
Epigenetics of Genes Preferentially Expressed in Dissimilar Cell Populations: Myoblasts and Cerebellum

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Genome-Wide Search for Genes Preferentially Expressed in Both Myoblasts and Cerebellum
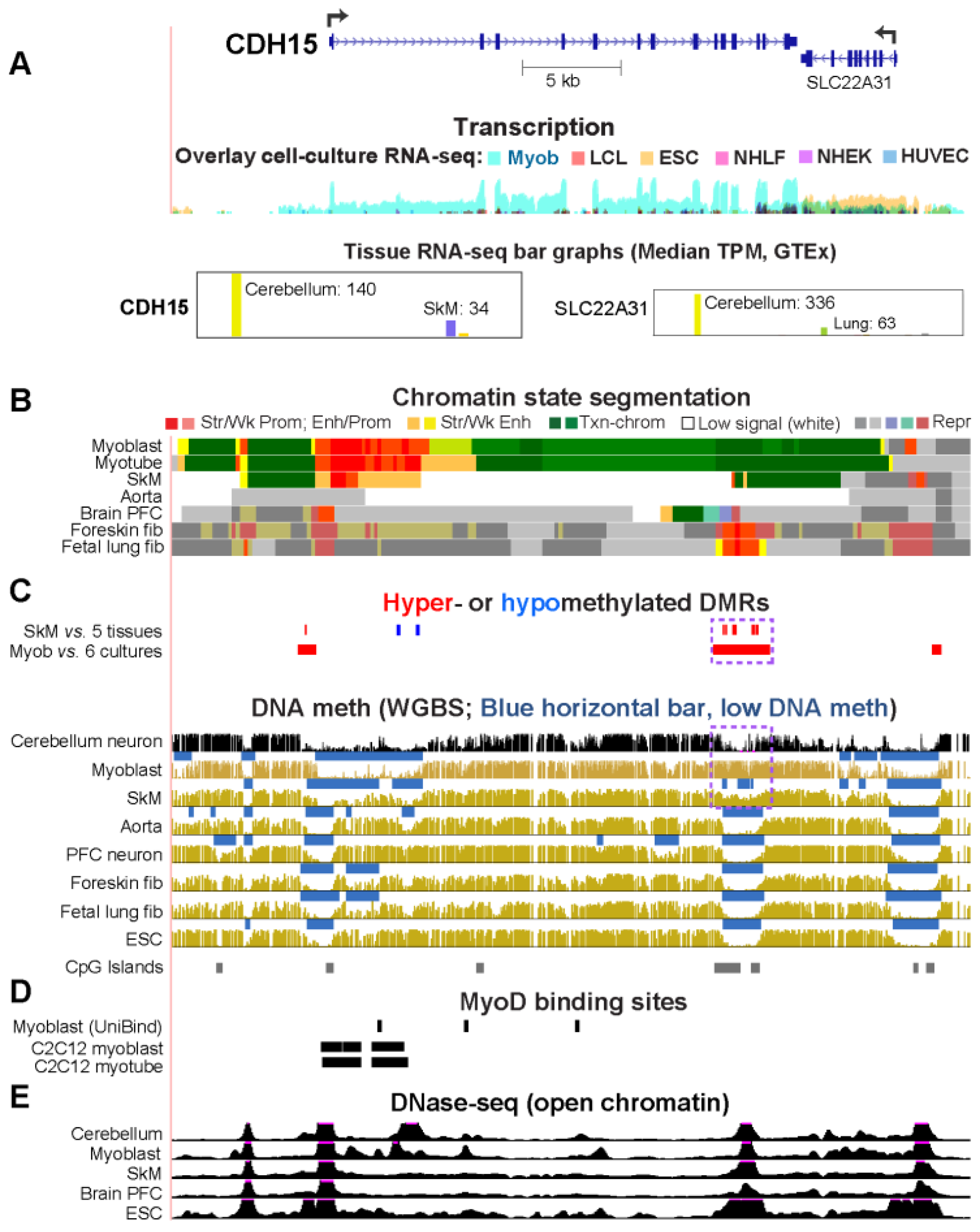
2.2. CDH15, Which Encodes a Myoblast/Cerebellum Cadherin, Has a Hypomethylated 5′ Region in Myoblasts and Cerebellum
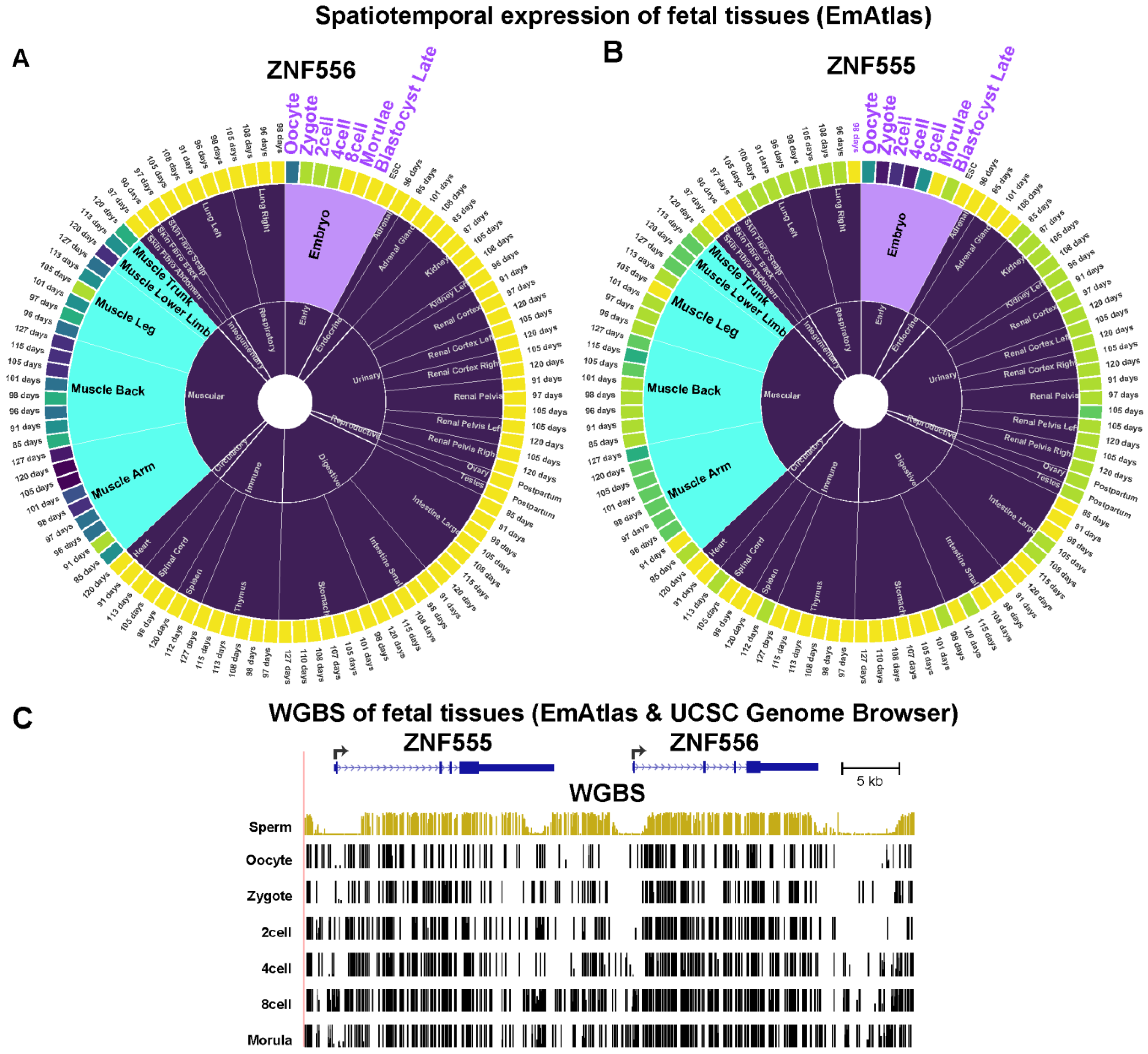
2.3. The Promoter Region of Transcription Factor-Encoding ZNF556 Is Hypomethylated Specifically in Tissues and Cells Expressing the Gene
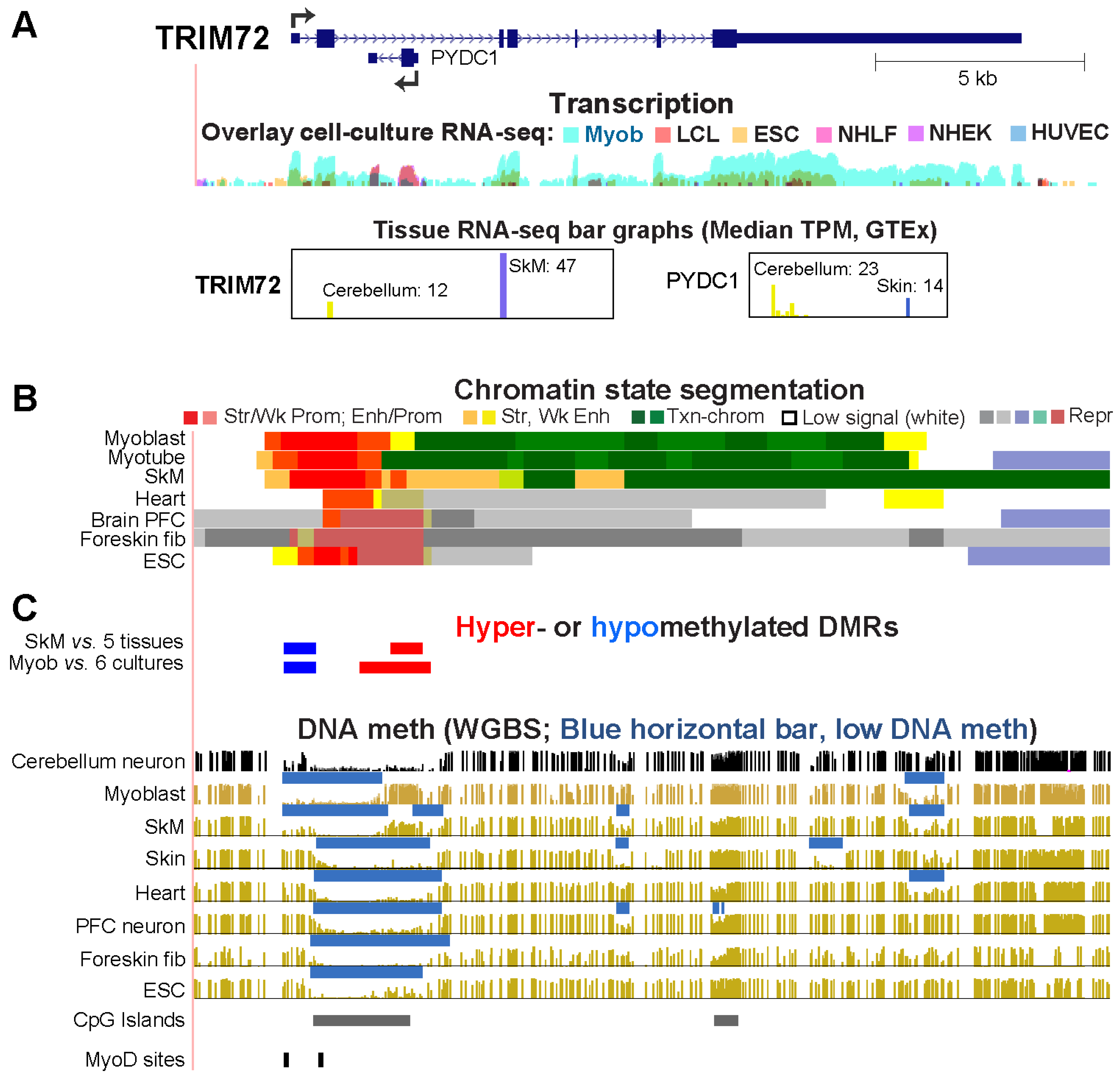
2.4. Differential Methylation of the Extended Promoter Regions of TRIM72 and Its Intronic Gene, PYDC, Correlates with Differential Expression in Myoblasts and Cerebellum
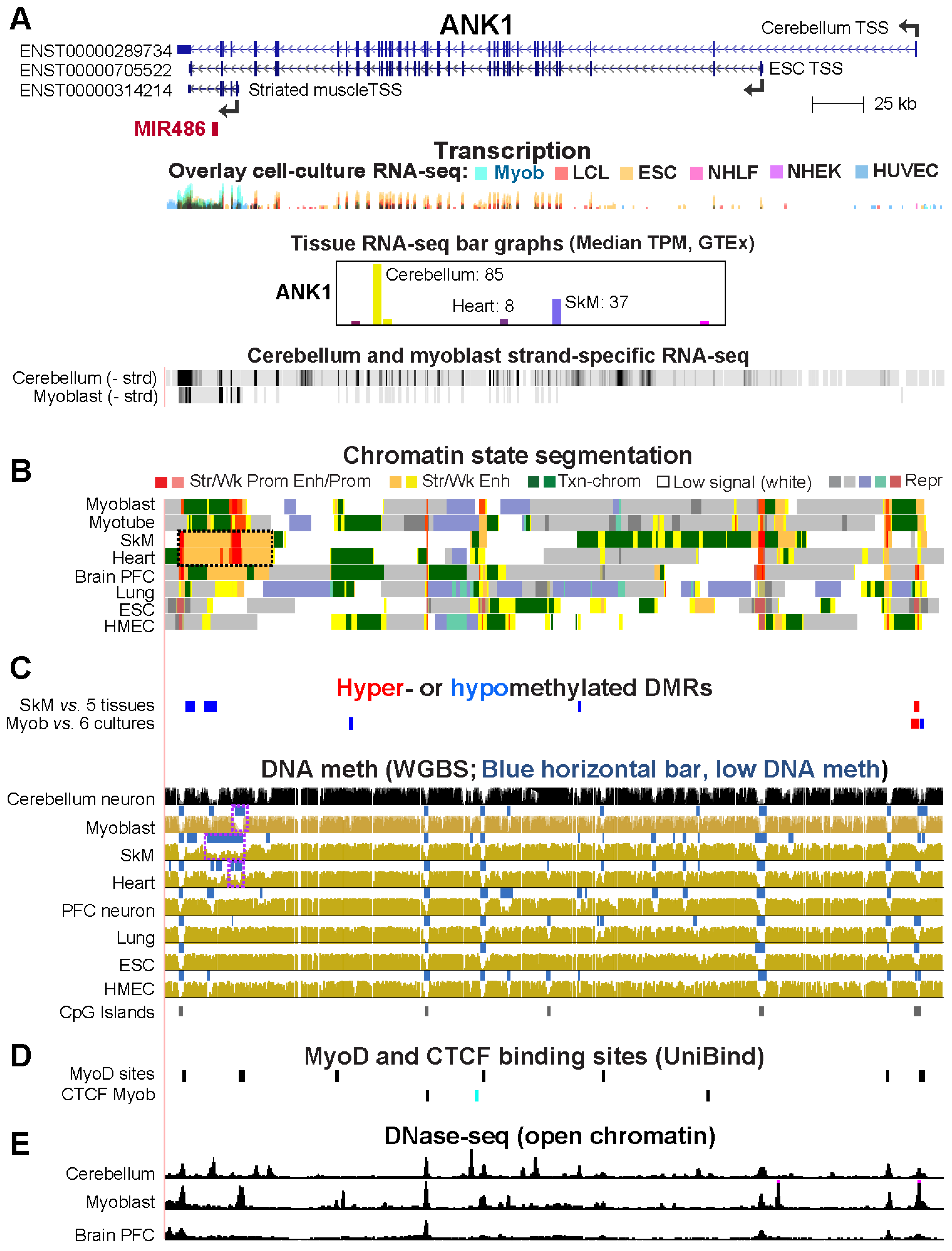
2.5. Alternate ANK1 Promoter Usage in Myoblasts vs. Cerebellum Is Associated with Differential Promoter Methylation
2.6. Myoblast DNA Hypermethylation of PAX3 May Be Downmodulating Gene Expression by Suppressing Super-Enhancer Formation
2.7. Other Genes Preferentially Expressed in Myoblasts and Cerebellum Profiles Display Myoblast- or Cerebellum-Associated Hypomethylation and Overlapping Transcription Factor Binding Sites
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Transcriptomics
5.2. Epigenomics
5.3. Reporter Gene Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fukada, S.I.; Higashimoto, T.; Kaneshige, A. Differences in muscle satellite cell dynamics during muscle hypertrophy and regeneration. Skelet. Muscle 2022, 12, 17. [Google Scholar] [CrossRef]
- Chal, J.; Pourquié, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Victor, P.; García-Prat, L.; Muñoz-Cánoves, P. Control of satellite cell function in muscle regeneration and its disruption in ageing. Nat. Rev. Mol. Cell Biol. 2022, 23, 204–226. [Google Scholar] [CrossRef]
- Murach, K.A.; Fry, C.S.; Dupont-Versteegden, E.E.; McCarthy, J.J.; Peterson, C.A. Fusion and beyond: Satellite cell contributions to loading-induced skeletal muscle adaptation. FASEB J. 2021, 35, e21893. [Google Scholar] [CrossRef] [PubMed]
- Sharples, A.P.; Turner, D.C. Skeletal muscle memory. Am. J. Physiol. Cell Physiol. 2023, 324, C1274–C1294. [Google Scholar] [CrossRef] [PubMed]
- Falick Michaeli, T.; Sabag, O.; Fok, R.; Azria, B.; Monin, J.; Nevo, Y.; Gielchinsky, Y.; Berman, B.P.; Cedar, H.; Bergman, Y. Muscle injury causes long-term changes in stem-cell DNA methylation. Proc. Natl. Acad. Sci. USA 2022, 119, e2212306119. [Google Scholar] [CrossRef]
- Robinson, D.C.L.; Dilworth, F.J. Epigenetic Regulation of Adult Myogenesis. Curr. Top. Dev. Biol. 2018, 126, 235–284. [Google Scholar] [CrossRef]
- Biressi, S.; Molinaro, M.; Cossu, G. Cellular heterogeneity during vertebrate skeletal muscle development. Dev. Biol. 2007, 308, 281–293. [Google Scholar] [CrossRef]
- Relaix, F.; Marcelle, C. Muscle stem cells. Curr. Opin. Cell Biol. 2009, 21, 748–753. [Google Scholar] [CrossRef]
