.3390/insects9040139
Submission received: 14 September 2018
/
Revised: 1 October 2018
/
Accepted: 8 October 2018
/
Published: 11 October 2018
(This article belongs to the Special Issue Mosquito – Arbovirus Interactions in Honor of Professor Walter J. Tabachnick)
Abstract
:The issue of typological versus population thinking in biology is briefly introduced and defined. It is then emphasized how population thinking is most relevant and useful in vector biology. Three points are made: (1) Vectors, as they exist in nature, are genetically very heterogeneous. (2) Four examples of how this is relevant in vector biology research are presented: Understanding variation in vector competence, GWAS, identifying the origin of new introductions of invasive species, and resistance to inbreeding. (3) The existence of high levels of vector genetic heterogeneity can lead to failure of some approaches to vector control, e.g., use of insecticides and release of sterile males (SIT). On the other hand, vector genetic heterogeneity can be harnessed in a vector control program based on selection for refractoriness.
1. Typological and Populational Thinking
There are two fundamental ways to view the biological world: As sets of nearly identical essential types (typology) or as populations of heterogeneous individuals (populational thinking). Typologists see the world as consisting of homogeneous populations in discrete groups with an idealized essence embodied by “normal” (wild type) individuals. The population view considers the living world to be composed of heterogeneous populations of unique individuals that can be assigned often only loosely to discrete groups or types. These nearly diametrically opposed views determine how research is designed and carried out and how data are interpreted and valued. While these views can be traced back to ancient Greece (e.g., Plato’s idealized types), we need only start around 1900 to appreciate how these views impact biology in general and vector biology in particular.
In 1900, Mendel’s principles of inheritance were rediscovered followed by the development of the field of genetics primarily by Thomas Hunt Morgan. An essential principal of genetics in this early stage was the recognition of a wild type and mutants. The wild type was the normal genotype that defined the species and predominated in all populations. Mutants were rare anomalies useful in genetic studies, but otherwise having little to do with the essence of the species. Morgan understood his genetic studies were important to understanding evolutionary processes and, in fact, wrote three books on the subject [1,2,3]. Morgan’s view of evolution was that it was driven by mutation. Most of the time, populations are composed of individuals all homozygous for wild type alleles. Rarely, a new favorable mutation occurs that quickly goes to fixation and a new set of homogeneous wild type is established. Morgan gave little role to natural selection except to weed out the unfit (discussed further in [4]). This mutation driven view of evolution persists [5].
Morgan’s books had little impact on the field of evolutionary biology primarily because they did not resonate with biologists who worked with natural populations (field biologists, taxonomists, ecologists, etc.). Such workers observed ample variation among individuals and populations in nature for a variety of evolutionarily important traits like morphology and behavior. Variation reigned rather than idealized types. For some time these workers doubted that the genetics as developed by Morgan and his school had much to do with what happens in real populations in nature. A rapprochement or sorts arose with the demonstration by R. A. Fisher, J. B. S. Haldane, and S. Wright in the 1920s and 1930s that, theoretically, Mendelian rules of inheritance were compatible with naturally occurring variation and Darwinian evolution. Dobzhansky [6] demonstrated how these mathematical theories are consistent with observations in natural populations. In contrast to Morgan’s mutation driven view of the evolutionary process, in this view, most evolutionary change is based on pre-existing genetic variation. Enough genetic variation is floating in natural populations for almost all traits to make adaptive evolutionary changes in the absence of any new mutations. Success by plant and animal breeders in selecting a plethora of diverse traits in many plants and animals confirmed this view. Population thinking began to replace typological thinking at least among those interested in natural history, ecology, and evolution, that is, workers interested in populations and species as they exist in nature, in contrast to laboratory cultures sitting on a shelf or in an incubator.
While population thinking was sweeping the world of ecology and evolution in the mid-20th Century, molecular biology was exploding as a discipline and most practitioners implicitly or explicitly embraced typology. T. H. Morgan was the intellectual father of many early molecular biologists. Biological problems could be reduced to a basic essence for which a physical/chemical explanation must hold across all of life. A popular quip at that time was “…anything found to be true for E. coli must also be true of Elephants” [7]. As R. C. Lewontin [8] pointed out, the two approaches to biology had fundamentally different goals and priorities. The molecular biologist sought constancy, general principles that held across the living world. Natural historians and population biologists consider variation to be the defining essence of biology, the basis of evolutionary change, and thus of all life.
2. Genetic Heterogeneity in Vectors
The problems inherent in vector-borne diseases can be fruitfully considered a problem in ecology and evolution, or more generally, population biology. Vector-borne diseases are most often a three species interaction problem: The vector (usually an arthropod), the pathogen (usually a microbe), and the vertebrate host (humans in cases of most interest to us). The population dynamics and interactions of three species need to be considered simultaneously. And, equally importantly, each level is heterogeneous, i.e., consisting of a heterogeneous grouping of individuals. Here I will focus on the vector side with the explicit caveat that this is only one-third of the overall problem; heterogeneity of pathogens and humans (or other vertebrate hosts) are equally crucial in reaching a full understanding of these diseases.
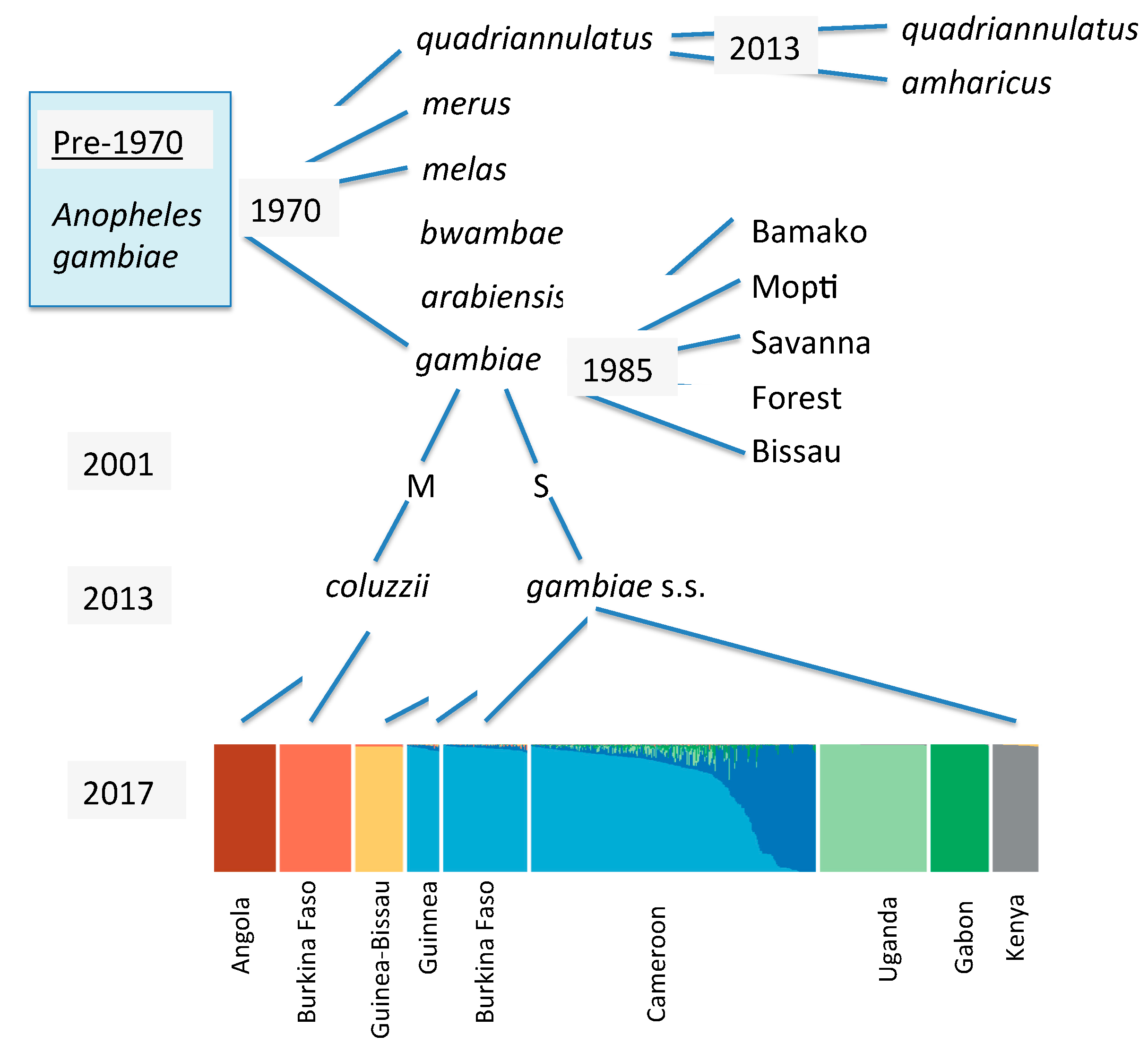
As just one prominent example of the importance of understanding the genetic heterogeneity of vectors we can consider the well-known and extremely important case of Anopheles gambiae, the primary vector of malaria in Africa where most deaths due to this disease occur. Mario Coluzzi and his collaborators studied chromosomal (inversion) polymorphism in this complex of species and uncovered a plethora of heterogeneity not distinguishable on the morphological level. Figure 1 is a quick historical overview of knowledge of this vector. What was considered a single species is now eight formally described species. In addition, various chromosomal forms were described [9], “form” being genetically distinct entities, but ambiguous with regard to taxonomic status. Furthermore, when two of the named species are examined in the detail provided by genome sequencing, individual populations are often found to be genetically distinguishable and unique [10]. And this is for inter-population variation. By far the most genetic variation exists within populations with nucleotide heterozygosity in these populations being in excess of 1% [10], i.e., an individual is heterozygous at >1% of all nucleotide sites which means that no two individuals are genetically identical.
