Hibernation Leads to Altered Gut Communities in Bumblebee Queens (Bombus terrestris)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Species
2.2. Experimental Design
2.3. Gut Dissection, DNA Extraction, PCR Amplification, and Illumina MiSeq Analysis
2.4. Determination of Bacterial Load and Pathogen Infection Using qPCR
2.5. Statistical Analyses
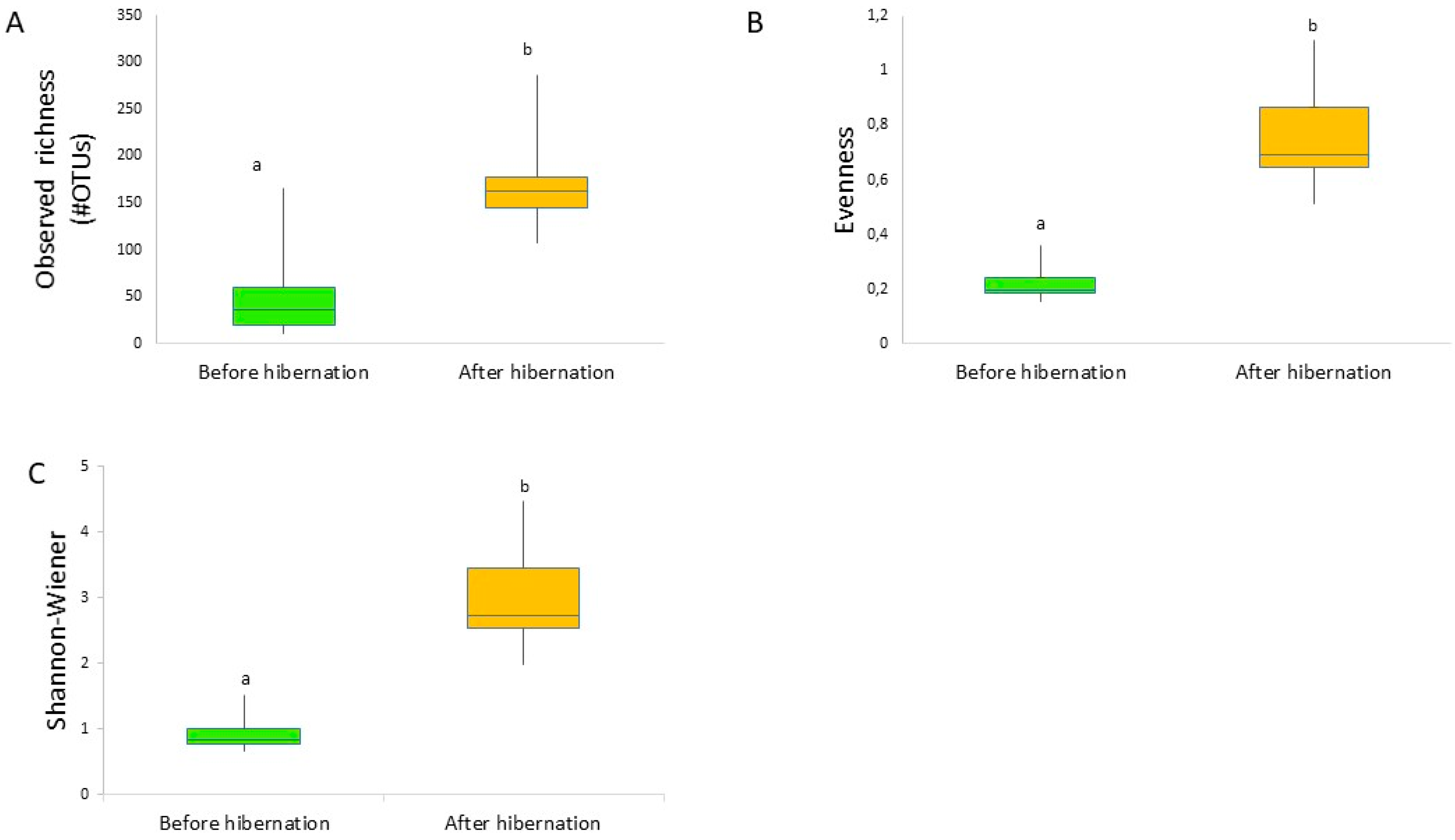
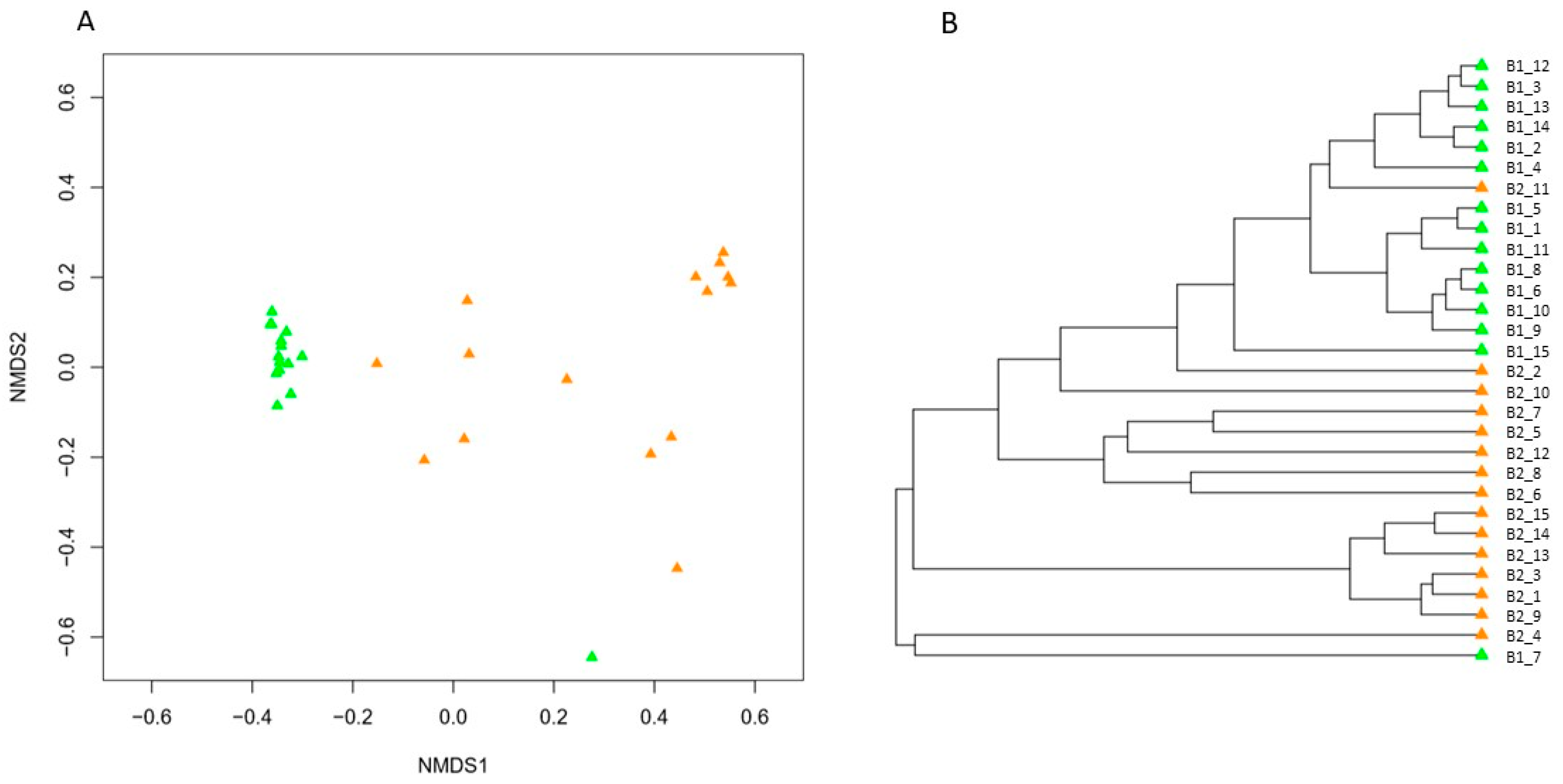
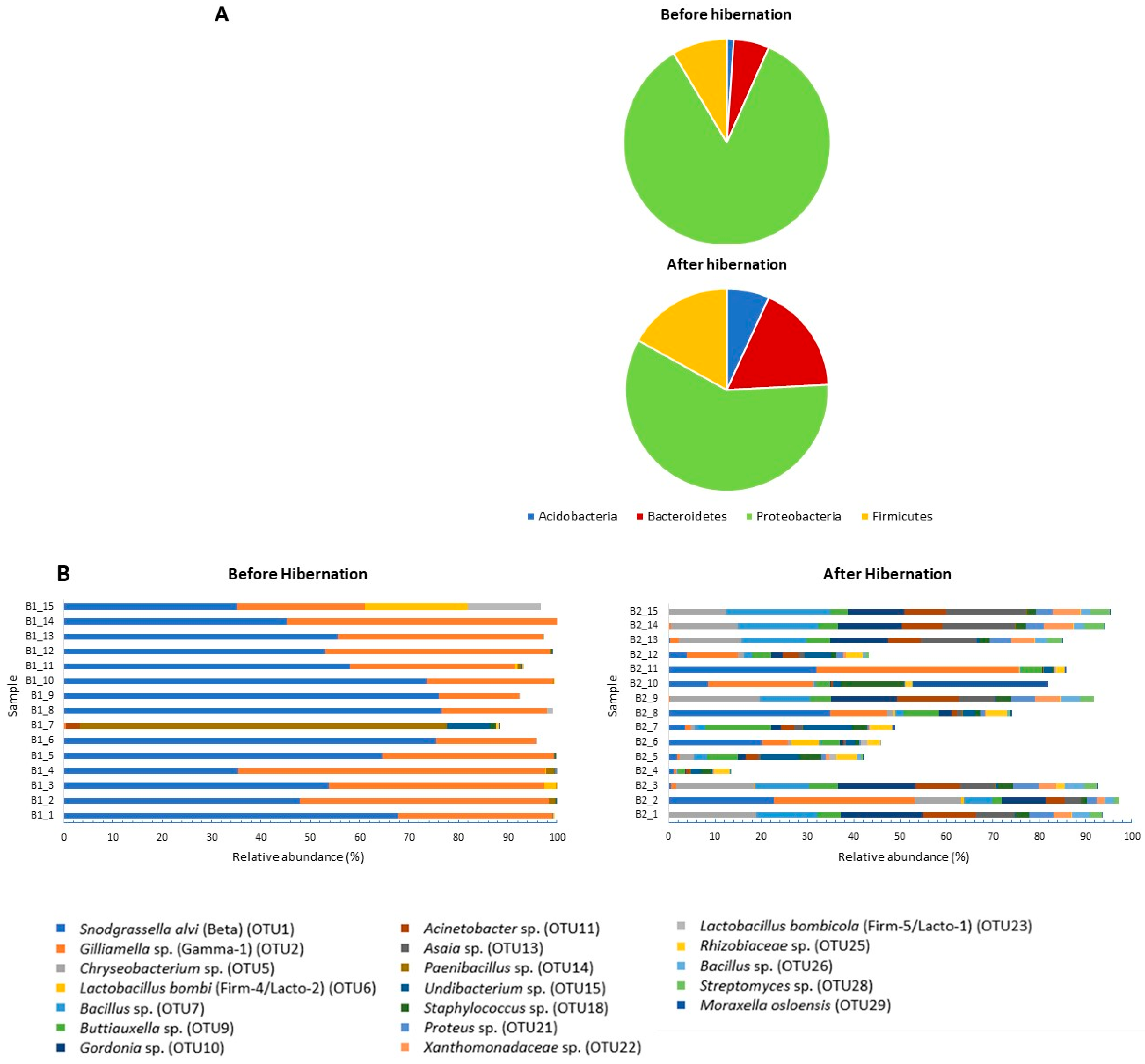
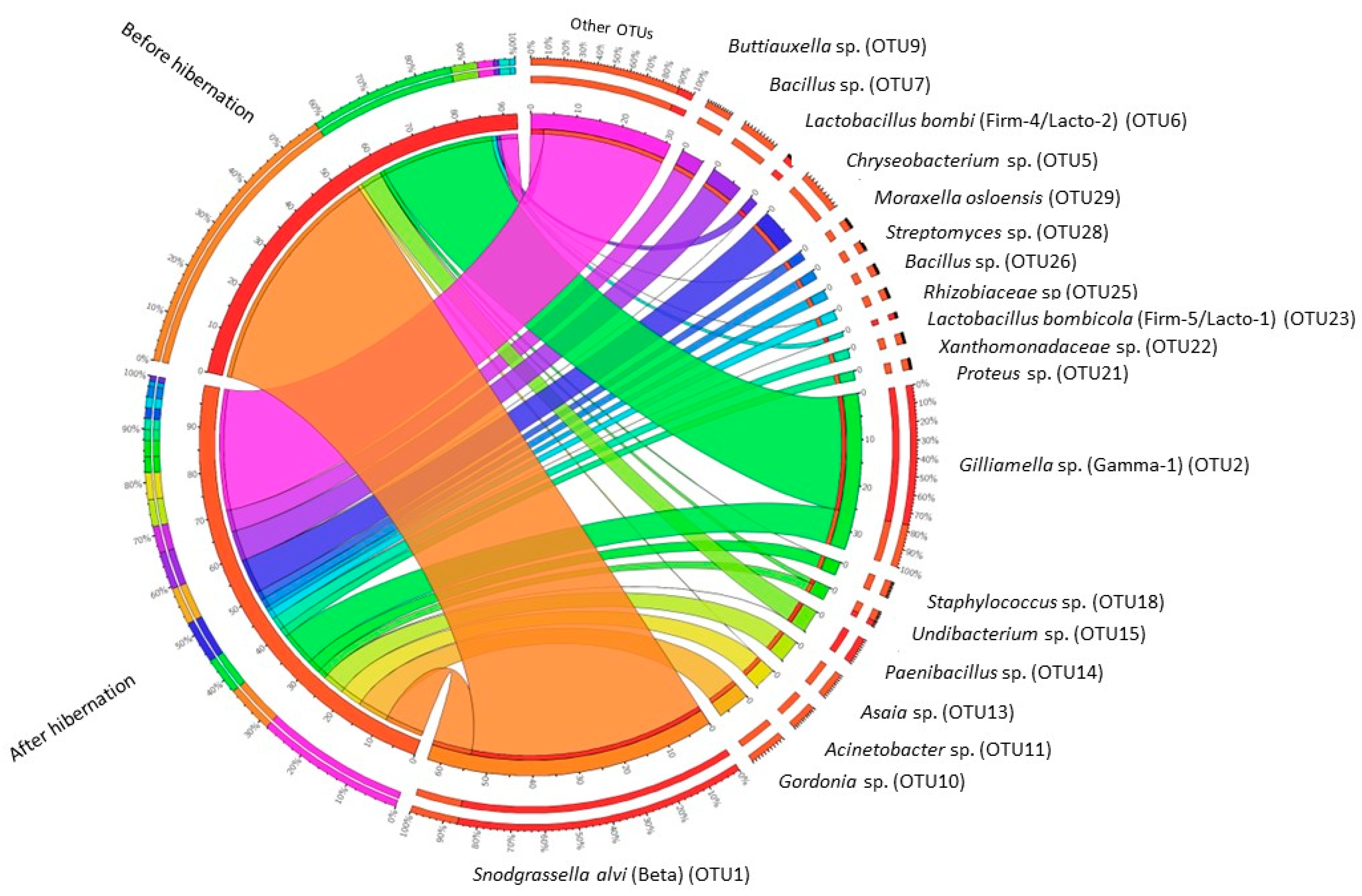
3. Results
4. Discussion
4.1. Community Structure of the Gut Bacterial Microbiota in Indoor-Reared Bumblebee Queens
4.2. Impact of Hibernation on the Gut Bacterial Community Composition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Neish, A.S. Microbes in gastrointestinal health and disease. Gastroenterology 2009, 136, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Madsen, E.L. Microorganisms and their roles in fundamental biogeochemical cycles. Curr. Opin. Biotechnol. 2001, 22, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.; Lee, S.M.; Shen, Y.; Khosravi, A.; Mazmanian, S.K. Host–bacterial symbiosis in health and disease. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2010; Volume 107, pp. 243–274. [Google Scholar]
- Hanning, I.; Diaz-Sanchez, S. The functionality of the gastrointestinal microbiome in non-human animals. Microbiome 2015, 3, 51. [Google Scholar] [CrossRef] [PubMed]
- West, S.A.; Griffin, A.S.; Gardner, A.; Diggle, S.P. Social evolution theory for microorganisms. Nat. Rev. Microbiol. 2006, 4, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Ruf, T.; Geiser, F. Daily torpor and hibernation in birds and mammals. Biol. Rev. 2015, 90, 891–926. [Google Scholar] [CrossRef] [PubMed]