- Bengtsen, M.; Winje, I.M.; Eftestøl, E.; Landskron, J.; Sun, C.; Nygård, K.; Domanska, D.; Millay, D.P.; Meza-Zepeda, L.A.; Gundersen, K. Comparing the epigenetic landscape in myonuclei purified with a PCM1 antibody from a fast/glycolytic and a slow/oxidative muscle. PLoS Genet. 2021, 17, e1009907. [Google Scholar] [CrossRef]
- Murach, K.A.; Dungan, C.M.; von Walden, F.; Wen, Y. Epigenetic evidence for distinct contributions of resident and acquired myonuclei during long-term exercise adaptation using timed in vivo myonuclear labeling. Am. J. Physiol. Cell Physiol. 2022, 322, C86–C93. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Dungan, C.M.; Mobley, C.B.; Valentino, T.; von Walden, F.; Murach, K.A. Nucleus type-specific DNA methylomics reveals epigenetic “memory” of prior sdaptation in skeletal muscle. Function 2021, 2, zqab038. [Google Scholar] [CrossRef] [PubMed]
- Ponnaluri, V.K.; Ehrlich, K.C.; Zhang, G.; Lacey, M.; Johnston, D.; Pradhan, S.; Ehrlich, M. Association of 5-hydroxymethylation and 5-methylation of DNA cytosine with tissue-specific gene expression. Epigenetics 2017, 12, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Marzban, H.; Del Bigio, M.R.; Alizadeh, J.; Ghavami, S.; Zachariah, R.M.; Rastegar, M. Cellular commitment in the developing cerebellum. Front. Cell Neurosci. 2014, 8, 450. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, S.; Badura, A.; Lutzu, S.; Pathak, S.S.; Thieme, A.; Verpeut, J.L.; Wagner, M.J.; Yang, Y.M.; Fioravante, D. Cognitive-Affective Functions of the Cerebellum. J. Neurosci. 2023, 43, 7554–7564. [Google Scholar] [CrossRef] [PubMed]
- The_GTEx_Consortium Human Genomics. The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Ehrlich, K.C.; Lacey, M.; Baribault, C.; Sen, S.; Esteve, P.O.; Pradhan, S.; Ehrlich, M. Promoter-Adjacent DNA Hypermethylation Can Downmodulate Gene Expression: TBX15 in the Muscle Lineage. Epigenomes 2022, 6, 43. [Google Scholar] [CrossRef]
- Loyfer, N.; Magenheim, J.; Peretz, A.; Cann, G.; Bredno, J.; Klochendler, A.; Fox-Fisher, I.; Shabi-Porat, S.; Hecht, M.; Pelet, T.; et al. A DNA methylation atlas of normal human cell types. Nature 2023, 613, 355–364. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef]
- Baribault, C.; Ehrlich, K.C.; Ponnaluri, V.K.C.; Pradhan, S.; Lacey, M.; Ehrlich, M. Developmentally linked human DNA hypermethylation is associated with down-modulation, repression, and upregulation of transcription. Epigenetics 2018, 13, 275–289. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Donalies, M.; Cramer, M.; Ringwald, M.; Starzinski-Powitz, A. Expression of M-cadherin, a member of the cadherin multigene family, correlates with differentiation of skeletal muscle cells. Proc. Natl. Acad. Sci. USA 1991, 88, 8024–8028. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Decato, B.; Hong, E.E.; Zhou, M.; Fang, F.; Qu, J.; Garvin, T.; Kessler, M.; Zhou, J.; Smith, A.D. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS ONE 2013, 8, e81148. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Rich, J.; Karoutas, A.; Tarlykov, P.; Cochet, E.; Malysheva, D.; Mamchaoui, K.; Ogryzko, V.; Pirozhkova, I. ZNF555 protein binds to transcriptional activator site of 4qA allele and ANT1: Potential implication in Facioscapulohumeral dystrophy. Nucleic Acids Res. 2015, 43, 8227–8242. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; O’Day, D.R.; Pliner, H.A.; Kingsley, P.D.; Deng, M.; Daza, R.M.; Zager, M.A.; Aldinger, K.A.; Blecher-Gonen, R.; Zhang, F.; et al. A human cell atlas of fetal gene expression. Science 2020, 370, eaba7721. [Google Scholar] [CrossRef] [PubMed]
