
Genetic Variability and Evidence of a New Subgroup in Watermelon Mosaic Virus Isolates


1
Department of Biological Science, The University of Tulsa, Tulsa, OK 74104, USA
2
Department of Plant Pathology, Faculty of Agriculture, Assiut University, Assiut 71526, Egypt
*
Author to whom correspondence should be addressed.
Pathogens 2021, 10(10), 1245; https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10101245
Submission received: 13 August 2021
/
Revised: 20 September 2021
/
Accepted: 21 September 2021
/
Published: 26 September 2021
(This article belongs to the Special Issue Evolution of Plant Viruses)
Abstract
:Watermelon mosaic virus (WMV) is one of the important Potyviruses that infect cucurbits worldwide. To better understand the population structure of WMV in the United States (U.S.), 57 isolates were collected from cucurbit fields located in nine southern states. The complete coat protein gene of all WMV isolates was cloned, sequenced and compared with 89 reported WMV isolates. The nucleotide and amino acid sequence identities among the U.S. WMV isolates ranged from 88.9 to 99.7% and from 91.5 to 100%, respectively. Phylogenetic analysis revealed that all the U.S. WMV isolates irrespective of their geographic origin or hosts belonged to Group 3. However, the fifty-seven isolates made three clusters in G3, where two clusters were similar to previously reported subgroups EM1 and EM2, and the third cluster, containing nine WMV isolates, formed a distinct subgroup named EM5 in this study. The ratio of non-synonymous to synonymous nucleotide substitution was low indicating the occurrence of negative purifying selection in the CP gene of WMV. Phylogenetic analysis of selected 37 complete genome sequences of WMV isolates also supported the above major grouping. Recombination analysis in the CP genes confirmed various recombinant events, indicating that purifying selection and recombination are the two dominant forces for the evolution of WMV isolates in the U.S.
1. Introduction
Cucurbits are important cash crops in the southern United States (U.S.). Unfortunately, more than 96 viruses [1] infect cucurbit crops naturally, and watermelon mosaic virus (WMV) is one of the important viruses that has a greater impact on the quality and quantity of cucurbits worldwide. WMV is a member of genus Potyvirus, family Potyviridae, which has flexuous rod-shaped particles of approximately 750 nm in length [2,3] and is transmitted by various aphids in a non-persistent manner [4,5]. Like other Potyviruses, the genome of WMV is a single-stranded positive-sense RNA and contains one large open reading frame, which translates into one large polypeptide of 3217 amino acids, including nine putative cleavage sites that produce ten different proteins [6,7]. WMV is most related to soybean mosaic virus (SMV) [8], suggesting that WMV has evolved as a result of recombination between SMV and bean common mosaic virus (BCMV) [9].
WMV is economically important and can infect more than 170 species, belonging to 27 different families, causing severe losses in important horticulture crops, especially cucurbits [9,10]. In cucurbits, WMV induces a variety of symptoms, according to the isolates and host cultivar that include: mosaic, vein banding, severe leaf deformation and filiformy, and some isolates induce discoloration and deformation, especially in squash fruits [4,5,11]. WMV is considered among the most important aphid-borne viruses infecting cucurbits worldwide, especially in temperate and Mediterranean climates [12,13,14].
In the U.S., WMV was first reported in 1965 [15] from southern Texas and is one of the main viruses infecting cucurbits in Texas [16], California [3], New Jersey [17], Illinois [18], Oklahoma [19] and other southern states [11]. Although WMV has been reported in the U.S. for more than 50 years, very little information is available about the genetic diversity of WMV in the U.S.
Most studies about WMV variability came from Europe, where emergent strains of WMV caused severe symptoms in cucurbits [20]. WMV isolates were classified into three molecular groups (G1-G3) based on the 3–5 amino acid (aa) motif deduced from the 218 nucleotides (nt) at the coat protein (CP) N-terminal region [21] which were recognized by monoclonal antibodies made against these specific amino acids. Group 1 (G1) (also known as classical (CL) isolates) and Group 2 (G2) isolates have KEA and KET aa motifs, respectively, at position 3–5 in the N-terminal part of the CP, while Group 3 (G3) isolates (also named emerging (EM) isolates) have a KEKET aa motif at position 3–7 (with the insertion of two aa) in the N-terminal part of the CP [21]. G3 (EM) isolates have been further divided into four subgroups (E1–E4) [20,22].
One study [23] reported the genetic diversity of 36 WMV isolates based on the CP N-terminal region from Slovakia and Iran, and showed that WMV isolates from Slovakia belong to G1 and G2 groups, while isolates from Iran belong to the G2 group only. Recently, genetic diversity of 56 WMV isolates from China [24], using complete CP gene nt sequences, showed that all Chinese WMV isolates belong to G3. In the U.S., the only available data about WMV isolates came from Quemada et al. [25], who reported that the deduced amino acid sequence of the CP gene of the WMV isolate (USA90-Accession no. D13913) shared 94.7% similarity with other WMV worldwide isolates and 82.6% with soybean mosaic virus (SMV). Desbiez et al. [21] used two U.S. WMV isolates (USA90 and USA92, Accession no. DQ845032) in comparison, and they were placed in G2 and G3, respectively.