- Carey, H.V.; Andrews, M.T.; Martin, S.L. Mammalian hibernation: Cellular and molecular responses to depressed metabolism and low temperature. Physiol. Rev. 2003, 83, 1153–1181. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Amaya, J.; Passement, C.A.; Dearing, M.D.; McCue, M.D. Unique and shared responses of the gut microbiota to prolonged fasting: A comparative study across five classes of vertebrate hosts. FEMS Microbiol. Ecol. 2014, 90, 883–894. [Google Scholar] [CrossRef]
- Sonoyama, K.; Fujiwara, R.; Takemura, N.; Ogasawara, T.; Watanabe, J.; Ito, H.; Morita, T. Response of gut microbiota to fasting and hibernation in Syrian hamsters. Appl. Environ. Microbiol. 2016, 75, 6451–6456. [Google Scholar] [CrossRef]
- Carey, H.V.; Walters, W.A.; Knight, R. Seasonal restructuring of the ground squirrel gut microbiota over the annual hibernation cycle. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2012, 304, R33–R42. [Google Scholar] [CrossRef]
- Sommer, F.; Ståhlman, M.; Ilkayeva, O.; Arnemo, J.M.; Kindberg, J.; Josefsson, J.; Bäckhed, F. The gut microbiota modulates energy metabolism in the hibernating brown bear Ursus arctos. Cell Rep. 2016, 14, 1655–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ultsch, G.R. Ecology and physiology of hibernation and overwintering among freshwater fishes, turtles, and snakes. Biol. Rev. 1989, 64, 435–515. [Google Scholar] [CrossRef]
- Storey, K.B.; Storey, J.M. Natural freezing survival in animals. Annu. Rev. Ecol. Syst. 1996, 27, 365–386. [Google Scholar] [CrossRef]
- Colston, T.J.; Jackson, C.R. Microbiome evolution along divergent branches of the vertebrate tree of life: What is known and unknown. Mol. Ecol. 2016, 25, 3776–3800. [Google Scholar] [CrossRef] [PubMed]