- Terragni, J.; Zhang, G.; Sun, Z.; Pradhan, S.; Song, L.; Crawford, G.E.; Lacey, M.; Ehrlich, M. Notch signaling genes: Myogenic DNA hypomethylation and 5-hydroxymethylcytosine. Epigenetics 2014, 9, 842–850. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Zheng, L.; Liang, P.; Long, C.; Li, H.; Li, H.; Liang, Y.; He, X.; Xi, Q.; Xing, Y.; Zuo, Y. EmAtlas: A comprehensive atlas for exploring spatiotemporal activation in mammalian embryogenesis. Nucleic Acids Res. 2023, 51, D924–D932. [Google Scholar] [CrossRef]
- Hammoud, S.S.; Low, D.H.; Yi, C.; Carrell, D.T.; Guccione, E.; Cairns, B.R. Chromatin and transcription transitions of mammalian adult germline stem cells and spermatogenesis. Cell Stem Cell 2014, 15, 239–253. [Google Scholar] [CrossRef]
- Mokhonova, E.I.; Avliyakulov, N.K.; Kramerova, I.; Kudryashova, E.; Haykinson, M.J.; Spencer, M.J. The E3 ubiquitin ligase TRIM32 regulates myoblast proliferation by controlling turnover of NDRG2. Hum. Mol. Genet. 2015, 24, 2873–2883. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Han, J.; Jeong, B.C.; Song, J.H.; Jang, S.H.; Jeong, H.; Kim, B.H.; Ko, Y.G.; Park, Z.Y.; Lee, K.E.; et al. Structure and activation of the RING E3 ubiquitin ligase TRIM72 on the membrane. Nat. Struct. Mol. Biol. 2023, 30, 1695–1706. [Google Scholar] [CrossRef]
- Benissan-Messan, D.Z.; Zhu, H.; Zhong, W.; Tan, T.; Ma, J.; Lee, P.H.U. Multi-Cellular Functions of MG53 in Muscle Calcium Signaling and Regeneration. Front. Physiol. 2020, 11, 583393. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.31–1.30.33. [Google Scholar] [CrossRef]
- Gallagher, P.G.; Forget, B.G. An alternate promoter directs expression of a truncated, muscle-specific isoform of the human ankyrin 1 gene. J. Biol. Chem. 1998, 273, 1339–1348. [Google Scholar] [CrossRef]
- Kodzius, R.; Kojima, M.; Nishiyori, H.; Nakamura, M.; Fukuda, S.; Tagami, M.; Sasaki, D.; Imamura, K.; Kai, C.; Harbers, M.; et al. CAGE: Cap analysis of gene expression. Nat. Methods 2006, 3, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Samani, A.; Hightower, R.M.; Reid, A.L.; English, K.G.; Lopez, M.A.; Doyle, J.S.; Conklin, M.J.; Schneider, D.A.; Bamman, M.M.; Widrick, J.J.; et al. miR-486 is essential for muscle function and suppresses a dystrophic transcriptome. Life Sci. Alliance 2022, 5, e202101215. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef]
- Wallén, A.; Perlmann, T. Transcriptional control of dopamine neuron development. Ann. N. Y. Acad. Sci. 2003, 991, 48–60. [Google Scholar] [CrossRef]
- Buckingham, M. Gene regulatory networks and cell lineages that underlie the formation of skeletal muscle. Proc. Natl. Acad. Sci. USA 2017, 114, 5830–5837. [Google Scholar] [CrossRef]