The aim of this present study is to provide more information about the genetic diversity of WMV isolates collected from cucurbit fields in nine southern states in the USA and determine their relationship with the worldwide WMV isolates, based on the CP gene. This information will be helpful to determine the evolutionary relationships of the U.S. WMV isolates, and to increase our understanding of how this virus evolved over time. Information about genetic diversity is essential for the control of plant viruses, using transgenic plants, and could reduce the impact of a virus on the quality and quantity of cucurbits.
2. Results
2.1. Genetic Diversity of WMV in the U.S.
The genetic diversity of the U.S. population of WMV was analyzed by comparing the nucleotide sequences of the CP gene. Sequencing of the CP gene showed that all the U.S. WMV isolates have 849 nt, which could be translated into 283 aa. No insertion or deletion was found but substitution was common among the isolates. Nucleotide sequence identities among the WMV isolates ranged from 88.9 to 99.7% at nt and from 91.5 to 100% at aa level, respectively. The DAG motif, which is responsible for aphid transmission in Potyviruses [26] was found in all the U.S. isolates.
Initially, phylogenetic trees were constructed based on the complete CP sequences of 57 WMV isolates according to the location (states) from where they were originally collected (Table 1). Phylogenetic trees of WMV isolates from AR, FL, MS, OK and TX states comprised two main groups (data not shown). The sequence identities among the WMV isolates according to the location ranged from 91.2 to 98.3% at nt and from 93.6 to 97.5% at aa (AR), 99.5–100% nt and 99.2–100% aa (FL), 91.6–99.2% nt and 93.9–98.9% aa (MS), 88.4–100% nt and 88.3–100% (OK), 89.5–100% nt and 91.8–100% aa, respectively. The number of isolates from other states (AL, GA, LA, and KY) was low and these were grouped together where the sequence identities among the WMV isolates ranged from 90.4 to 99.5% nt and from 92.2 to 99.5% aa, respectively.