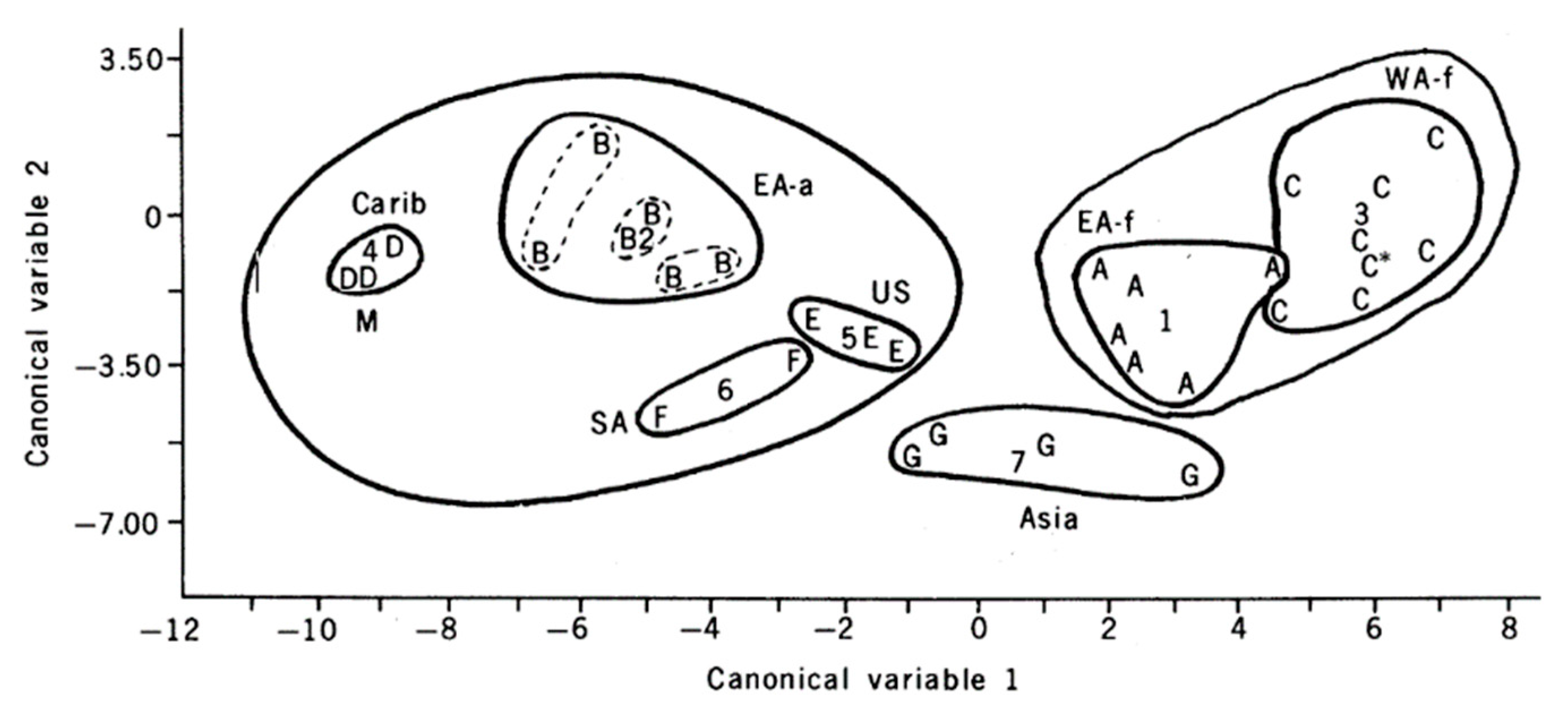
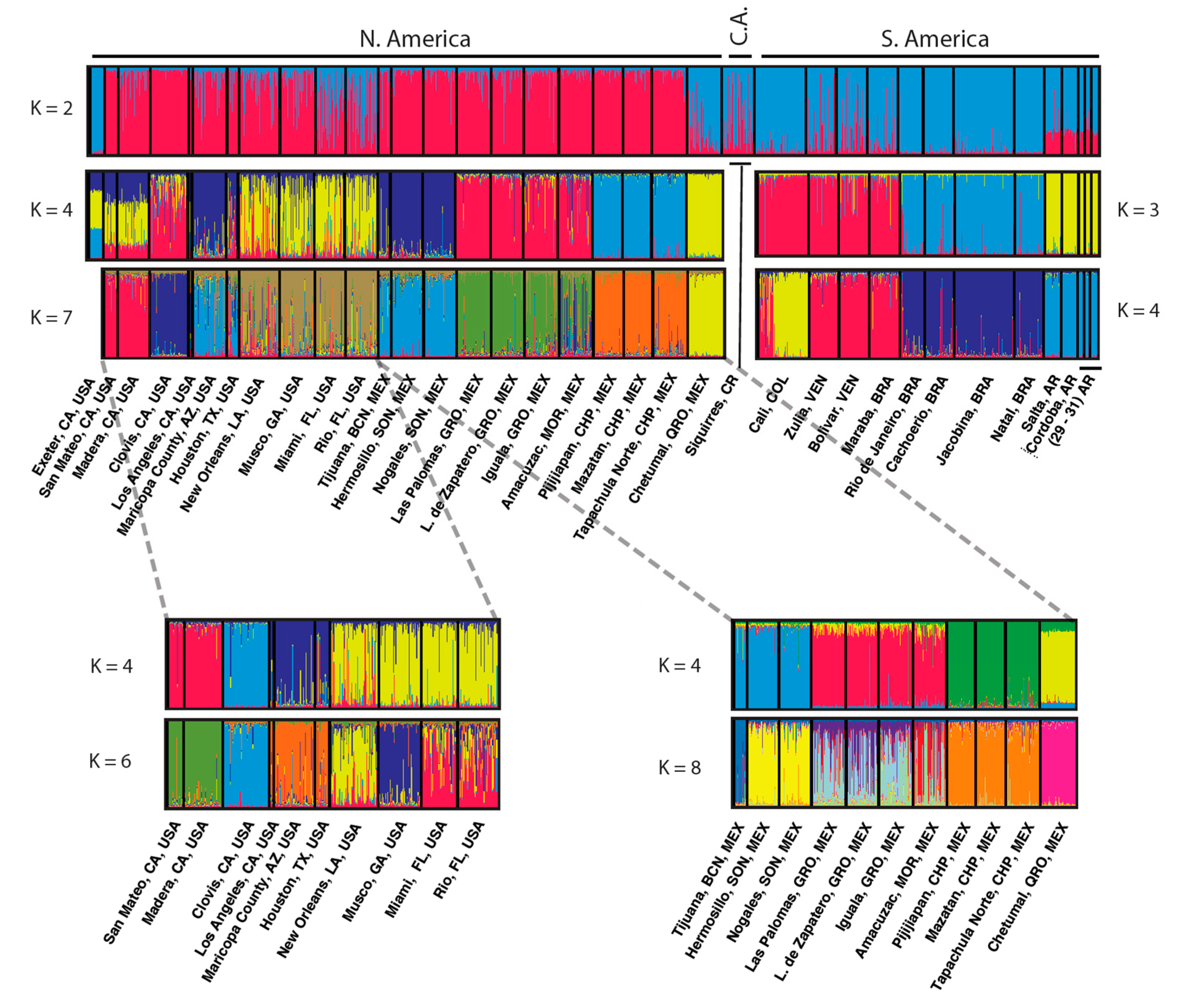
A similar story holds for another well-studied vector, Aedes aegypti. This species is composed of two named sub-species that differ in morphology and behavior with a darker zoophilic form (Ae. aegypti formosus) occupying the ancestral sub-Saharan Africa region and a light anthropophilic form (Ae. aegypti aegypti) that has colonized the tropical and subtropical world outside Africa [16,17]. Even with the relatively crude method of electrophoresis of proteins, this species was found to be genetically differentiated into discrete populations throughout its distribution, Figure 2 [18,19]. More recent DNA-based technologies have only led to confirmation and further resolution of the discrete genetic patterns exhibited by this vector (Figure 3). While not every population is genetically distinct using these genetic tools, clearly the genetic diversity across populations is remarkable.
It is not only genetic variation detectable by molecular technologies that is ubiquitous in vectors. Chromosomal inversion polymorphisms are well known especially in anopheline mosquitoes with favorable polytene chromosomes. As noted earlier, M. Coluzzi’s work on An. gambiae is a monumental demonstration of the importance of inversion polymorphism in vectors (see references in [21]). In an early work, Craig et al. [22] showed that strains of Ae. aegypti from different localities varied in sex-ratio from about 22% to 52% females. Inbreeding revealed about 40 different morphologically identifiable mutations and that each of 30 strains examined carried on average one recessive morphologically identifiable mutation. McClelland [23] documents extensive morphological variation in the patterns of scales on Ae. aegypti both within and between the named subspecies. Jupp et al. [24] documented considerable variation among S. African samples of Ae. aegypti for the scaling patterns used to distinguish the subspecies, so much so that offspring of single wild-caught females displayed variation ranging from “pure” Ae. aegypti formosus to “pure” Ae. aegypti aegypti. This seriously questions the validity of the typological subspecies designations.