- Medic, S.; Rizos, H.; Ziman, M. Differential PAX3 functions in normal skin melanocytes and melanoma cells. Biochem. Biophys. Res. Commun. 2011, 411, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Tsumagari, K.; Baribault, C.; Terragni, J.; Varley, K.E.; Gertz, J.; Pradhan, S.; Badoo, M.; Crain, C.M.; Song, L.; Crawford, G.E.; et al. Early de novo DNA methylation and prolonged demethylation in the muscle lineage. Epigenetics 2013, 8, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.M.; Stamatoyannopoulos, J.; Snyder, M.; Dunham, I.; Hardison, R.C.; Bernstein, B.E.; Gingeras, T.R.; Kent, W.J.; Birney, E.; Wold, B.; et al. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9, e1001046. [Google Scholar]
- Roth, R.B.; Hevezi, P.; Lee, J.; Willhite, D.; Lechner, S.M.; Foster, A.C.; Zlotnik, A. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics 2006, 7, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Suzuki, M.; Sato, S.; Arai, Y.; Shinohara, T.; Tanaka, S.; Greally, J.M.; Hattori, N.; Shiota, K. A new class of tissue-specifically methylated regions involving entire CpG islands in the mouse. Genes. Cells 2007, 12, 1305–1314. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef]
- Rebollo, R.; Miceli-Royer, K.; Zhang, Y.; Farivar, S.; Gagnier, L.; Mager, D.L. Epigenetic interplay between mouse endogenous retroviruses and host genes. Genome Biol. 2012, 13, R89. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes. Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Shen, L.; Kondo, Y.; Guo, Y.; Zhang, J.; Zhang, L.; Ahmed, S.; Shu, J.; Chen, X.; Waterland, R.A.; Issa, J.P. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007, 3, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Gruhn, W.H.; Tang, W.W.C.; Dietmann, S.; Alves-Lopes, J.P.; Penfold, C.A.; Wong, F.C.K.; Ramakrishna, N.B.; Surani, M.A. Epigenetic resetting in the human germ line entails histone modification remodeling. Sci. Adv. 2023, 9, eade1257. [Google Scholar] [CrossRef] [PubMed]
- Tessema, M.; Yingling, C.M.; Picchi, M.A.; Wu, G.; Ryba, T.; Lin, Y.; Bungum, A.O.; Edell, E.S.; Spira, A.; Belinsky, S.A. ANK1 Methylation regulates expression of MicroRNA-486-5p and discriminates lung tumors by histology and smoking status. Cancer Lett. 2017, 410, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Peng, H.Y.; Mo, K.C.; Hsu, Y.M.; Wu, G.H.; Hsiao, J.R.; Lin, S.F.; Wang, H.D.; Shiah, S.G. MicroRNA-486-3p functions as a tumor suppressor in oral cancer by targeting DDR1. J. Exp. Clin. Cancer Res. 2019, 38, 281. [Google Scholar] [CrossRef] [PubMed]
- Lev Maor, G.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef]
- Wang, Q.; Xiong, F.; Wu, G.; Liu, W.; Chen, J.; Wang, B.; Chen, Y. Gene body methylation in cancer: Molecular mechanisms and clinical applications. Clin. Epigenetics 2022, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Janssen, S.M.; Lorincz, M.C. Interplay between chromatin marks in development and disease. Nat. Rev. Genet. 2022, 23, 137–153. [Google Scholar] [CrossRef]