Similarly, average evolutionary divergence within and between the WMV isolates collected from the respective states was calculated using MEGA7. The divergence in the complete CP nt sequences was highest within isolates from OK (0.0631 ± 0.0057), followed by isolates from other states (AL, GA, LA and KY) (0.0624 ± 0.0066), AR isolates (0.0572 ± 0.0049), TX isolates (0.0559 ± 0.0049), MS isolates (0.0487 ± 0.0056), and FL isolates (0.0057 ± 0.0018). The divergence among the 57 WMV isolates from nine different states varied from 0.05 to 0.07.
2.2. Phylogenetic Analysis of the CP Gene
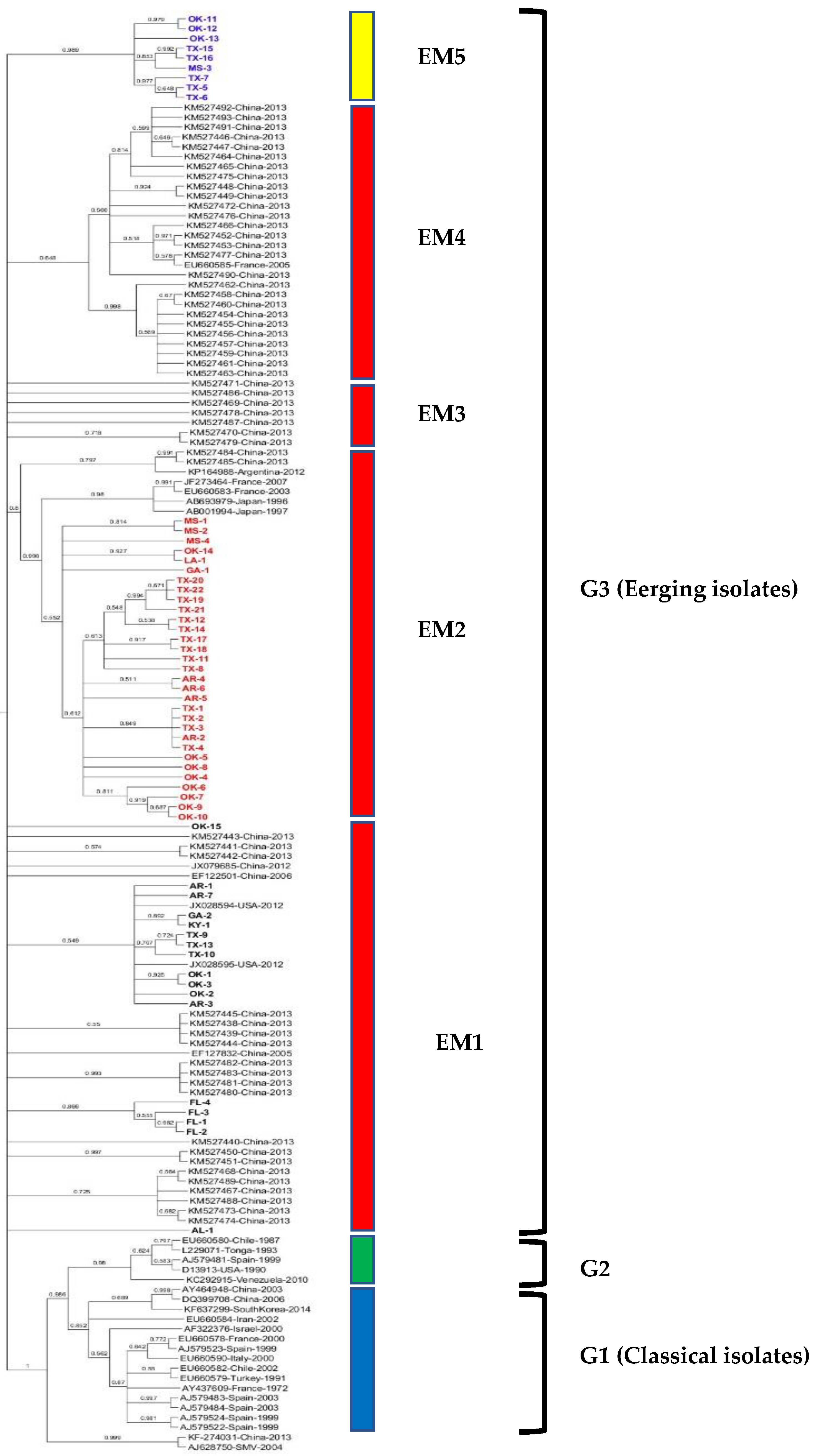
The maximum likelihood tree was constructed from the complete CP gene nt sequence of all 57 WMV isolates along with 89 WMV isolates (Table S1) reported worldwide. The phylogenetic tree shows that all 57 WMV isolates clustered with G3 (EM isolates) (Figure 1) but in various subgroups within G3. For example, subgroup EM1 included 17 WMV isolates (1 each from AL, GA and KY, 3 each from AR, and TX, and 4 each from FL and OK). Similarly, subgroup EM2 contained 31 WMV isolates (1 each from LA, and GA, 3 from MS, 4 from AR, 8 from OK, and 14 isolates from TX). Subgroup EM5 (named in this study) contained nine WMV isolates (one from MS, three from OK, and five from TX) and formed a separate distinct cluster from the published WMV isolates used in the phylogenetic analysis (Figure 1).
Intragroup variability in the complete CP nt sequence in subgroup EM5 was higher (0.0337 ± 0.0039) than subgroups EM1 (0.0218 ± 0.0023) and EM2 (0.0218 ± 0.0024). Similarly, intergroup variability was higher between subgroups EM5 and EM1 (0.09), EM5 and EM2 (0.09) than subgroups EM1 and EM2 (0.08).
2.3. Amino Acid Sequence Comparison
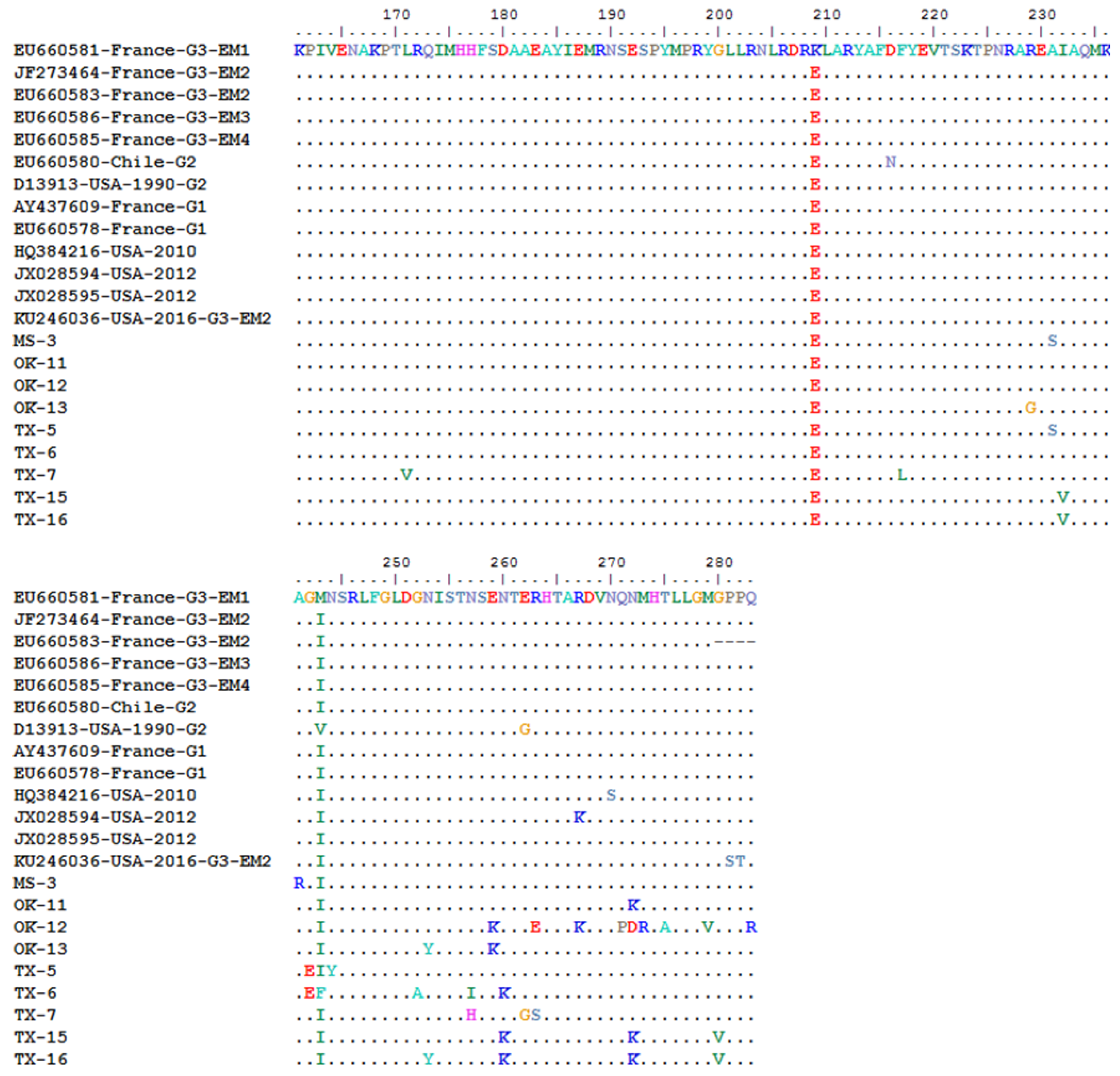
