
A New Atomistic Mechanism for Heterogeneous Nucleation in the Systems with Negative Lattice Misfit: Creating a 2D Template for Crystal Growth

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Simulation and Experimental Methods
2.1. Simulation Approaches
- Allowing systematic investigation of systems with varying lattice misfit, i.e., many different substrates.
- Elimination of the effects of chemistry and surface condition of the substrate.
- Pinned substrate represents a variety of high temperature substrates, such as oxides, nitrides and borides practice (e.g., TiB2 with Tl = 3498 K [36]).
2.2. Experimental Methods
3. Results
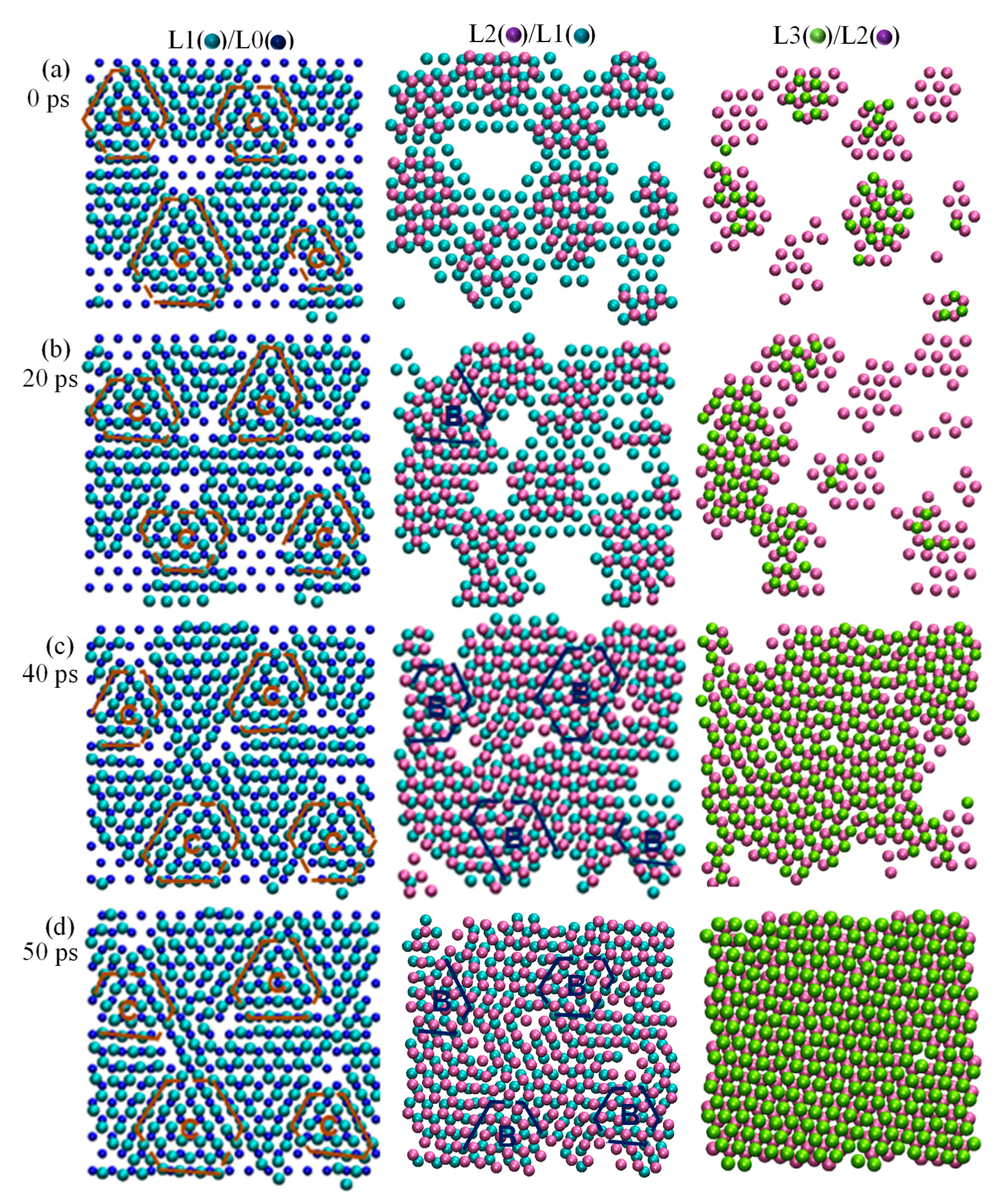
3.1. Heterogeneous Nucleation Process
3.2. Effect of Lattice Misfit
3.3. A New Atomistic Mechanism for Heterogeneous Nucleation
- Prenucleation creating a 2D ordered structure that acts as a precursor for nucleation.