3. Importance of Variation Vector Biology
I will present four contexts in which heterogeneity of vector populations are important in vector biology. The first is how the heterogeneity of vectors is reflected in the relative importance of different subgroups and genotypes in transmitting diseases. The second, linkage disequilibrium, might seem a basic property of the genetic structure of populations, but it has an important use in understanding the genetic basis of many traits of particular interest to vector biology. The third context is determining the origin of new introductions of invasive species that continue to spread today. Finally, I discuss the difficulty in inbreeding vectors in attempts to reduce genetic variation, implying that much of the standing genetic variation in vectors contributes to their fitness and adaptive flexibility.
Vector competence: Vector competence, the relative efficiency with which a vector can acquire a pathogen and transmit it, is a fundamental factor in understanding any vector borne disease. This trait displays considerable variation among species, populations, and individuals. A few illustrative examples are presented here; Tabachnick [25] should be a consulted for a more detailed authoritative review of this issue.
On the species level, we can again use the example of An. gambiae complex. Some members of the complex (An. gambiae s.s., An. arabiensis, and An. coluzzii) are extremely efficient carriers of human malaria parasites, whereas others (An. quadriannulatus, An. bwambae) are poor carriers, while still others (An. merus and An. melas) are intermediate. Thus knowledge of which of these morphologically identical species prevails in a location allows assessment of the threat they pose. It is not always clear if the relative importance of these sibling species is actually due to the relative physiological ability to support development of the parasite (competence) or other behavioral (e.g., host preference) or ecological factors (not living closely associated with human habitats). Regardless, the various subunits of An. gambiae s.l. pose very different public health threats.
On the intraspecific level, there is also evidence of considerable variation in ability to transmit pathogens among individual genotypes. Aedes aegypti is the best studied in this regard primarily because it is easy to rear in the laboratory and the primary vector of several viral diseases of contemporary concern. Table 1 shows results for three viruses. Different mosquito populations were studied in the same laboratory using identical methods, so pairs of populations in each study are comparable. Souza-Neto et al. [26] review many more such studies for several other strains of viruses and Ae. aegypti populations. In some cases, individual genetic factors have sufficiently strong effects on competence they have been identified and mapped [27,28,29]. Lambrechts et al. [30] demonstrated local adaptation between Ae. aegypti populations and dengue virus isolates from their localities.
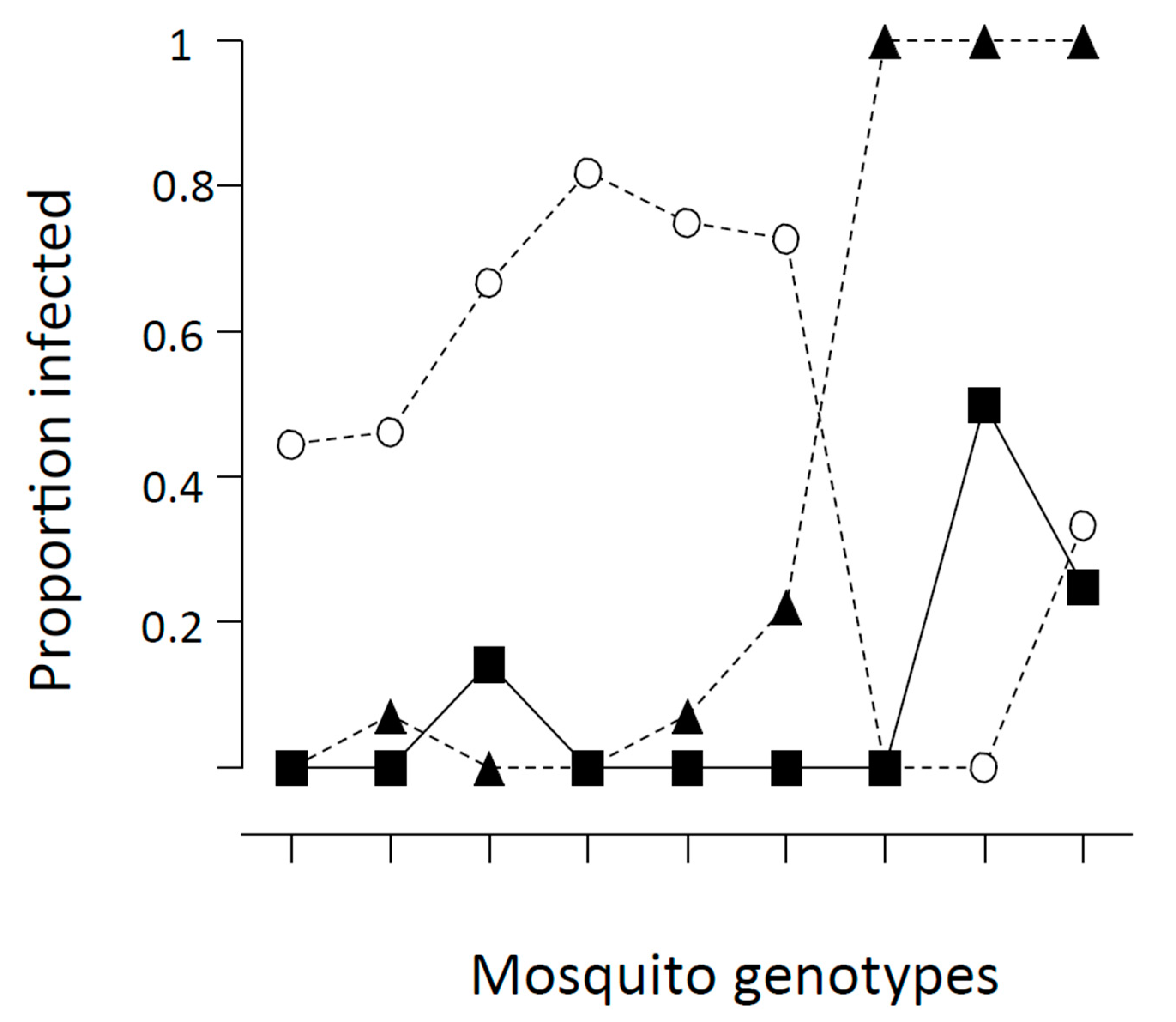
While not so easily studied in the laboratory, An. gambiae s.s. has been shown to have similar variation in ability to be infected with Plasmodium falciparum. Figure 4 shows the results of infecting nine different families of An. gambiae derived from the same population from Kenya for three different isolates of the malaria parasite (also from Kenya) [30]. Obviously, there is considerable variation even within a mosquito population for susceptibility to infection and different Plasmodium isolates also vary considerably, confirming that heterogeneity of the pathogen is important in this regard.