In order to determine any subgroup’s specific amino acid pattern in the CP gene, the 283 aa sequences of the 57 WMV isolates were compared with the representative isolates that were previously classified into G1, G2 and G3 isolates (Figure 2). All G3 isolates carry a specific motif (KEKET) at position 3–7 in the N-terminal of CP gene, while G1 and G2 isolates have KET and KEA at the same positions (Figure 2). All of the 57 WMV isolates had the KEKET aa motif except for two isolates (OK-11 and OK-12), which were unique and have “A” at position 7 instead of “T” (KEKEA instead of KEKET) (Figure 2). In addition, they also had a unique aa: V at position 28, which did not exist in any WMV isolates (Figure 2). Both OK-11 and OK-12 WMV isolates clustered in the new subgroup EM5, which was identified in this work.
Further analysis of the aa sequence comparison showed that some of the WMV isolates in subgroup EM5 had 10 unique and conserved aa: V, S, I, A, A, Y, K, K, K, and V at positions 13, 28, 43, 45, 107, 253, 259, 260, 272, and 280, respectively (Figure 2). Among these 10 unique aa, four aa (V, S, I and A) at positions 13, 28, 43, and 45 were located at the N-terminal of the CP gene while the remaining were located at the C-terminal of CP gene except one at positions 107 which was in the core region of CP gene. Apart from the specific aa of WMV isolates in subgroup EM5, there were four aa: E, D, N, and V, at positions 16, 23, 26, 33, which were common between EM5 and G2 isolates (Figure 2), while there was one aa (T) which was common between EM5, G1, and G2 isolates.
The U.S. isolate from Texas (Accession no. KU246036) that was classified as G3-EM2 isolate by phylogenetic analysis (Figure 3) shared three aa (V, D and A) at positions 58, 75 and 114 with G3-EM2 isolates (Figure 2), which further confirmed that it is the G3-EM2 isolate. The other U.S. isolate (Accession no. HQ384216) collected from a weed and classified as a G1 isolate based on the presence of KEA at the N-terminal of CP gene (Figure 2) showed similar aa (V, D, and A) at positions 58, 75, and 114, just like G3-EM2 isolates which were similar as shown in the phylogenetic analysis (Figure 3) where they clustered with G3 isolates.