- Accommodation of misfit by forming a partial edge dislocation network in L1.
- Relaxation of the lattice distortion by twisting L2 by an angle (θ) through the formation of a partial screw dislocation network in L2.
- Completion of nucleation by creating a crystal plane of the solid (L3) that provides a template for further growth.
3.4. Experimental Validation
4. Discussion
- The liquid phase: The CNT assumes that the liquid is a disordered phase containing instantaneous short-range order (unstable). In contrast, the new concept acknowledges the importance of prenucleation that provides a stable 2D ordered structure as a precursor for heterogeneous nucleation (Figure 3).
- The interface: The CNT assumes that all the interfaces are sharp with zero thickness (the capillarity assumption), while the new concept suggests that all the interfaces are diffuse with finite thickness (Figure 6). Such diffuse interface can be quantified by atomic density profile (ρ(z)) and in-plane order parameter (S(z)).
- Nucleation outcome: The outcome of classical heterogeneous nucleation is a spherical cap of the solid with a critical radius (3D), while in the new concept heterogeneous nucleation provides a crystalline plane of the solid (2D).
- Energy barrier: In the CNT, there may be an energy barrier for heterogeneous nucleation depending on the nucleation undercooling (ΔTn), while in the new concept heterogeneous nucleation is a down-hill process with no energy barrier.
- Atomic ordering mechanism: the CNT relies on structural fluctuation to provide the critical nucleus and thus is a stochastic process, while in the new concept nucleation starts from the precursor provided by prenucleation, proceeds layer-by-layer through structural templating (Figure 9), and therefore is a deterministic process.
5. Summary
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kelton, K.F.; Greer, A.L. Introduction. In Nucleation in Condensed Mater: Applications in Materials and Biology; Pergamon, Elsevier: Oxford, UK, 2010. [Google Scholar]
- Kashchiev, D. Nucleation: Basic Theory with Applications; Butterworth-Heinemann: Oxford, UK, 2000. [Google Scholar]
- Bartels-Rausch, T. Chemistry: Ten things we need to know about ice and snow. Nature 2013, 494, 27–29. [Google Scholar] [CrossRef]
- Sosso, G.C.; Chen, J.; Cox, S.J.; Fitzner, M.; Pedevilla, P.; Zen, A.; Michaelides, A. Crystal nucleation in liquids: Open questions and future challenges in molecular dynamics simulations. Chem. Rev. 2016, 116, 7078–7116. [Google Scholar] [CrossRef] [Green Version]
- Greer, A.L. Overview: Application of heterogeneous nucleation in grain-refining of metals. J. Chem. Phys. 2016, 145, 211704. [Google Scholar] [CrossRef]
- Erdemir, D.; Lee, A.Y.; Myerson, A.S. Polymorph selection: The role of nucleation, crystal growth and molecular modeling. Curr. Opin. Drug Discov. Dev. 2007, 10, 746–755. [Google Scholar]
- Michaels, T.C.T.; Šarić, A.; Curk, S.; Bernfur, K.; Arosio, P.; Meisl, G.; Dear, A.J.; Cohen, S.I.A.; Dobson, C.M.; Vendruscolo, M.; et al. Dynamics of oligomer populations formed during the aggregation of Alzheimer’s Aβ42 peptide. Nat. Chem. 2020, 12, 445–451. [Google Scholar] [CrossRef]
- Easton, M.; Qian, M.; Prasad, A.; StJohn, D. Recent advances in grain refinement of light metals and alloys. Curr. Opin. Solid State Mater. Sci. 2016, 20, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, J.W. On the equilibrium of heterogeneous substances. Am. J. Sci. 1878, s3-16, 441–458. [Google Scholar] [CrossRef]
- Volmer, M.; Weber, A.Z. Nucleus formation in supersaturated systems. Zeitschrift für Physikalische Chemie 1926, 119, 277–301. [Google Scholar]
- Becker, R.; Döring, W. Kinetic treatment of nucleation in supersaturated vapors. Ann. Phys. (Leipzig) 1935, 416, 719–752. [Google Scholar] [CrossRef]
- Zeldovich, Y.B. On the theory of new phase formation. Cavitation. Acta Physicochem 1943, 18, 1–22. [Google Scholar]