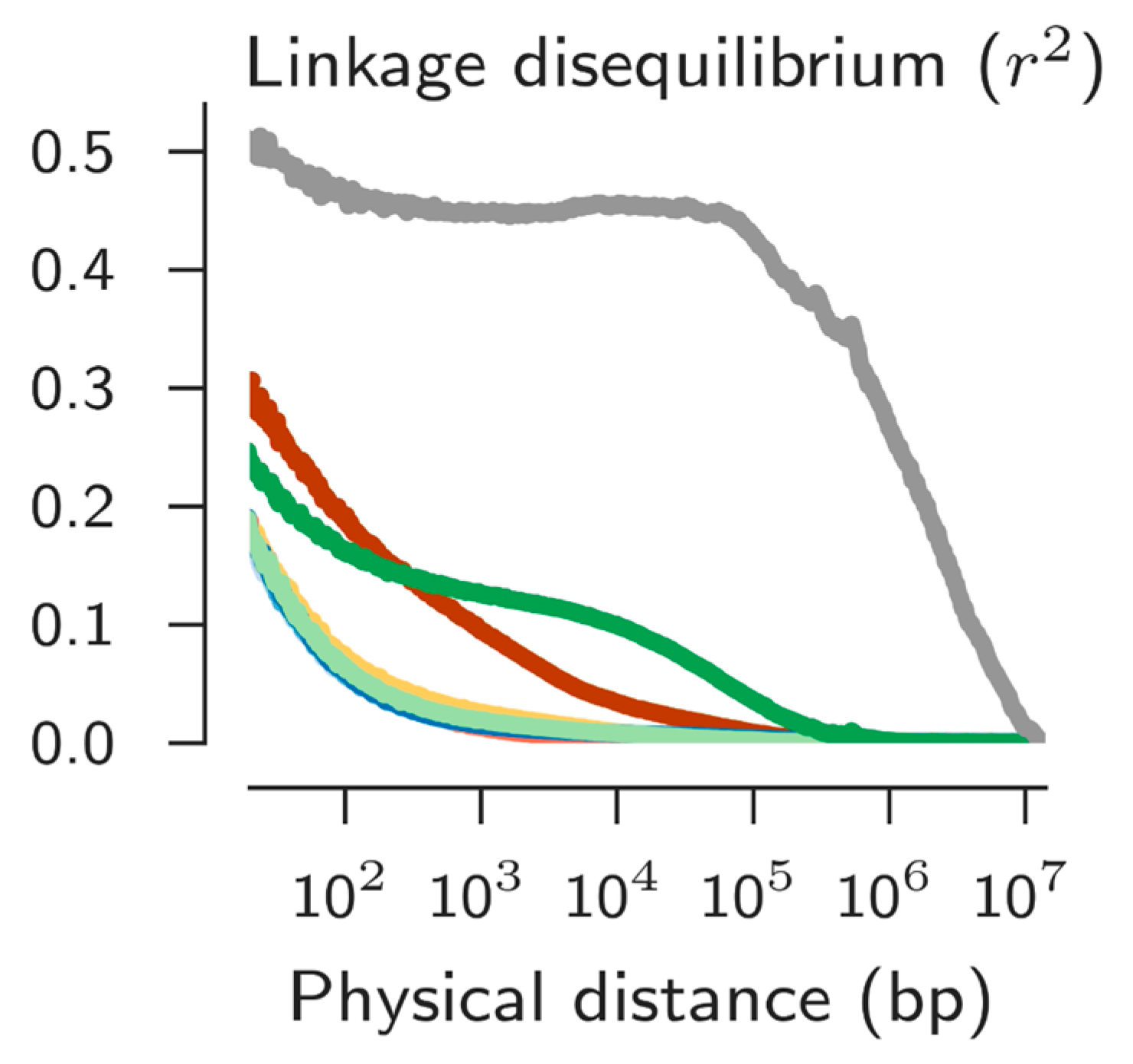
GWAS: A relatively new methodology in genetic analyses has arisen as the result of advances in acquiring genome-level data (reviewed in [35]). It is now possible to identify genes responsible for different phenotypes, i.e., bridge the genotype-phenotype chasm, even in non-model organisms that are not easily reared in the laboratory. This method is called GWAS genome wide association studies. If one has a dense enough set of polymorphic markers (usually single nucleotide polymorphisms, SNPs) distributed along a genome in samples that differ in a phenotype, it is possible to make statistical associations of the SNPs to the phenotypes. SNPs exhibiting such associations are unlikely to be directly causally related to the phenotype of interest, but rather they are non-randomly linked to the causative genetic factors. Thus the degree of non-random association between physically linked positions in a genome (called linkage disequilibrium, LD) is important in determining the success of this methodology. Too little LD means extremely high density of markers and very large sample sizes are needed to detect significant associations, too much LD means any association will mark such a large region of the genome as to be of little guidance as to number or identity of candidate causative genes.
Initial studies of An. gambiae and its sibling An. arabiensis reported very low LD in these species, generally not greater than extending over 200 bps [36,37]. Such observations would discourage trying to perform GWAS with these important vectors. However, the recent complete genome sequences of 765 individuals of these species across Africa revealed the existence of significant LD is some populations (Figure 5). Like so many other traits, LD is not a set parameter in a species. Importantly, this means GWAS may well be feasible if one chooses the right population to study. In fact GWAS has been successful in this group of mosquitoes for at least two traits, insecticide resistance [38] and desiccation resistance [39].
A similar situation occurs for Ae. aegypti. The level of LD found in populations is expected to be affected by the number of individuals that found a population (affecting effective population size) and the age of the population. Given the history of Ae. aegypti [17,40] it is expected that the ancestral populations that still exist in Africa should have less LD than populations outside Africa. Africa populations are relatively old and, at least historically, their habitat (tropical forest) was continuous implying large populations. Outside Africa, populations are likely to have been started with relatively few individuals, exist in discontinuous habitats, and are relatively young, not more than 500 years old, many much younger. One measure of LD is the distance at which the maximum observed correlation between loci drops to one-half. In a population from Mexico this occurs at 198 Kb while in an African population (Gabon) this occurs at 13 Kb [29].
A basic feature of genomes closely related to LD is recombination rate. When comparing two well-studied vectors, An. coluzzii and Ae. aegypti, we see a considerable difference in recombination rate per physical distance. Recombination in An. coluzzii is about 1.3 cM/Mb [41] while for Ae. aegypti it is 0.33 cM/Mb [29], a four-fold difference.
Thus, like so many other traits, there is no single level of LD for a species, it varies among populations dependent primarily on age and size of populations. At least between species, rates of recombination also varies considerably directly affecting levels of LD.
Origins of New Introductions: Many vectors are invasive species [42], species that have ongoing shifts in their distributions. These are often species closely associated with human habitats which enhances their spread by human activities as well as increasing their medical importance by being in close proximity to humans. Aedes aegypti is a classical well-studied invasive vector. In the last ~600 years it has spread from its native Africa around the tropical and subtropical world and continues to expand its distribution [43]. Due to the population-specific genetic heterogeneity of this species (Figure 3), it is possible to trace the origin of new introductions or what are thought to be new introductions.
Table 2 summarizes the power of using microsatellites or a battery of ~16,000 SNPs from a SNP chip [44]. Three studies where these methods have been applied are for recent introductions into The Netherlands [45], Washington, DC [46], and Las Vegas [47].
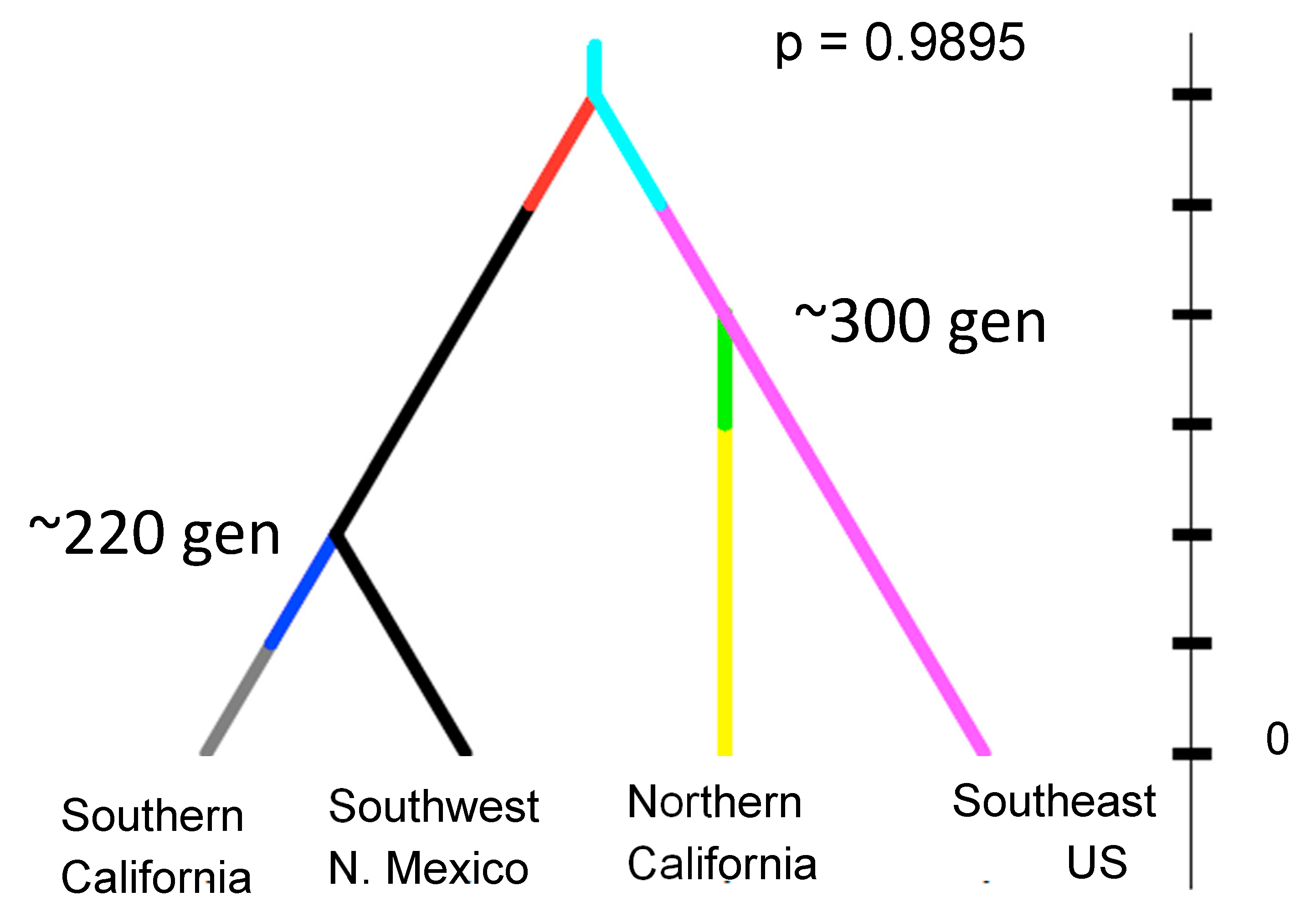
Perhaps the most revealing and potentially disturbing example of genetic analyses of presumed new introductions is California. Aedes aegypti was first reported in California in 2013. Initial work on samples from northern California indicated a likely origin as the southeast of the US, possibly New Orleans [49]. Subsequent collecting in California revealed the species to be quite widespread including in southern California. It became clear that the northern populations closely related to the southeast US are genetically distinct from southern California populations that are closely related to southwest US populations (Arizona and New Mexico) and northern Mexico [50]. The disturbing result from these genetic analyses is that estimates of the age of the populations in California date to ~300 generations ago (Figure 6). If we assume about ten generations per year, possibly fewer given the relatively cool temperatures in northern California, this implies the populations have been there at least 30 years, much before their “discovery” in 2013. California has an extensive and active mosquito surveillance program, yet did not detect this species.