- Meehan, R.R.; Pennings, S. Shoring up DNA methylation and H3K27me3 domain demarcation at developmental genes. EMBO J. 2017, 36, 3407–3408. [Google Scholar] [CrossRef]
- James, S.J.; Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.P. Elevated 5-hydroxymethylcytosine in the Engrailed-2 (EN-2) promoter is associated with increased gene expression and decreased MeCP2 binding in autism cerebellum. Transl. Psychiatry 2014, 4, e460. [Google Scholar] [CrossRef]
- Wang, M.; Xie, H.; Shrestha, S.; Sredni, S.; Morgan, G.A.; Pachman, L.M. Methylation alterations of WT1 and homeobox genes in inflamed muscle biopsies from untreated juvenile dermatomyositis suggests self-renewal capacity. Arthritis Rheum. 2012, 64, 3478–3485. [Google Scholar] [CrossRef]
- Van Dyck, L.; Güiza, F.; Derese, I.; Pauwels, L.; Casaer, M.P.; Hermans, G.; Wouters, P.J.; Van den Berghe, G.; Vanhorebeek, I. DNA methylation alterations in muscle of critically ill patients. J. Cachexia Sarcopenia Muscle 2022, 13, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Li, X.; Looso, M.; Liu, H.; Ding, D.; Günther, S.; Kuenne, C.; Liu, S.; Weissmann, N.; Boettger, T.; et al. Spurious transcription causing innate immune responses is prevented by 5-hydroxymethylcytosine. Nat. Genet. 2023, 55, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xie, L.; Wei, H.; Li, X.J.; Lin, L. Dynamic Regulation of DNA Methylation and Brain Functions. Biology 2023, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Nestor, C.E.; Ottaviano, R.; Reinhardt, D.; Cruickshanks, H.A.; Mjoseng, H.K.; McPherson, R.C.; Lentini, A.; Thomson, J.P.; Dunican, D.S.; Pennings, S.; et al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol. 2015, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.P. Complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl. Psychiatry 2013, 3, e232. [Google Scholar] [CrossRef] [PubMed]
- Soltani, A.; Lebrun, S.; Carpentier, G.; Zunino, G.; Chantepie, S.; Maïza, A.; Bozzi, Y.; Desnos, C.; Darchen, F.; Stettler, O. Increased signaling by the autism-related Engrailed-2 protein enhances dendritic branching and spine density, alters synaptic structural matching, and exaggerates protein synthesis. PLoS ONE 2017, 12, e0181350. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef]
- Ehrlich, M.; Ehrlich, K.C. DNA cytosine methylation and hydroxymethylation at the borders. Epigenomics 2014, 6, 563–566. [Google Scholar] [CrossRef]
- Angiari, S.; D’Alessandro, G.; Paolicelli, R.C.; Prada, I.; Vannini, E. Editorial: Cell-Cell Interactions Controlling Neuronal Functionality in Health and Disease. Front. Integr. Neurosci. 2022, 16, 968029. [Google Scholar] [CrossRef]
- Krauss, R.S.; Joseph, G.A.; Goel, A.J. Keep Your Friends Close: Cell-Cell Contact and Skeletal Myogenesis. Cold Spring Harb. Perspect. Biol. 2017, 9, a029298. [Google Scholar] [CrossRef]