- Cantor, B. Heterogeneous nucleation and adsorption. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2003, 361, 409–417. [Google Scholar] [CrossRef]
- Russo, J.; Tanaka, H. Crystal nucleation as the ordering of multiple order parameters. J. Chem. Phys. 2016, 145, 211801. [Google Scholar] [CrossRef] [Green Version]
- Lutsko, J.F. Novel paradigms in nonclassical nucleation theory. In New Perspectives on Mineral Nucleation and Growth: From Solution Precursors to Solid Materials; Van Diessche, A.E.S., Kellermeier, M., Benning, L.G., Gebauer, D., Eds.; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Fan, Z.; Wang, Y.; Zhang, Y.; Qin, T.; Zhou, X.R.; Thompson, G.E.; Pennycook, T.; Hashimoto, T. Grain refining mechanism in the Al/Al-Ti-B system. Acta Mater. 2015, 84, 292–304. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, Y.; Yang, Y.; Kim, D.S.; Yuan, A.; Tian, X.; Ophus, C.; Sun, F.; Schmid, A.K.; Nathanson, M.; et al. Observing crystal nucleation in four dimensions using atomic electron tomography. Nat. Cell Biol. 2019, 570, 500–503. [Google Scholar] [CrossRef] [Green Version]
- Shibuta, Y.; Sakane, S.; Miyoshi, E.; Okita, S.; Takaki, T.; Ohno, M. Heterogeneity in homogeneous nucleation from billion-atom molecular dynamics simulation of solidification of pure metal. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable prenucleation calcium carbonate clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vekilov, P.G. The two-step mechanism of nucleation of crystals in solution. Nanoscale 2010, 2, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, E.; Liu, J.; Zhang, S.; Lin, Z.; Duan, X.; Heinz, H.; Huang, Y.; De Yoreo, J.J. Building two-dimensional materials one row at a time: Avoiding the nucleation barrier. Science 2018, 362, 1135–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, W.D.; Kauffmann, Y. Structural order in liquids induced by interfaces with crystals. Annu. Rev. Mater. Res. 2006, 36, 1–48. [Google Scholar] [CrossRef]
- Oh, S.H.; Kauffmann, Y.; Scheu, C.; Kaplan, W.D.; Rühle, M. Ordered liquid aluminium at the interface with sapphire. Science 2005, 310, 661–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffmann, Y.; Oh, S.H.; Koch, C.T.; Hashibon, A.; Scheu, C.; Rühle, M.; Kaplan, W.D. Quantitative analysis of layering and in-plane ordering at an alumina-aluminium solid-liquid interface. Acta Mater. 2011, 59, 4378–4386. [Google Scholar] [CrossRef]
- Geysermans, P.; Gorse, D.; Pontikis, V. Molecular dynamics study of the solid–liquid interface. J. Chem. Phys. 2000, 113, 6382–6389. [Google Scholar] [CrossRef]
- Hashibon, A.; Adler, J.; Finnis, M.W.; Kaplan, W.D. Atomistic study of structural correlations at a liquid–solid interface. Comput. Mater. Sci. 2002, 24, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Olmsted, D.L.; Asta, M.; Laird, B.B. Atomistic characterization of the chemically heterogeneous Al–Pb solid–liquid interface. Acta Mater. 2012, 60, 4960–4971. [Google Scholar] [CrossRef]
- Men, H.; Fan, Z. Prenucleation induced by crystalline substrates. Metall. Mater. Trans. A 2018, 49, 2766–2777. [Google Scholar] [CrossRef]
- Palafox-Hernandez, J.P.; Laird, B.B.; Asta, M. Atomistic characterization of the Cu-Pb solid-liquid interface. Acta Mater. 2011, 59, 3137–3144. [Google Scholar] [CrossRef]
- Fang, C.M.; Men, H.; Fan, Z. Effect of substrate chemistry on prenucleation. Met. Mater. Trans. A 2018, 49, 6231–6242. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Men, H.; Fan, Z. Atomic ordering in the liquid adjacent to an atomically rough solid surface. Comput. Mater. Sci. 2018, 153, 73–81. [Google Scholar] [CrossRef]
- Wang, L.; Lu, W.; Hu, Q.; Xia, M.; Wang, Y.; Li, J.-G. Interfacial tuning for the nucleation of liquid AlCu alloy. Acta Mater. 2017, 139, 75–85. [Google Scholar] [CrossRef]