Similar to California where previously undetected populations are reported as new introductions is the case of the Black Sea. Beginning in the early 2000s, Ae. aegypti was reported as new introductions after an apparent absence of about 50 years (e.g., [52]). Genetic analyses of these “new” introductions showed them to be older than all other Asian populations dating to about 200 years [48].
Inbreeding: For many studies of genetics of vectors, e.g., genome sequencing and assembly, reducing genetic variation is desirable. The usual way this is done is to inbreed, produce lines from matings of close relatives. In many vectors this has proven problematic.
One clear example is the easily-reared Ae. aegypti which is quite amenable to most genetic work. However, when lines with reduced genetic variation are attempted to be derived through inbreeding, they are only partially successful. One recent example is Powell and Evans [53]. Heterozygosity of the founding populations is expected to be reduced to (0.75)n after n generations of full-sibling inbreeding, 13% after seven generations. The observed reduction after seven generations of full-sib matings was only to 72% of the starting heterozygosity. Similar results have been observed in other Aedes [54] and Anopheles [55,56].
This implies the genetic variation in natural populations of vectors imparts high fitness through such mechanisms as heterosis or overdominance; the expected frequencies of homozygotes are not obtained due to low fitness. This means the genetic variation so ubiquitous in vector populations is adaptive and almost certainly accounts for their ability to occupy diverse niches (e.g., reference [57]) and to respond quickly to evolutionary challenges (see next section). Reduction in fitness due to loss of genetic variation is almost inevitable with laboratory rearing and colonization of vectors. This can affect fitness traits in males with implications for genetic modification programs using releases of males [55].
4. Importance of Vector Variation in Control Programs
Controlling diseases through control of vector populations can be quantitative or qualitative. Quantitative control means reducing the number of vectors, qualitative control means genetically changing vectors to be less dangerous to humans. The standing genetic variation in vector populations plays a big role in determining the effectiveness of both approaches.
Quantitative control: Quantitative control methods may reduce either the absolute numbers of vectors or frequency of vectors biting human hosts. Destruction of habitat, usually larval habitats, is well known and often effective. Screens and bed nets are effective especially for nighttime biters like many of the most important anopheline vectors of malaria. While genetic variation in vector populations may affect such control programs, for example, due to the propensity of different genotypes to take blood meals indoors or outdoors [58], it is much more obvious how genetic variation affects control programs based on insecticides and release of sterile vectors.
Wherever insecticides have been used, resistance almost invariably arises. Resistance is most often due to genetic changes in populations by increasing already present genetic variants that allow individuals to survive the chemicals being applied. This is exactly the expectation of the populational view of diversity: So much variation exists in natural populations that almost any trait can be selected.
Less well appreciated is the role genetic variation in populations plays in sterile insect release (SIT) programs. This method has had limited application to vectors and more experience comes from agricultural applications. One issue directly related to heterogeneity of vectors is evolution of mating discrimination against the release strain. Because females that mate with released sterile males leave no offspring, there is strong selection favoring females that do not mate with the released males. Over time, the release strain males lose their effectiveness to suppress the population. This was documented in the Mediterranean fruitfly SIT control program [59]. While not documented in the longest such program, that for the screwworm, this may be due to the frequent changing of the release strain, some 17 times over the about 60 years [60].
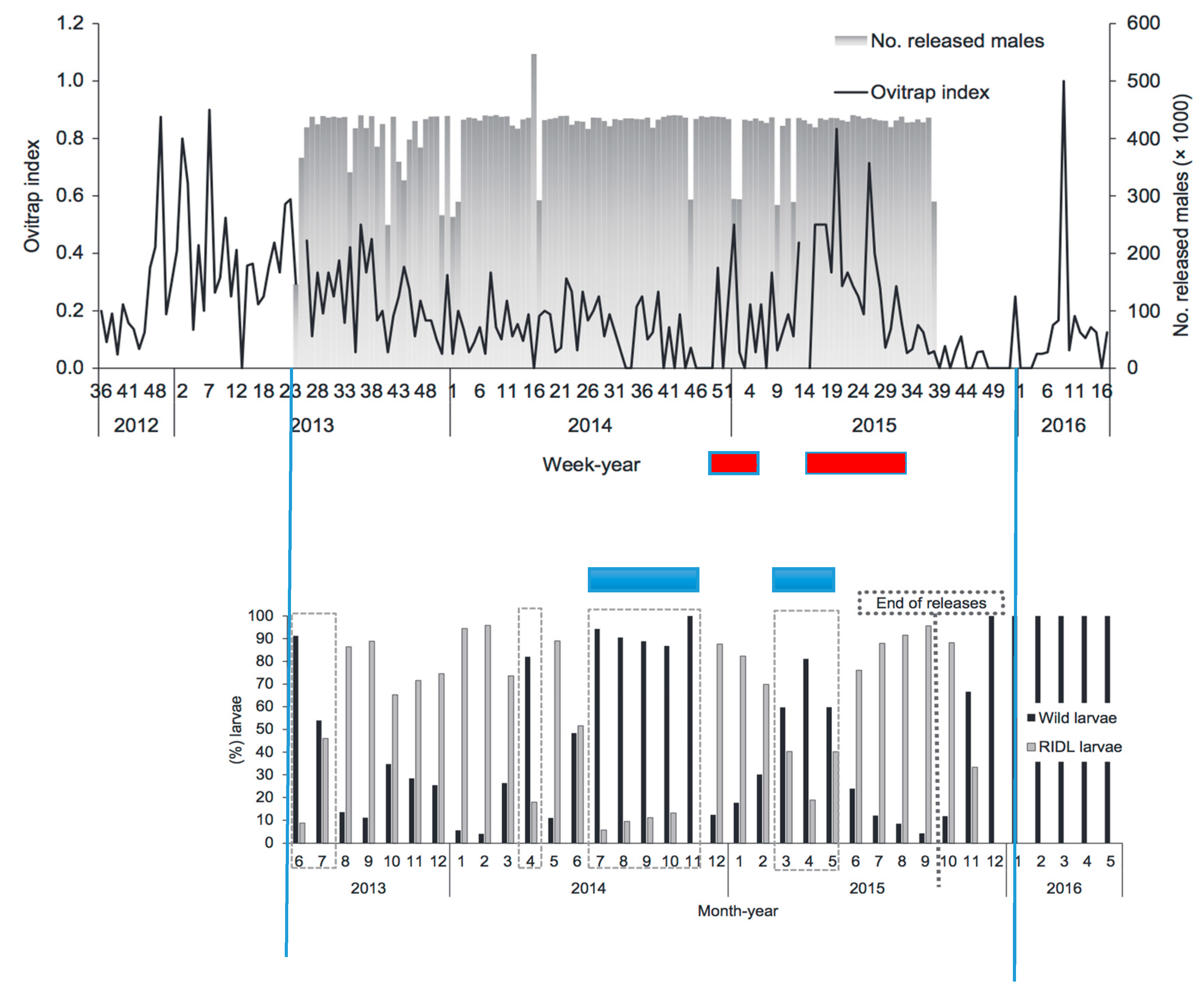
The most extensive SIT program for a vector of human diseases has been for Aedes aegypti employing a release strain with males carrying a dominant lethal gene, a method dubbed RIDL (Release of Insects with a Dominant Lethal) [61,62]. The most intensive releases have been done in Brazil, in particular in a city in Bahia, Jacobina. This release program began to lose its effectiveness after about 18 months of releases, Figure 7 [63]. Blue bars have been added in this figure indicating when released males had significantly reduced effectiveness in producing F1 eggs followed (lower graph), temporally, by the red bars when the adult female population significantly increased (upper graph). While not explicitly tested, this pattern is consistent with mating discrimination against the released males as a likely explanation for this breakdown of the effectiveness of control of numbers.
Qualitative control: The idea of genetically altering vectors to render them less harmful to humans has a long history dating to at least 50 years. This requires two major steps. (a) Identifying a genetic variant (some DNA change) that results in a vector phenotype that is less capable of transmitting a pathogen. This may take the form of causing a physiological change that reduces or eliminates the growth of the pathogen in the vector, or other trait such as reducing or eliminating a preference for taking blood meals from humans. (b) The second step is to somehow replace the target vector population with the variant. Many studies have identified genetic changes that satisfy the first step, the major stumbling block has been the second step. To date no such program has been successful.
Despite this lack of progress, new optimism and hope arose with the discovery of the CRISPR/Cas9 system that seemed to hold great promise (reviewed in Reference [64]). In this system, DNA regions heterozygous for the CRISPR/Cas9 endonuclease convert the alternative allele into itself. This produces a distinct advantage to the spread of the gene through a population. Linking this system to a desirable allele (e.g., one conferring resistance to transmitting a pathogen) to the endonuclease would “drive” the resistant gene construct through a population. In theory, this should work very efficiently [65,66].