- Guppy, M.; Withers, P. Metabolic depression in animals: Physiological perspectives and biochemical generalizations. Biol. Rev. 1999, 74, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Danks, H.V. Insect adaptations to cold and changing environments. Can. Entomol. 2006, 138, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.E. Principles of insect low temperature tolerance. In Insects at Low Temperature; Springer: Boston, MA, USA, 1991; pp. 17–46. [Google Scholar]
- Koch, H.; Schmid-Hempel, P. Bacterial communities in central European bumblebees: Low diversity and high specificity. Microb. Ecol. 2011, 62, 121–133. [Google Scholar] [CrossRef]
- Kleijn, D.; Winfree, R.; Bartomeus, I.; Carvalheiro, L.G.; Henry, M.; Isaacs, R.; Ricketts, T.H. Delivery of crop pollination services is an insufficient argument for wild pollinator conservation. Nat. Commun. 2015, 6, 7414. [Google Scholar] [CrossRef] [Green Version]
- Meeus, I.; Mommaerts, V.; Billiet, A.; Mosallanejad, H.; Van de Wiele, T.; Wäckers, F.; Smagghe, G. Assessment of mutualism between Bombus terrestris and its microbiota by use of microcolonies. Apidologie 2013, 44, 708–719. [Google Scholar] [CrossRef]
- Lanan, M.C.; Rodrigues, P.A.P.; Agellon, A.; Jansma, P.; Wheeler, D.E. A bacterial filter protects and structures the gut microbiome of an insect. ISME J. 2016, 10, 1866–1876. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, S. Insect pathogenic fungi: Genomics, molecular interactions, and genetic improvements. Annu. Rev. Entomol. 2017, 62, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Edgar, R. SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. BioRxiv 2016, 074161. [Google Scholar] [CrossRef]
- Beccati, A.; Gerken, J.; Quast, C.; Yilmaz, P.; Glöckner, F.O. SILVA tree viewer: Interactive web browsing of the SILVA phylogenetic guide trees. BMC Bioinformat. 2017, 18, 433:1–433:4. [Google Scholar] [CrossRef] [PubMed]