- Schülli, T.U.; Daudin, R.; Renaud, G.; Vaysset, A.; Geaymond, O.; Pasturel, A. Substrate-enhanced supercooling in AuSi eutectic droplets. Nature 2010, 464, 1174–1177. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Horsfield, A.; Schwingenschlögl, U.; Lee, P.D. Heterogeneous nucleation of solid Al from the melt by TiB2 and Al3Ti: An ab initio molecular dynamics study. Phys. Rev. B 2010, 82, 184203. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Men, H. A molecular dynamics study of heterogeneous nucleation in generic liquid/substrate systems with positive lattice misfit. Mater. Res. Express 2020, 7, 126501. [Google Scholar] [CrossRef]
- Munro, R.G. Material properties of titanium diboride. J. Res. Natl. Inst. Stand. Technol. 2000, 105, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Structure. Iida, T.; Guthrie, R.I.L. (Eds.) In The Physical Properties of Liquid Metals; Clarendon Press: Oxford, UK; Oxford University Press: New York, NY, USA, 1988. [Google Scholar]
- Zope, R.R.; Mishin, Y. Interatomic potentials for atomistic simulations of the Ti-Al system. Phys. Rev. B 2003, 68, 024102. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.S.; Horsfield, A.; Lee, P.D.; Brommer, P. Heterogeneous nucleation of solid Al from the melt by Al3Ti: Molecular dynamics simulations. Phys. Rev. B 2010, 82, 144203. [Google Scholar] [CrossRef]
- Todorov, I.T.; Smith, W.; Trachenko, K.; Dove, M.T. DL_POLY_3: New dimensions in molecular dynamics simulations via massive parallelism. J. Mater. Chem. 2006, 16, 1911–1918. [Google Scholar] [CrossRef] [Green Version]
- Hashibon, A.; Adler, J.; Finnis, M.W.; Kaplan, W.D. Ordering at solid-liquid interfaces between dissimilar materials. Interface Sci. 2001, 9, 175–181. [Google Scholar] [CrossRef]
- Hook, J.R.; Hall, H.E. Scattering of neutrons and electrons from solids. In Solid State Physics, 2nd ed.; Wiley: Chichester, UK, 1991. [Google Scholar]
- Jackson, K.A. The interface kinetics of crystal growth processes. Interface Sci. 2002, 10, 159–169. [Google Scholar] [CrossRef]
- Steinhardt, P.J.; Nelson, D.R.; Ronchetti, M. Bond-orientational order in liquids and glasses. Phys. Rev. B 1983, 28, 784–805. [Google Scholar] [CrossRef]
- Baumgartner, J.; Dey, A.A.; Bomans, P.H.H.; Le Coadou, C.; Fratzl, P.; Sommerdijk, N.A.J.M.; Faivre, D. Nucleation and growth of magnetite from solution. Nat. Mater. 2013, 12, 310–314. [Google Scholar] [CrossRef]
- Fan, Z.; Wang, Y.; Xia, M.; Arumuganathar, S. Enhanced heterogeneous nucleation in AZ91D alloy by intensive melt shearing. Acta Mater. 2009, 57, 4891–4901. [Google Scholar] [CrossRef]
- Hirth, J.; Pond, R.; Hoagland, R.; Liu, X.-Y.; Wang, J. Interface defects, reference spaces and the Frank–Bilby equation. Prog. Mater. Sci. 2013, 58, 749–823. [Google Scholar] [CrossRef]
- Fan, Z. An epitaxial model for heterogeneous nucleation on potent substrates. Met. Mater. Trans. A 2012, 44, 1409–1418. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, D.; Vonnegut, B. Nucleation catalysis. Ind. Eng. Chem. 1952, 44, 1292–1298. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, C.; Zhou, L.; Hashimoto, T.; Zhou, X.; Ramasse, Q.; Fan, Z. Mechanism for Zr poisoning of Al-Ti-B based grain refiners. Acta Mater. 2019, 164, 428–439. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Z.; Men, H.; Wang, Y.; Que, Z. A New Atomistic Mechanism for Heterogeneous Nucleation in the Systems with Negative Lattice Misfit: Creating a 2D Template for Crystal Growth. Metals 2021, 11, 478. https://0-doi-org.brum.beds.ac.uk/10.3390/met11030478
Fan Z, Men H, Wang Y, Que Z. A New Atomistic Mechanism for Heterogeneous Nucleation in the Systems with Negative Lattice Misfit: Creating a 2D Template for Crystal Growth. Metals. 2021; 11(3):478. https://0-doi-org.brum.beds.ac.uk/10.3390/met11030478
Chicago/Turabian StyleFan, Zhongyun, Hua Men, Yun Wang, and Zhongping Que. 2021. "A New Atomistic Mechanism for Heterogeneous Nucleation in the Systems with Negative Lattice Misfit: Creating a 2D Template for Crystal Growth" Metals 11, no. 3: 478. https://0-doi-org.brum.beds.ac.uk/10.3390/met11030478