2.4. Selection Pressure in the CP Gene
The selection pressure on the CP gene of WMV isolates for the three subgroups (EM1, EM2 and EM5) was determined by calculating dN/dS (ω) ratio (Table 2). The dN/dS ratio for the three subgroups, EM1 isolates (0.219), EM2 isolates (0.3089) and EM5 isolates (0.2075), indicated that negative (or purifying) selection is occurring across WMV isolates in the CP gene. The strength of the negative selection was higher among the subgroup EM2 isolates as compared to subgroups EM1 and EM5 isolates, indicating that stronger constraints are operating in the CP genes of WMV isolates among the later two groups. Similarly, the dN/dS ratio in the CP gene was also estimated according to the origin of collection of WMV isolates. The dN/dS ratio was 0.0859 (AR isolates), 0.3170 (FL isolates), 0.0936 (MS isolates), 0.0940 (OK isolates), 0.0827 (TX isolates), and 0.0847 for WMV isolates (Table S2) collected from AL, GA, LA, and KY states, and also showed that negative selection occurred in WMV isolates collected from nine different states. However, the strength of negative selection was stronger in all WMV isolates except FL WMV isolates, which showed weaker negative selection compared with the isolates from other southern states.
Selection pressure calculated at the level of individual codons by the three codon-based methods (SLAC, FEL, and REL) among the three subgroups mostly showed that a number of codons are under negative selection (Table 2). However, a total of 16 codons in EM2 and 3 codons in EM5 were under positive selection as detected by the REL algorithm only. The ENC indicates a positive correlation with the rate of synonymous substitution. For example, EM1 and EM5 showed stronger codon bias (ENC of 50) compared with the EM2 subgroup isolates (ENC 51), confirming a stronger purifying selection in these two group isolates.
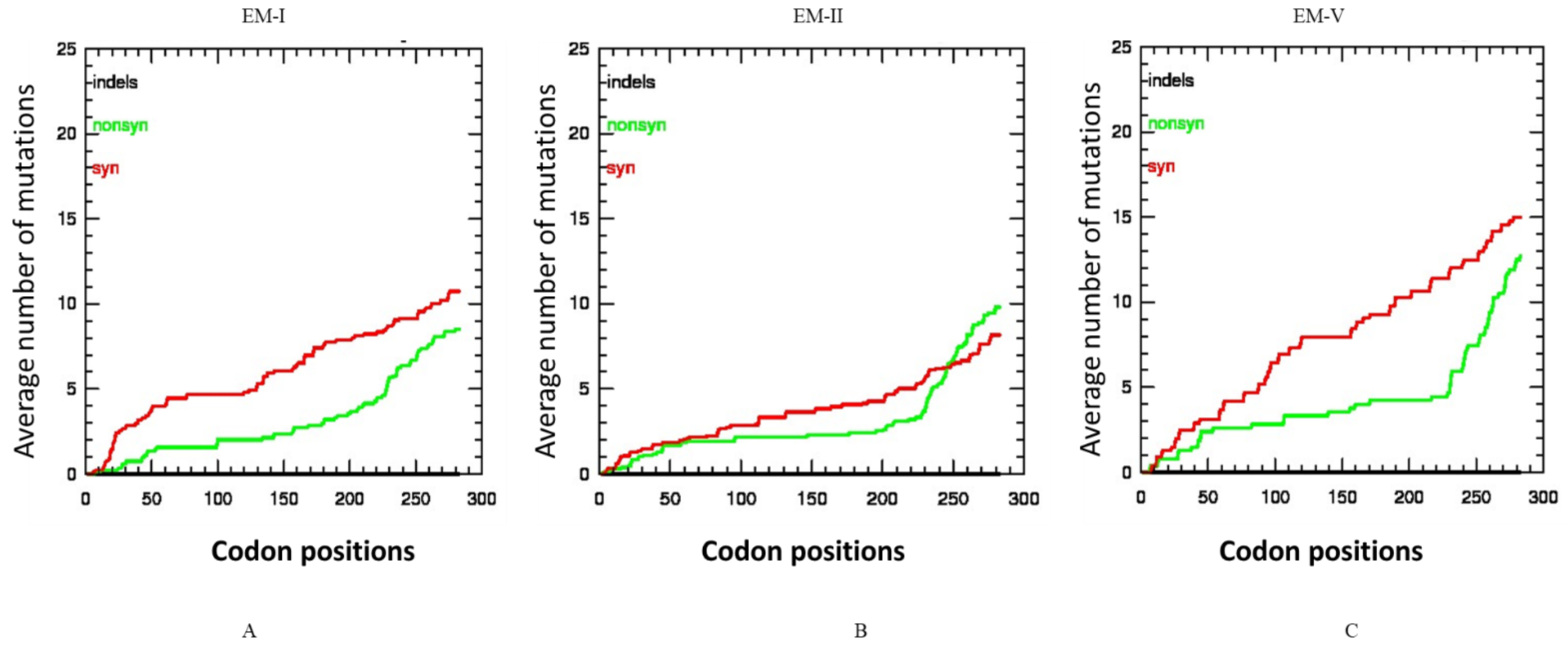
The selective constraints in the CP gene of the three subgroup isolates were also determined by analyzing the distribution of synonymous and non-synonymous and indel mutations. In the EM2 subgroup the number of synonymous and non-synonymous mutations was not significantly different (Figure 4A), but in the case of EM1 and EM5 subgroups (Figure 4B,C), the synonymous mutations were higher than the non-synonymous mutations. No indel was present in any WMV isolates among the three subgroups.