- Cariveau, D.P.; Powell, J.E.; Koch, H.; Winfree, R.; Moran, N.A. Variation in gut microbial communities and its association with pathogen infection in wild bumblebees (Bombus). ISME J. 2014, 8, 2369–2379. [Google Scholar] [CrossRef]
- Meeus, I.; Parmentier, L.; Billiet, A.; Maebe, K.; Van Nieuwerburgh, F.; Deforce, D.; Smagghe, G. 16S rRNA amplicon sequencing demonstrates that indoor-reared bumblebees (Bombus terrestris) harbor a core subset of bacteria normally associated with the wild host. PLoS ONE 2015, 10, e0125152. [Google Scholar] [CrossRef] [Green Version]
- Mockler, B.K.; Kwong, W.K.; Moran, N.A.; Koch, H. Microbiome structure influences infection by the parasite Crithidia bombi in bumble bees. Appl. Environ. Microbiol. 2018, 84, AEM-02335. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G.; Ramsing, N.B. Molecular methods to study the organization of microbial communities. Water Sci. Technol. 1995, 32, 1–9. [Google Scholar] [CrossRef]
- Huang, Q.; Evans, J.D. Identification of microRNA-like small RNAs from fungal parasite Nosema ceranae. J. Invertebr. Pathol. 2016, 133, 107–109. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Jost, L. Entropy and diversity. Oikos 2016, 13, 363–375. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, N.A.; Hansen, A.K.; Powell, J.E.; Sabree, Z.L. Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS ONE 2012, 7, e36393. [Google Scholar] [CrossRef] [PubMed]
- Martinson, V.G.; Danforth, B.N.; Minckley, R.L.; Rueppell, O.; Tingek, S.; Moran, N.A. A simple and distinctive microbiota associated with honey bees and bumblebees. Mol. Ecol. 2011, 20, 619–628. [Google Scholar] [CrossRef]