- Ozawa, M. E-cadherin cytoplasmic domain inhibits cell surface localization of endogenous cadherins and fusion of C2C12 myoblasts. Biol. Open 2015, 4, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.; Luo, Y.; Buchan, T.; Beachem, M.A.; Guzauskas, G.F.; Ladd, S.; Bratcher, S.J.; Schroer, R.J.; Balsamo, J.; DuPont, B.R.; et al. Alterations in CDH15 and KIRREL3 in patients with mild to severe intellectual disability. Am. J. Hum. Genet. 2008, 83, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Redies, C.; Hertel, N.; Hübner, C.A. Cadherins and neuropsychiatric disorders. Brain Res. 2012, 1470, 130–144. [Google Scholar] [CrossRef]
- Jankowski, J.; Holst, M.I.; Liebig, C.; Oberdick, J.; Baader, S.L. Engrailed-2 negatively regulates the onset of perinatal Purkinje cell differentiation. J. Comp. Neurol. 2004, 472, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.A.; Gnocchi, V.F.; White, R.B.; Boldrin, L.; Perez-Ruiz, A.; Relaix, F.; Morgan, J.E.; Zammit, P.S. Integrated functions of Pax3 and Pax7 in the regulation of proliferation, cell size and myogenic differentiation. PLoS ONE 2009, 4, e4475. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Katahira, T.; Matsunaga, E.; Sato, T. Isthmus organizer for midbrain and hindbrain development. Brain Res. Brain Res. Rev. 2005, 49, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Schinzel, F.; Seyfer, H.; Ebbers, L.; Nothwang, H.G. The Lbx1 lineage differentially contributes to inhibitory cell types of the dorsal cochlear nucleus, a cerebellum-like structure, and the cerebellum. J. Comp. Neurol. 2021, 529, 3032–3045. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.D.; Barber, M.C.; Tomescu, O.; Barr, F.G.; Ruben, S.; Friedman, T.B. Identification of target genes regulated by PAX3 and PAX3-FKHR in embryogenesis and alveolar rhabdomyosarcoma. Genomics 2002, 79, 278–284. [Google Scholar] [CrossRef]
- Buckingham, M.; Bajard, L.; Chang, T.; Daubas, P.; Hadchouel, J.; Meilhac, S.; Montarras, D.; Rocancourt, D.; Relaix, F. The formation of skeletal muscle: From somite to limb. J. Anat. 2003, 202, 59–68. [Google Scholar] [CrossRef]
- Cao, Y.; Yao, Z.; Sarkar, D.; Lawrence, M.; Sanchez, G.J.; Parker, M.H.; MacQuarrie, K.L.; Davison, J.; Morgan, M.T.; Ruzzo, W.L.; et al. Genome-wide MyoD binding in skeletal muscle cells: A potential for broad cellular reprogramming. Dev. Cell 2010, 18, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, K.C.; Lacey, M.; Ehrlich, M. Epigenetics of Skeletal Muscle-Associated Genes in the ASB, LRRC, TMEM, and OSBPL Gene Families. Epigenomes 2020, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Huang, C.; Xu, C.; Qiu, L.; Yang, F. Dysfunction of ZNF554 promotes ROS-induced apoptosis and autophagy in Fetal Growth Restriction via the p62-Keap1-Nrf2 pathway. Placenta 2023, 143, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Sirmaci, A.; Edwards, Y.J.; Akay, H.; Tekin, M. Challenges in whole exome sequencing: An example from hereditary deafness. PLoS ONE 2012, 7, e32000. [Google Scholar] [CrossRef]