Unfortunately, there is increasing evidence that the genetic variation inherent in natural populations of vectors is likely to frustrate any such CRISPR/Cas9 type replacement programs. Both theory [67] and experimental data in laboratory populations of insects, Drosophila [68] and Tribolium beetles [69], indicate that resistance to the drive mechanism is very likely to arise quickly in any such program. This is due to a combination of naturally existing variation in the target site for the endonuclease as well as the fact that the endonuclease itself is mutation-inducing with error prone repair where it cuts, leading to destruction of the target site.
While not a modification of a vector’s genome, there is a successful modification of a vector’s microbiome that renders the carriers less efficient vectors of human diseases. This is based on the endosymbiotic bacteria, Wolbachia. This bacterium is ubiquitous in insects [70] but does not exist naturally in Ae. aegypti [71]. However, when artificially infected with Wolbachia in the laboratory, Ae. aegypti become less efficient in transmitting dengue virus and other pathogens [72]. Release of Ae. aegypti with Wolbachia has led to establishment in the field [73]. While the infection remains stable where locally established, so far it has only spread at a very slow rate [74], much less than in other insects like Drosophila [75]. In another locality (Rio de Janeiro, Brazil) release of the same strain derived from Australian mosquitoes did not establish, possibly due to it being particularly susceptible to the insecticide being used locally. Outcrossing to native Rio Ae. aegypti produced a strain that could establish in that city (L. Moreira, personal communication). This is strong evidence that any attempts to genetically modify populations should be based on the genotypes prevalent in the target population.
Finally, there is one proposed method of genetic control of vectors that takes advantage of the genetic diversity inherent in populations. This is to select from the natural population to be controlled for a strain or strains of mosquitoes refractory to transmission of a pathogen [76]. By releasing such strains repeatedly, the target population could be reduced significantly in competence. Because no new genetic material is introduced, just increasing frequencies of already existing refractory alleles, this method has been called “genetic shifting”. Recent modeling has indicated that size and number of releases needed to significantly lower the ability of the target population to transmit are realistically achievable [77]. Because the release strain is identical to genotypes already present, mating discrimination is less likely to evolve and if it does, new strains for release could be re-selected from the target population. The replacement of the release strain is much less laborious than reconstructing transgenic modified strains.
5. Concluding Remarks
The forgoing is necessarily biased by the author’s familiarity with the field and has been confined to mosquito examples for the most part. Nevertheless, the major point should be clear: It is safe to conclude that arthropod vectors in general harbor large amounts of genetic variation that affect both how research is carried out and what control programs are most likely to be effective. While it may seem that I have set up a “straw man” and that the points explicated here are obvious, much vector biology research still proceeds with typological concepts at the forefront. I have purposely not cited such work to avoid irritating colleagues. Suffice it to end with one anecdote. Mario Coluzzi spent his life promoting populational thinking in vector biology. Near the end of his life (in 2012), in referring to a very prominent vector biologist, he asked me “Do you think he finally gets it?”.
Funding
Research in my laboratory cited in this paper is supported by the NIH, National Institute of Allergies and Infectious Diseases.
Acknowledgments
Walter Tabachnick has been a friend and colleague for more than 40 years. We have coauthored papers from 1977 to 2014. The theme of this contribution in his honor is one that he has championed throughout his career. Alessandra della Torre kindly contributed to developing Figure 1.
Conflicts of Interest
The author declares no conflict of interest.
References
- Morgan, T.H. Evolution and Adaptation; Macmillan: New York, NY, USA, 1903. [Google Scholar]
- Morgan, T.H. Evolution and Genetics; Princeton University Press: Princeton, NJ, USA, 1925. [Google Scholar]
- Morgan, T.H. The Scientific Basis of Evolution; W. W. Norton: New York, NY, USA, 1932. [Google Scholar]
- Powell, J.R. “In the air”—Theodosius Dobzhansky’s Genetics and the Origin of Species. Genetics 1987, 117, 363–366. [Google Scholar] [PubMed]
- Nei, M. Mutation-Driven Evolution; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Dobzhansky, T. Genetics and the Origin of Species, 1st ed.; Columbia University Press: New York, NY, USA, 1937. [Google Scholar]
- Monod, J.; Jacob, F. General conclusions: Teleonomic mechanisms in cellular metabolism, growth and differentiation. Cold Spring Harb. Symp. Quant. Biol. 1961, 26, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Lewontin, R.C. The Genetic Basis of Evolutionary Change; Columbia University Press: New York, NY, USA, 1974; ISBN 0-231-03392-3. [Google Scholar]
- Coluzzi, M.; Petrarca, V.; DiDeco, M.A. Chromosomal inversion intergradation and incipient speciation in Anopheles gambiae. Ital. J. Zool. 1985, 52, 45–63. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Bonnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- The Anopheles gambiae 1000s Genomes Consortium. Genetic diversity of the African malaria vector Anopheles gambiae. Nature 2017, 552, 96–100. [CrossRef]
- Coluzzi, M. Sibling species in Anopheles and their importance in malariaology. Misc. Publ. Entomol. Soc. Am. 1970, 7, 63–77. [Google Scholar]
- della Torre, A.; Fanello, C.; Akogbeto, M.; Dossau-yovo, J.; Favia, F.; Peterarca, V.; Coluzzi, M. Molecular evidence of incipient speciation within Anopheles gambiae s.s. in West Africa. Insect Mol. Biol. 2001, 10, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, M.; Hunt, R.H.; Wilderson, R.; della Torre, A.; Coulibaly, M.B.; Besansky, N.J. Anopoheles coluzzii and Anopheles amharicus, new members of the Anopheles gambiae complex. Zootaxa 2013, 3619, 246–274. [Google Scholar] [CrossRef] [PubMed]
- Neafsey, D.E.; Lawniczak, M.K.N.; Park, D.J.; Redmond, S.N.; Coulibaly, M.B.; Traore, S.F.; Sagnon, N.; Costantini, C.; Johnson, C.; Wiegand, R.C.; et al. SNP genotyping defines comples gene-flow boudaries among African malaria vector mosquitoes. Science 2010, 333, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Tabachnick, W.J. Evolutionary genetics and the yellow fever mosquito. Am. Entomol. 1991, 37, 14–26. [Google Scholar] [CrossRef]
- Powell, J.R.; Tabachnick, W.J. History of domestication and spread of Aedes aegypti. Mem. Inst. Oswaldo Cruz 2013, 108, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Tabachnick, W.J.; Powell, J.R. A worldwide survey of genetic variation in the yellow fever mosquito, Aedes aegypti. Genet. Res. 1979, 34, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R.; Tabachnick, W.J.; Arnold, J. Genetics and the origin of a vector population: Aedes aegypti, a case study. Science 1980, 208, 1385–1387. [Google Scholar] [CrossRef] [PubMed]
- Gloria-Soria, A.; Ayala, D.; Bheecarry, A.; Calderon-Arguedas, O.; Chadee, D.D.; Chiappero, M.; Coetzee, M.; Bin Elahee, K.; Fernandez-Salas, I.; Kamal, H.A.; et al. Global genetic diversity of Aedes aegypti. Mol. Ecol. 2016, 25, 5377–5395. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R.; Besansky, N.J.; della Torre, A.; Petrarca, V. Mario Coluzzi (1938-2012). Malar. J. 2014, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Craig, G.B.; Vendehey, R.C.; Hickey, W.A. Genetic variability in populations of Aedes aegypti. Bull. World Health Organ. 1961, 24, 527–539. [Google Scholar] [PubMed]
- McClelland, G.A.H. A worldwide survey of variation in scale pattern of the abdominal tergum of Aedes aegypti (L.) (Diptera: Culicidae). Trans. R. Entomol. Soc. Lond. 1974, 126, 239–259. [Google Scholar] [CrossRef]
- Jupp, P.G.; Kemp, A.; Frangos, C. The potential for dengue in South Africa: Morphology and taxonomic status of Aedes aegypti populations. Mosq. Syst. 1991, 23, 182–190. [Google Scholar]
- Tabachnick, W.J. Nature, nurture and evolution of intra-species variation in mosquito arbovirus transmission competence. Int. J. Environ. Res. Public Health 2013, 10, 249–277. [Google Scholar] [CrossRef] [PubMed]
- Souza-Neto, J.A.; Powell, J.R.; Bonizzoni, M. Aedes aegypti vector competence studies: A review. Infect. Genet. Evol. 2018, in press. [Google Scholar]
- Bosio, C.F.; Fulton, R.E.; Salasek, M.L.; Beaty, B.J.; Black, W.C. Quantiative trait loci that control vecor competence for dengue-2 virus in the mosquito Aedes aegypti. Genetics 2000, 156, 687–698. [Google Scholar] [PubMed]