- Killer, J.; Dubná, S.; Sedláček, I.; Švec, P. Lactobacillus apis sp. nov., from the stomach of honeybees (Apis mellifera), having an in vitro inhibitory effect on the causative agents of American and European foulbrood. Int. J. Syst. Evolut. Microbiol. 2014, 64, 152–157. [Google Scholar] [CrossRef]
- Praet, J.; Meeus, I.; Cnockaert, M.; Aerts, M.; Smagghe, G.; Vandamme, P. Bifidobacterium commune sp. nov. isolated from the bumble bee gut. Antonie Van Leeuwenhoek 2015, 107, 1307–1313. [Google Scholar] [CrossRef]
- Martinson, V.G.; Moy, J.; Moran, N.A. Establishment of characteristic gut bacteria during development of the honey bee worker. Appl. Environ. Microbiol. 2012, AEM-07810. [Google Scholar] [CrossRef]
- Kwong, W.K.; Moran, N.A. Gut microbial communities of social bees. Nat. Rev. Microbiol. 2016, 14, 374–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, W.K.; Medina, L.A.; Koch, H.; Sing, K.W.; Soh, E.J.Y.; Ascher, J.S.; Moran, N.A. Dynamic microbiome evolution in social bees. Sci. Adv. 2017, 3, e1600513. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, T.J.; Duddleston, K.N.; Buck, C.L. Effects of season and host physiological state on the diversity, density, and activity of the arctic ground squirrel cecal microbiota. Appl. Environ. Microbiol. 2014, 80, 5611–5622. [Google Scholar] [CrossRef] [PubMed]
- Weng, F.C.H.; Yang, Y.J.; Wang, D. Functional analysis for gut microbes of the brown tree frog (Polypedates megacephalus) in artificial hibernation. BMC Genom. 2016, 17, 1024. [Google Scholar] [CrossRef] [PubMed]
- Margesin, R.; Miteva, V. Diversity and ecology of psychrophilic microorganisms. Res. Microbiol. 2011, 162, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Tarpy, D.R.; Mattila, H.R.; Newton, I.L. Characterization of the honey bee microbiome throughout the queen-rearing process. Appl. Environ. Microbiol. 2015, AEM-00307. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OUT b | Taxonomic Affiliation | Before Hibernation | After Hibernation | |||||