- Aruga, J.; Shimoda, K.; Mikoshiba, K. A 5′ segment of the mouse Zic1 gene contains a region specific enhancer for dorsal hindbrain and spinal cord. Brain Res. Mol. Brain Res. 2000, 78, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Aruga, J.; Millen, K.J. ZIC1 function in normal cerebellar development and human developmental pathology. Adv. Exp. Med. Biol. 2018, 1046, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Vandervore, L.V.; Schot, R.; Hoogeboom, A.J.M.; Lincke, C.; de Coo, I.F.; Lequin, M.H.; Dremmen, M.; van Unen, L.M.A.; Saris, J.J.; Jansen, A.C.; et al. Mutated zinc finger protein of the cerebellum 1 leads to microcephaly, cortical malformation, callosal agenesis, cerebellar dysplasia, tethered cord and scoliosis. Eur. J. Med. Genet. 2018, 61, 783–789. [Google Scholar] [CrossRef]
- Pan, H.; Gustafsson, M.K.; Aruga, J.; Tiedken, J.J.; Chen, J.C.; Emerson, C.P., Jr. A role for Zic1 and Zic2 in Myf5 regulation and somite myogenesis. Dev. Biol. 2011, 351, 120–127. [Google Scholar] [CrossRef]
- Correns, A.; Zimmermann, L.A.; Baldock, C.; Sengle, G. BMP antagonists in tissue development and disease. Matrix Biol. Plus 2021, 11, 100071. [Google Scholar] [CrossRef]
- Mikawa, S.; Sato, K. Chordin expression in the adult rat brain. Neuroscience 2014, 258, 16–33. [Google Scholar] [CrossRef]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef]
- Alfano, G.; Shah, A.Z.; Jeffery, G.; Bhattacharya, S.S. First insights into the expression of VAX2 in humans and its localization in the adult primate retina. Exp. Eye Res. 2016, 148, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Schnaar, R.L.; Gerardy-Schahn, R.; Hildebrandt, H. Sialic acids in the brain: Gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol. Rev. 2014, 94, 461–518. [Google Scholar] [CrossRef]
- Go, S.; Go, S.; Veillon, L.; Ciampa, M.G.; Mauri, L.; Sato, C.; Kitajima, K.; Prinetti, A.; Sonnino, S.; Inokuchi, J.I. Altered expression of ganglioside GM3 molecular species and a potential regulatory role during myoblast differentiation. J. Biol. Chem. 2017, 292, 7040–7051. [Google Scholar] [CrossRef] [PubMed]
- Sarnat, H.B.; Benjamin, D.R.; Siebert, J.R.; Kletter, G.B.; Cheyette, S.R. Agenesis of the mesencephalon and metencephalon with cerebellar hypoplasia: Putative mutation in the EN2 gene—Report of 2 cases in early infancy. Pediatr. Dev. Pathol. 2002, 5, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Sassoon, D.A. A role for Engrailed-2 in determination of skeletal muscle physiologic properties. Dev. Biol. 2001, 231, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.P.; Gross, M.K.; Kioussi, C. Muscle development: Forming the head and trunk muscles. Acta Histochem. 2008, 110, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Sdek, P.; Oyama, K.; Angelis, E.; Chan, S.S.; Schenke-Layland, K.; MacLellan, W.R. Epigenetic regulation of myogenic gene expression by heterochromatin protein 1 alpha. PLoS ONE 2013, 8, e58319. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, L.; Wang, J.; Li, Y.; Chen, Y. SP1-induced lncRNA MCF2L-AS1 promotes cisplatin resistance in ovarian cancer by regulating IGF2BP1/IGF2/MEK/ERK axis. J. Gynecol. Oncol. 2022, 33, e75. [Google Scholar] [CrossRef]
- Burden, S.J.; Huijbers, M.G.; Remedio, L. Fundamental Molecules and Mechanisms for Forming and Maintaining Neuromuscular Synapses. Int. J. Mol. Sci. 2018, 19, 490. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FPKM or TPM (Expression Ratio ≥5) a | No. of Myob DMRs b | Probable Function of Differential Methylation c | ||||