- Bennett, K.E.; Flick, D.; Fleming, K.H.; Beaty, B.J.; Black, W.C., IV. Quantitative trait loci that control Dengue-2 virus dissemination in the mosquito Aedes aegypti. Genetics 2005, 170, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.J.; Dudchenko, O.; Kingan, S.; Koren, S.; Redmond, S.; Glassford, W.J.; Rose, N.H.; Antoshechkin, I.; Batra, S.S.; Brito-Sierra, C.A.; et al. Improved Aedes aegypti mosquito reference genome assembly enables biological discovery and vector control. Nature 2018, in press. [Google Scholar]
- Lambrechts, L.; Chevellon, C.; Albright, R.G.; Thaisomboonsuk, B.; Richardson, J.H.; Jarman, R.G.; Scott, T.W. Genetic specificity and potential for local adaptation between dengue viruses and mosquito vectors. BMC Evol. Biol. 2009, 9, 160. [Google Scholar] [CrossRef] [PubMed]
- Roundy, C.M.; Azar, S.R.; Rossi, S.L.; Huang, J.H.; Leal, G.; Yun, R.; Fernandez-Salas, I.; Vitek, C.J.; Paploski, I.A.; Kitron, U.; et al. Variation in Aedes aegypti mosquito competence for Zika virus transmission. Emerg. Infect. Dis. 2017, 23, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.; Jupatanakul, N.; Ramirez, J.L.; Kang, S.; Romero-Vivas, C.M.; Mohammed, H.; Dimopoulos, G. Transcriptomic profiling of diverse Aedes aegypti strains reveals increased basal-level immune activation in dengue virus-refractory populations and identifies novel virus-vector molecular interactions. PLoS Negl. Trop. Dis. 2013, 7, e2295. [Google Scholar] [CrossRef] [PubMed]
- Tabachnick, W.J.; Wallis, G.P.; Aitken, T.H.; Miller, B.R.; Amato, G.D.; Lorenz, L.; Powell, J.R.; Beaty, B.J. Oral infection of Aedes aegypti with yellow fever virus: Geographic variation and genetic considerations. Am. J. Trop. Med. Hyg. 1985, 34, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, L.; Halbert, J.; Durand, P.; Gouagna, L.C.; Koella, J.C. Host genotype by parasite genotype interactions underlying the resistance of anopheline mosquitoes to Plasmodium falciparum. Malar. J. 2005, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvin, A.; Craddock, N.; Sullivan, P.F. Genome-wide association stdies: A primer. Psychol. Med. 2010, 40, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.; Rousset, F.; Morlais, I.; Fontenille, D.; Cohuet, A. Low linkage disequilibrium in wild Aonpheles gambiae s.l. populations. BMC Genet. 2010, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D.; Lee, Y.; Kreppel, K.; Weakley, A.; Cornel, A.; Ferguson, H.M.; Eskn, E.; Lanzaro, G.C. Diversity, differentiation, and likage disequiblibrium: Prospects for association mapping the in the malaria vector Anopheles arabiensis. G3 2014, 4, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Weetman, D.; Wilding, C.S.; Steen, K.; Morgan, J.C.; Simard, F. Association mapping of insecticide resistance in wild Anopheles gambiae populations: Major variants identified in a low-linkage disequilibrium genome. PLoS ONE 2010, 5, e13140. [Google Scholar] [CrossRef] [PubMed]
- Ayala, D.; Zhang, S.; Chateau, M.; Fouet, C.; Moriais, I.; Costantini, C.; Hahn, M.W.; Besansky, N.J. Association mapping diesiccation resistance within chromosomal inversions in the African malaria vector Anopheles gambiae. Mol. Ecol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R.; Gloria-Soria, A.; Kotsakiozi, P. Recent history of Aedes aegypti: Vector genomics and epidemiology recods. BioScience 2018, in press. [Google Scholar]
- Pombi, M.; Stump, A.D.; della Torre, A.; Besansky, N.J. Variation in recombination rate across the X chromosome of Anopheles gambiae. Am. J. Trop. Med. Hyg. 2006, 75, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Lounibos, L.P. Invasions by insect vectors of human disease. Ann. Rev. Entomol. 2002, 47, 233–266. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R. Mosquitoes on the move. Science 2016, 354, 971–972. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.R.; Gloria-Soria, A.; Hou, L.; McBride, C.; Bonizzoni, M.; Zhao, H.; Powell, J.R. A multipurpose high-throughput SNP chip for the dengue and yellow fever mosquito, Aedes aegypti. G3 2015, 5, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.E.; Scholte, E.-J.; Dik, M.; Hartog, W.D.; Beeuwkes, J.; Powell, J.R. Aedes aegypti mosquitoes imported into The Netherlands, 2010. Emerg. Infect. Dis. 2011, 17, 2335–2337. [Google Scholar] [CrossRef] [PubMed]
- Gloria-Soria, A.; Lima, A.; Lovin, D.D.; Cunningham, J.M.; Severson, D.W.; Powell, J.R. Origin of a high latitude population of Aedes aegypti in Washington, DC. Am. J. Trop. Med. Hyg. 2018, 98, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Pless, E.; Raman, V. Origin of Aedes aegypti in Clark County, Nevada. J. Am. Mosq. Control Assoc. 2018, in press. [Google Scholar]
- Kotsakiozi, P.; Gloria-Soria, A.; Schaffner, F.; Robert, V.; Powell, J.R. Aedes aegypti in the Black Sea: Recent introduction or ancient remnant? Parasites Vectors 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Gloria-Soria, A.; Brown, J.E.; Kramer, V.; Hardstone Yoshimizu, M.; Powell, J.R. Origin of the dengue fever mosquito, Aedes aegypti, in California. PLoS Negl. Trop. Dis. 2014, 8, E3029. [Google Scholar] [CrossRef] [PubMed]
- Pless, E.; Gloria-Soria, A.; Evans, B.R.; Kramer, V.; Bolling, B.G.; Tabachnick, W.J.; Powell, J.R. Multiple introductions of the dengue vector, Aedes aegypti, into California. PLOS Negl. Trop. Dis. 2017, 11, e0005718. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.A.; Zhang, W.; Balding, D.J. Approximate Bayesian computation in population genetics. Genetics 2002, 162, 2025–2035. [Google Scholar] [PubMed]
- Akiner, M.M.; Demirci, B.; Babuadze, G.; Robert, V.; Schaffner, F. Spread of the invasive mosuqitoes Aedes aegypti and Aedes albopictus in the Black Sea region increases rist of chikungunya, dengue, and Zika outbreaks in Europe. PLOS Negl. Trop. Dis. 2016, 10, e0004664. [Google Scholar]
- Powell, J.R.; Evans, B.R. How much does inbreeding reduce heterozygosity? Empirical results from Aedes aegypti. Am. J. Trop. Med. Hyg. 2017, 96, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.C.; Craig, G.B. Heterozygosity I inbred strains of the tree-hole mosquito Aedes triseriatus. Biochem. Genet. 1987, 25, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Baeshen, R.; Ekechukwu, N.E.; Toure, M.; Paton, D.; Coulibaly, M.; Traore, S.F.; Tripet, F. Differential effects of inbreeding and selection on male reproductive phenotypes associated with the colonization and laboratory maintenance of Anopheles gambiae. Malar. J. 2014, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Turissini, D.A.; Gamez, S.; White, B.J. Genome-wide patterns of polymorphism in inbred line of the African malaria mosquito Anopheles gambiae. Genome Biol. Evol. 2014, 6, 3094–3204. [Google Scholar] [CrossRef] [PubMed]
- Toure, Y.T.; Petrarca, V.; Trauore, S.F.; Coulibaly, A.; Malga, H.M.; Sankare, O.; Sow, M.; Di Deco, M.A.; Coluzzi, M. Distribution and inversion polymorphism of chromosomally recognized taxa of the Anopheles gambiae complex in Male, West Africa. Parassitologia 1998, 40, 477–511. [Google Scholar] [PubMed]
- Coluzzi, M.; Sabatini, A.; Petrarca, V.; De Deco, M.A. Behavioral divergences between mosquitoes with different inversion karyotypes in polymorphic populations of of the Anopheles gambiae complex. Nature 1977, 266, 832–833. [Google Scholar] [CrossRef] [PubMed]
- McInnis, D.O.; Lance, D.R.; Jackson, C.G. Behavioral resistance to the sterile insect technique by Mediterranean fruit fly (Diptera: Tephritidae) in Hawaii. Ann. Entomol. Soc. Am. 1996, 89, 739–744. [Google Scholar] [CrossRef]
- Scott, M.; Concha, C.; Welch, J.B.; Philllips, P.L.; Skoda, S.R. Review of research advances in the screwworm eradication program over the past 25 years. Entomol. Exp. Appl. 2017, 164, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.D.; Donnell, C.A.; Wood, J.J.; Alphey, L.S. Insect population control using a dominant, repressible, lethal genetic system. Science 2000, 287, 2474–2476. [Google Scholar] [CrossRef] [PubMed]
- Phuc, H.K.; Andreasen, M.H.; Burton, R.S.; Vass, C.; Epton, M.J.; Pape, G.; Fu, G.; Condon, K.C.; Scaife, S.; Donnelly, C.A.; et al. Late-acting dominant lethal genetic systems and mosquito control. BMC Biol. 2007, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Graziera, L.; Pedrosa, M.C.; Almeida de Souza, F.; Gomez, M.; Moreira, M.B.; Verginio, J.F.; Capurro, M.L.; Carvalho, D.O. Effect of interruption of over-flooding releases of trans genic mosquitoes over wild population of Aedes aegypti: Two case studies in Brazil. Entomol. Exp. Appl. 2017, 164, 327–339. [Google Scholar] [CrossRef]
- Wade, M. High-throughput silencing using the CRISPR-Cas9 system: A review of the benefits and challenges. J. Biomol. Screen. 2015, 20, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Dederec, A.; Burt, A.; Godfray, H.C.J. The population genetics of using homing endonuclease genes in vector and pest management. Genetics 2008, 179, 2013–2026. [Google Scholar] [CrossRef] [PubMed]