|---|---|---|---|---|---|---|---|---|
| Phylum | Family | Species c | Name OTU in Literature d | Relative Abundance (%) | Present in B. terrestris (n = 15) | Relative Abundance (%) | Present in B. terrestris (n = 15) | |
| OTU_1 | Proteobacteria | Neisseriaceae | Snodgrassella alvi (98.8%) | Beta | 54.525 | 15 | 8.668 | 15 |
| OTU_2 | Proteobacteria | Orbaceae | Gilliamella apicola, G. bombi, G. bombicola; G. mensalis (100%) | Gamma-1 | 33.805 | 15 | 8.753 | 15 |
| OTU_5 * | Bacteroidetes | Flavobacteriaceae | Chryseobacterium daecheongense (100%) | 0.001 | 2 | 7.593 | 15 | |
| OTU_10 * | Actinobacteria | Nocardiaceae | Gordonia polyisoprenivorans, G. soli (100%) | 0 | 0 | 7.131 | 12 | |
| OTU_7 * | Firmicutes | Bacillaceae | Several Bacillaceae spp. (100%) | 0.001 | 2 | 6.999 | 15 | |
| OTU_14 | Firmicutes | Paenibacillaceae | Paenibacillus amylolyticus, P. pabuli, P. taichungensis, P. tundra, P. tylopili, P. xylanexedens and P. xylanilyticus (100%) | 5.3 | 15 | 0.116 | 13 | |
| OTU_9 * | Proteobacteria | Enterobacteriaceae | Several Enterobacteriaceae spp., including Buttiauxella agrestis (100%) | Gamma-E1 | 0.004 | 7 | 5.124 | 15 |
| OTU_13 * | Proteobacteria | Acetobacteraceae | Asia bogorensis, A. siamensis, A. prunella, A. lannensis (100%) | 0 | 0 | 5.112 | 15 | |
| OTU_11 * | Proteobacteria | Moraxellaceae | Acinetobacter vivianii, A. proteolyticus, A. modestus, A. courvalinii (100%) | 0.186 | 7 | 4.996 | 14 | |
| OTU_18 * | Firmicutes | Staphylococcaceae | Several Staphylococcaceae spp. (100%) | 0.111 | 9 | 2.954 | 15 | |
| OTU_15 | Proteobacteria | Oxalobacteraceae | Undibacterium oligocarboniphilum (100%) | 0.618 | 12 | 2.417 | 15 | |