|---|---|---|---|---|---|---|
| Gene | Myob | Cbl | Hypom | Hyperm | Myob DMRs | Cbl Hypom or Hyperm |
| ZNF556 | 12 (1076) | 2 (27) | 1 | 0 | Prom hypom allowing txn d | Prom hypom allowing txn d |
| CDH15 | 164 (560) | 141 (1441) | 1 | 2 | Prom dnstm hypom ↑ txn d | Prom dnstm hypom ↑ txn d |
| TRIM72 | 43 (127) | 12 (53) | 1 | 1 | Prom hypom ↑ txn d; hyperm repr intronic PYDC1 | Prom hypom ↑ TRIM72 txn d and intronic PYDC1 |
| ANK1 | 29 (15) | 85 (25) | 2 | 1 | Alt prom usage | Different prom use from Myob |
| MCF2L | 21 (5) | 110 (9) | 2 | 3 | Alt prom usage d; many RNA splicing isoforms | Alt prom and splicing d; hypom ↑ txn from enhs |
| DOK7 | 45 (220) | 21 (27) | 0 | 1 | Alt prom usage d | Alt prom usage d |
| CNPY1 | 2 (39) | 26 (158) | 0 | 2 | Alt prom usage | Alt prom usage |
| KCNJ12 | 5 (6) | 81 (40) | 0 | 3 | Prom-dnstm hyperm ↓ txn | Prom-adjacent hypom ↑ txn |
| ST8SIA5 | 4 (6) | 51 (14) | 0 | 1 | Prom-upstm hyperm ↓ txn | Prom-dnstm hypom ↑ txn |
| ZIC1 | 5 (39) | 311 (57) | 0 | 14 | Prom-upstm/dnstm hyperm ↓ txn and repressing adj ZIC4 | Whole-gene hypom ↑ txn of both ZIC1 and ZIC4 |
| VAX2 | 4 (6) | 19 (15) | 0 | 7 | Prom-upstm hyperm ↓ txn; Intron-1 hyperm may block formation of repr chrom d | Intron-1 hyperm may block formation of repr chrom d |
| EN2 | 3 (56) | 68 (201) | 0 | 4 | Prom-upstm/dnstm hyperm may ↓ txn | Hyperm far upstm and dnstm of 7 kb EN2 may ↑ txn |
| LBX1 | 1 (230) | 3 (78) | 0 | 4 | Hyperm upstm/dnstm of 2 kb gene may ↑ txn | Methylation profile similar to those of most tissues |
| PAX3 | 1 (23) | 4 (28) | 0 | 11 | Prom-upstm/dnstm hyperm ↓ txn d | Prom-upstm/dnstm hyperm ↓ txn d |
| CHRD | 4 (7) | 243 (22) | 1 | 0 | Intergenic hypom may precede Enh formation | Hypom at different intergenic regions ↑ txn |
| FNDC5 | 64 (88) | 109 (8) | 0 | 0 | NA | 3′ gene hypom ↑ txn |
| PLCB4 | 54 (22) | 57 (18) | 0 | 0 | NA | Prom-dnstm hypom ↑ txn |
| MPP4 | 9 (9) | 3 (16) | 0 | 0 | NA | Uncertain |
| PTPRR | 2 (6) | 22 (9) | 0 | 0 | NA | Alt prom usage |
| IL11 | 6 (6) | 6 (14) | 0 | 0 | NA | Uncertain |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ehrlich, M.; Ehrlich, K.C.; Lacey, M.; Baribault, C.; Sen, S.; Estève, P.-O.; Pradhan, S. Epigenetics of Genes Preferentially Expressed in Dissimilar Cell Populations: Myoblasts and Cerebellum. Epigenomes 2024, 8, 4. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes8010004
Ehrlich M, Ehrlich KC, Lacey M, Baribault C, Sen S, Estève P-O, Pradhan S. Epigenetics of Genes Preferentially Expressed in Dissimilar Cell Populations: Myoblasts and Cerebellum. Epigenomes. 2024; 8(1):4. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes8010004
Chicago/Turabian StyleEhrlich, Melanie, Kenneth C. Ehrlich, Michelle Lacey, Carl Baribault, Sagnik Sen, Pierre-Olivier Estève, and Sriharsa Pradhan. 2024. "Epigenetics of Genes Preferentially Expressed in Dissimilar Cell Populations: Myoblasts and Cerebellum" Epigenomes 8, no. 1: 4. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes8010004