2.5. Genetic Differentiation and Gene Flow Analysis of WMV Isolates
Genetic differentiation and gene flow analysis (Table 3) estimated from F statistics (Fst) indicated that there is frequent gene flow of WMV isolates among different states (Fst < 0.33) except between Arkansas and Florida (Fst = 0.439), Mississippi and Florida (Fst = 0.620), Oklahoma and Florida (Fst = 0.470), Texas and Florida (Fst = 0.569) and Florida and other states (Fst = 0.270). Similarly, the Nm value for all states pairs was >1 except different states vs. Florida (<1) indicating frequent gene flow among WMV isolates. The p values for all permutation-based tests Ks, Ks*, Z* and Snn were <0.01 for all WMV isolates from states pairs involving Florida, showing significant genetic differentiation (Table 3).
2.6. Co-Evolution Analysis of CP Gene Sequences
Analysis of BGM showed 16 co-evolving codon pairs with a Bayesian posterior probability of at least 50% in the CP gene of WMV isolates (Table S4). Most of the co-evolving sites were located on the C-terminal of the CP gene.
2.7. Phylogenetic Analysis of the Complete Genome WMV Isolates
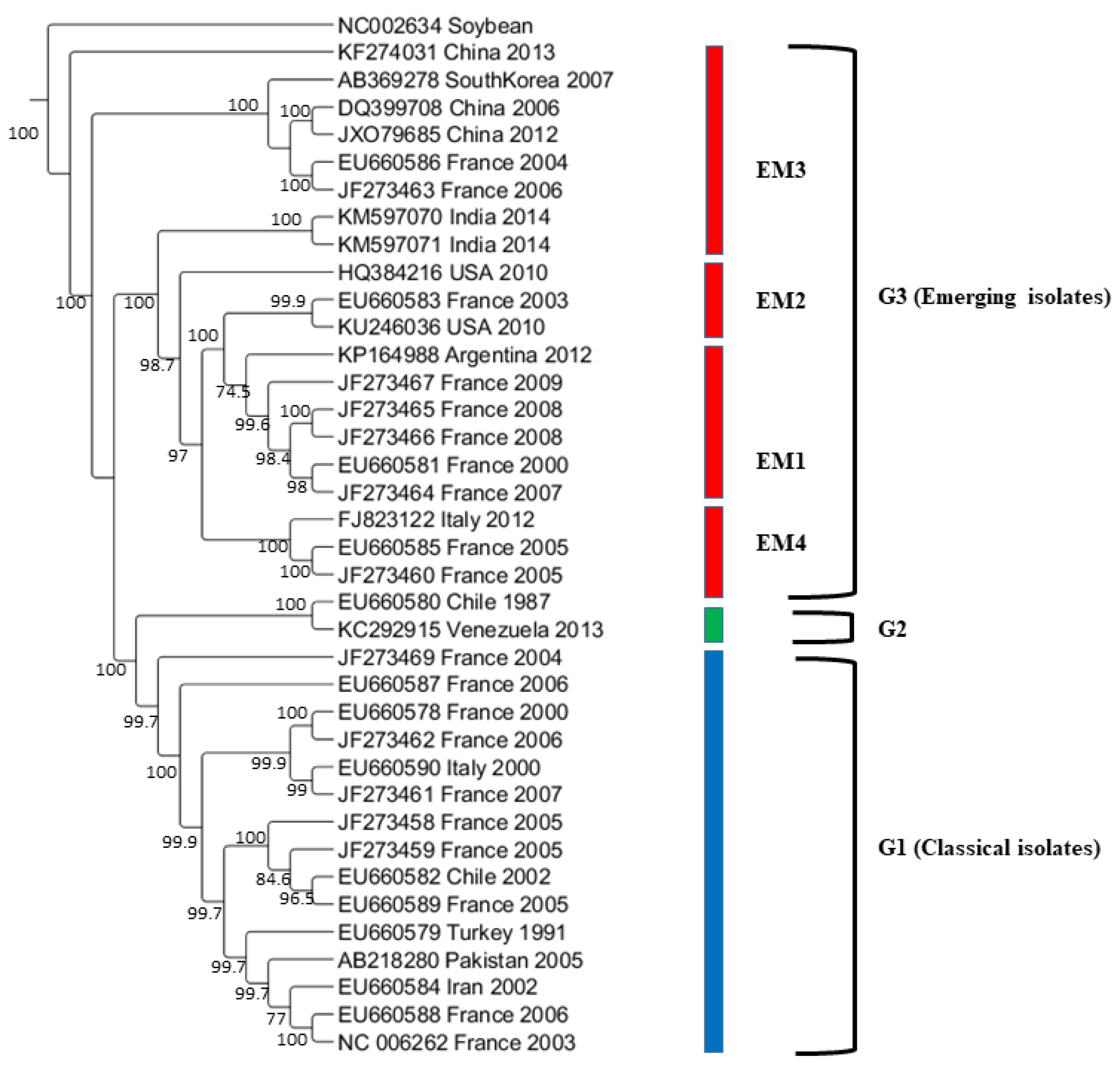
The maximum likelihood tree was constructed from the complete genome sequences of 37 representatives WMV isolates (Figure 3). The phylogenetic tree shows that that the 37 isolates formed three major groups (G1, G2, and G3) as obtained from the CP gene sequences [21]. In G3, WMV isolates were further divided into four subgroups that included EM1, EM2, EM3, and EM4 (Figure 3) as described previously [20]. Our phylogenetic analysis showed that U.S. WMV isolates from Texas belonged to the EM2 subgroup within G3. Classification based on the complete genome sequences showed additional evidence that grouping obtained on the basis of CP gene sequences and complete genome sequences are somewhat similar.