- Unckless, R.L.; Messer, P.W.; Connallon, T.; Clark, A.G. Modeling the manipulation of natural populations by the mutagenic chain reaction. Genetics 2015, 201, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Unckless, R.L.; Clark, A.G.; Messer, P.W. Evolution of resistance against CRISPR/Cas9 gene drive. Genetics 2017, 205, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Champer, J.; Reeves, R.; Oh, S.Y.; Liu, C.; Liu, J.; Clark, A.G.; Messer, P.W. Novel CRISPR/Cas9 gene drive constructs reveal insights into mechanisms of resistance allele formation and drive efficiency in genetically diverse populations. PLOS Genet. 2017, 13, e1006796. [Google Scholar] [CrossRef] [PubMed]
- Drury, D.W.; Dapper, A.L.; Siniard, D.J.; Zentner, G.E.; Wade, M.J. CRISPR/Cas9 gene drives in genetically variable and nonrandomly mating wild populations. Sci. Adv. 2017, 3, e1601910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgenboeker, K.; Hammerstein, P.; Schalattmann, P.; Telschow, A.; Werren, J.H. How many species are infected with Wolbachia? A statistical analysis of current data. FEMS Microbiol. Lett. 2008, 281, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Gloria-Soria, A.; Chiodo, T.; Powell, J.R. Lack of evidence for natural Wolbachia infections in Aedes aegypti (Diptera: Culicidae). J. Med. Entomol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.A.; Iturbe-Ormaetxe, I.; Jeffery, J.A.; Lu, G.; Pyke, A.T.; Hedges, L.M.; Rocha, B.C.; Hall-Mendelin, S.; Day, A.; Riegler, M.; et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, chikungunya, and Plasmodium. Cell 2009, 139, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.A.; Montgomery, B.L.; Popovici, I.; Iturbe-Ormaetxe, P.H.; Johnson, F.; Muzzi, F.; Greenfield, M.; Durkan, M.; Leong, Y.; Dong, Y. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature 2011, 476, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.L.; Barton, N.H.; Rasic, G.; Turley, A.P.; Montgomery, B.L.; Iturbe-Ormaetxe, I.; Cook, P.E.; Ryan, P.A.; Ritchie, S.A.; Hoffmann, A.A.; et al. Local introduction and heterogeneous spatial spread of dengue-suppressing Wolbachia through an urban population of Aedes aegypti. PLoS Biol. 2017, 15, e2001894. [Google Scholar] [CrossRef] [PubMed]
- Turelli, M.; Hoffmann, A.A. Rapid spread of an inherited incompatibility factor in California Drosophila. Nature 1991, 353, 440. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R.; Tabachnick, W.J. Genetic shifting: A novel approach for controlling vector-borne diseases. Trends Parasitol. 2014, 30, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Baskett, M.L.; Powell, J.R. Quantifying the efficacy of genetic shifting in mosquito control. Evol. Appl. submitted.
Figure 1.
Schematic history of the taxonomy and genetic diversity of Anopheles gambiae. The light blue box in upper left indicates it was considered a single species up to the 1960s. Italicized names are formally described species; non-italicized designations are informal “forms”. The bottom colored block is a STRUCTURE plot [10] based on whole genome sequencing. Summarized from work reported in [11,12,13,14,15].
Figure 1.
Schematic history of the taxonomy and genetic diversity of Anopheles gambiae. The light blue box in upper left indicates it was considered a single species up to the 1960s. Italicized names are formally described species; non-italicized designations are informal “forms”. The bottom colored block is a STRUCTURE plot [10] based on whole genome sequencing. Summarized from work reported in [11,12,13,14,15].

Figure 2.
Step-wise linear discriminant analysis based on allozyme variation in populations of Aedes aegypti. Letters indicate population samples, numbers the center of lettered groups. A: East Africa formosus subspecies, B: East Africa aegypti subspecies, C: West Africa formosus, D: Caribbean, E: United States, F: South America, G: Asia. From Reference [19].
Figure 2.
Step-wise linear discriminant analysis based on allozyme variation in populations of Aedes aegypti. Letters indicate population samples, numbers the center of lettered groups. A: East Africa formosus subspecies, B: East Africa aegypti subspecies, C: West Africa formosus, D: Caribbean, E: United States, F: South America, G: Asia. From Reference [19].

Figure 3.
Genetic structure of population samples of Aedes aegypti from the Americas based on microsatellites. Bayesian STRUCTURE plots [10] are shown for different groups of populations. K is the number of subpopulations assumed. From [20].

Figure 4.
Proportion of Anopheles gambiae females infected after taking Plasmodium falciparum-infected blood meals. Three lines are for three different isolates of the parasite, nine ticks along the X-axis represents nine different families of the mosquito. From [30,34].

Figure 5.
Linkage disequilibrium across populations of An. coluzzii and An. gambiae s.s. Four indistinguishable lines with lowest LD are from West Africa. Maroon line is A. coluzzii from Angola, dark green line is An. gambiae s.s. from Gabon, and the grey is An. gambiae s.s. from Kenya. From Reference [11].
Figure 5.
Linkage disequilibrium across populations of An. coluzzii and An. gambiae s.s. Four indistinguishable lines with lowest LD are from West Africa. Maroon line is A. coluzzii from Angola, dark green line is An. gambiae s.s. from Gabon, and the grey is An. gambiae s.s. from Kenya. From Reference [11].

Figure 6.
Estimates of the time of splits (in generations) of populations of Aedes aegypti in major regions of western North America based on microsatellites. The topology of the tree is supported over alternative topologies by the Bayesian posterior probability shown (methods in [51]). From [50].

Figure 7.
Results of releasing Aedes aegypti males carrying a dominant lethal gene in Jacobina, Bahia, Brazil. Blue lines were added to line up the same time in the two graphs taken from [63]. (Upper graph) shows the numbers released each week (grey bars, right scale). Dark line in upper graph is the ovitrap index as a measure of fertile female adults present. (Lower graph) shows the frequencies of wild type larvae reared from eggs in ovitraps (dark black lines) and F1 offspring from mating with the RIDL release strain (grey lines).
Figure 7.
Results of releasing Aedes aegypti males carrying a dominant lethal gene in Jacobina, Bahia, Brazil. Blue lines were added to line up the same time in the two graphs taken from [63]. (Upper graph) shows the numbers released each week (grey bars, right scale). Dark line in upper graph is the ovitrap index as a measure of fertile female adults present. (Lower graph) shows the frequencies of wild type larvae reared from eggs in ovitraps (dark black lines) and F1 offspring from mating with the RIDL release strain (grey lines).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Examples of vector competence studies on Aedes aegypti for three of the major viruses this species transmits, Zika, dengue, and yellow fever. The geographic origin of mosquitoes tested is in first column with the virus in the second. Infection rate (third column) is the percent of females that blood fed on infective blood that became infected.
Table 1.
Examples of vector competence studies on Aedes aegypti for three of the major viruses this species transmits, Zika, dengue, and yellow fever. The geographic origin of mosquitoes tested is in first column with the virus in the second. Infection rate (third column) is the percent of females that blood fed on infective blood that became infected.
| Geographic Origin Mosquito | Virus | Infection Rate | Reference |
|---|---|---|---|
| Salvador, Brazil | Zika DAK AR | 100% | [31] |
| Rio Grande, Texas | “ | 40% | |
| Singapore | Dengue Guinea C | 90% | [32] |
| Bangkok | “ | 10% | |
| Guatemala | Yellow Fever Asibi | 2% | [33] |
| Kwa Dzivo Kenya | “ | 57% |
Note that in this table are presented studies using the same strain of virus and assayed in.
Table 2.
Probabilities of correctly assigning an individual Aedes aegypti to its population of origin using either microsatellite variation or ~16,000 SNPs from a chip. From [48].
Table 2.
Probabilities of correctly assigning an individual Aedes aegypti to its population of origin using either microsatellite variation or ~16,000 SNPs from a chip. From [48].
| Genetic Data Type | Probability of Assignment to Correct Continent | Probability of Assignment to Correct Population |
|---|---|---|
| Microsatellites | 99.9% | 80.9% |
| SNPs from chip | 100% | 99.7% * |
* An exception is Florida and Costa Rica which are not distinguishable.
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Powell, J.R. Genetic Variation in Insect Vectors: Death of Typology? Insects 2018, 9, 139. https://0-doi-org.brum.beds.ac.uk/10.3390/insects9040139
AMA Style
Powell JR. Genetic Variation in Insect Vectors: Death of Typology? Insects. 2018; 9(4):139. https://0-doi-org.brum.beds.ac.uk/10.3390/insects9040139
Chicago/Turabian StylePowell, Jeffrey R. 2018. "Genetic Variation in Insect Vectors: Death of Typology?" Insects 9, no. 4: 139. https://0-doi-org.brum.beds.ac.uk/10.3390/insects9040139
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.