| OTU_21 * | Proteobacteria | Enterobacteriaceae | Proteus mirabilis, P. penneri, Cosenzaea myxofaciens (100%) | 0 | 0 | 2.340 | 15 | |
| OTU_22 * | Proteobacteria | Xanthomonadaceae | Thermomonas haemolytica (99%) | 0 | 0 | 2.287 | 14 | |
| OTU_29 | Proteobacteria | Moraxellaceae | Moraxella osloensis (100%) | 0.007 | 4 | 2.064 | 11 | |
| OTU_25 | Proteobacteria | Rhizobiaceae | Agrobacterium tumefaciens, A. fabrum (100%) | 0.049 | 7 | 1.958 | 15 | |
| OTU_6 | Firmicutes | Lactobacillaceae | Lactobacillus bombi (100%) | Firm-4/Lacto-2 | 1.612 | 9 | 0.507 | 10 |
| OTU_28 | Actinobacteria | Streptomycetaceae | Several Streptomycetaceae spp. (100%) | 0 | 0 | 1.567 | 14 | |
| OTU_26 | Firmicutes | Bacillaceae | Several Bacillaceae spp. (100%) | 0 | 0 | 1.518 | 13 | |
| OTU_23 | Firmicutes | Lactobacillaceae | Lactobacillus bombicola (100%) | Firm-5/Lacto-1 | 1.092 | 11 | 0.213 | 7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosmans, L.; Pozo, M.I.; Verreth, C.; Crauwels, S.; Wäckers, F.; Jacquemyn, H.; Lievens, B. Hibernation Leads to Altered Gut Communities in Bumblebee Queens (Bombus terrestris). Insects 2018, 9, 188. https://0-doi-org.brum.beds.ac.uk/10.3390/insects9040188
Bosmans L, Pozo MI, Verreth C, Crauwels S, Wäckers F, Jacquemyn H, Lievens B. Hibernation Leads to Altered Gut Communities in Bumblebee Queens (Bombus terrestris). Insects. 2018; 9(4):188. https://0-doi-org.brum.beds.ac.uk/10.3390/insects9040188
Chicago/Turabian StyleBosmans, Lien, María I. Pozo, Christel Verreth, Sam Crauwels, Felix Wäckers, Hans Jacquemyn, and Bart Lievens. 2018. "Hibernation Leads to Altered Gut Communities in Bumblebee Queens (Bombus terrestris)" Insects 9, no. 4: 188. https://0-doi-org.brum.beds.ac.uk/10.3390/insects9040188