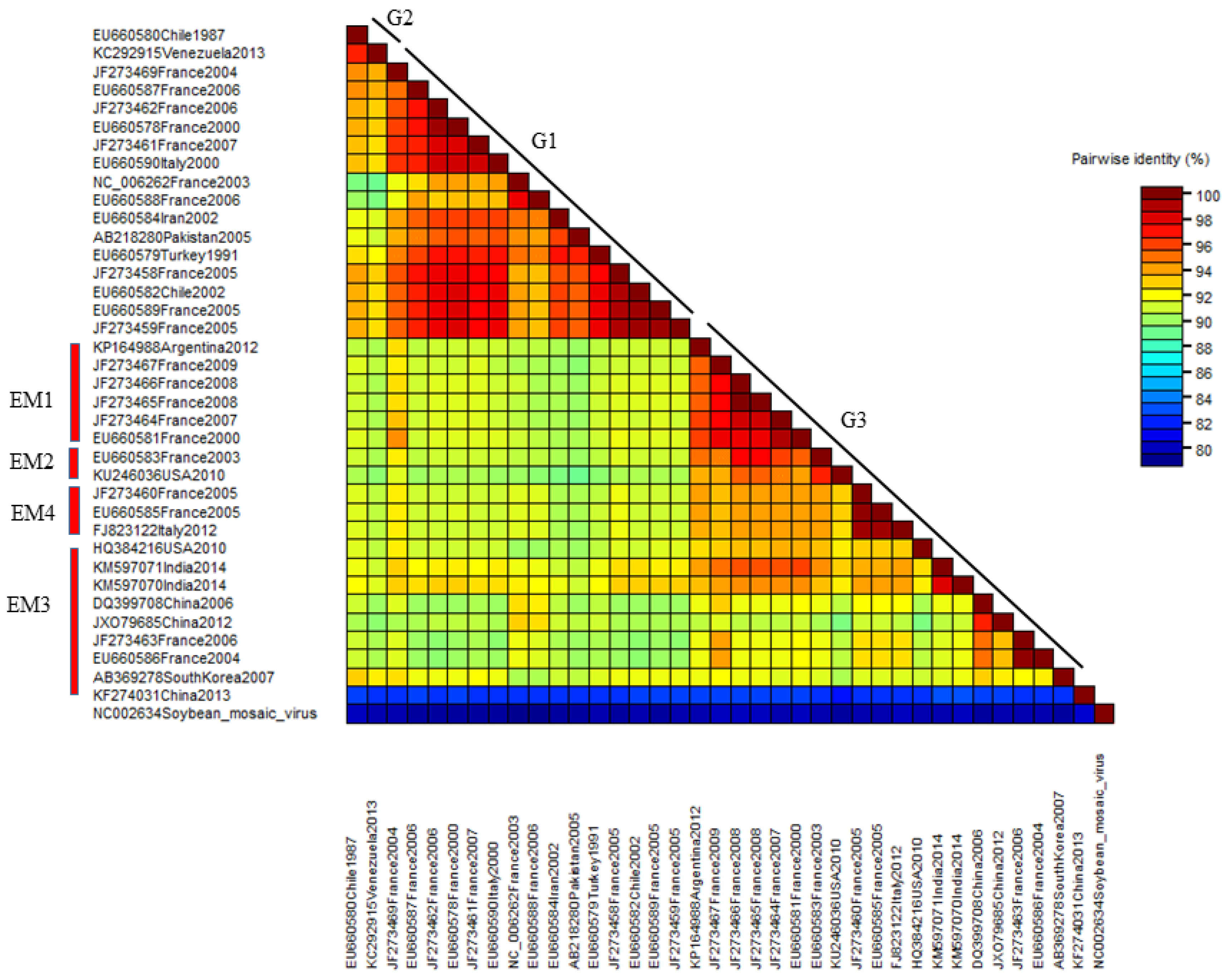
The nucleotide pairwise genetic identities between the complete genome sequences of 37 WMV isolates was calculated by SDT software (Figure 5). WMV isolates in G2 showed >98% pairwise identities followed by G1 isolates that ranged from 88 to 98%. G3 isolates showed the most diversity, and the lowest pairwise identity ranged from 82 to 98%.
2.8. Recombination Events within the CP Gene
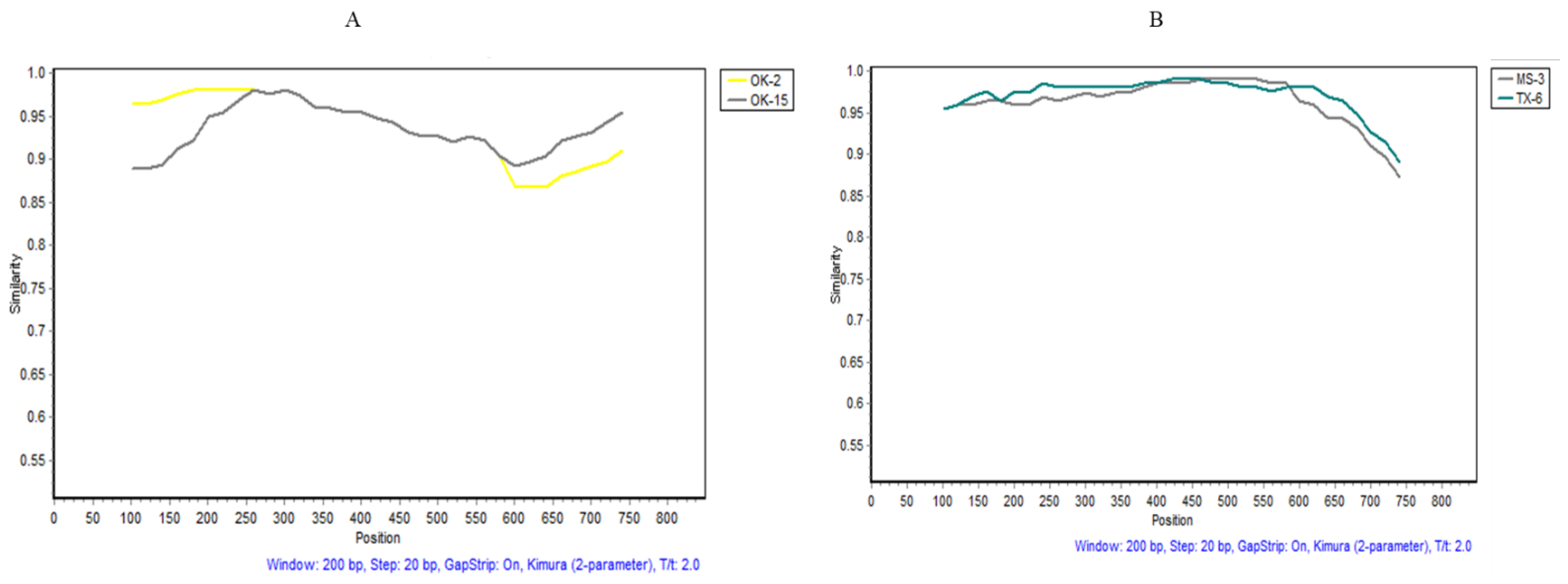
The recombination events were examined in the 57 WMV isolates differentiated into three subgroups. Various recombination events (Table 4) were observed in subgroups EM1 and EM5 WMV isolates, but none in EM2 isolates, by different algorithms employed in RDP4 (Table 4). Recombination events that were detected by four or more algorithms [27,28] were considered to be real, while the one detected by two or three algorithms was ignored and removed from the analysis (Table 4). In subgroup EM1 isolates, recombinant event #3 was statistically significant in OK-15 isolate as confirmed by Simplot software (Figure 6A). The potential breakpoints of the recombination event were located at 298–810 nt. The major parent was the OK-2 isolate, while the minor parent was unknown (possible AL-1 isolate) (Table 4). Similarly, in subgroup EM5, recombination event #3 (Table 4) was observed in the MS-3 isolate and the potential breakpoints of the recombination event were located at ~155–724 nt (Figure 6B). The major parent was TX-6 isolates and the minor parent was unknown (possible AL-1) (Table 4).
3. Discussion
We have shown for the first time the genetic diversity of 57 WMV isolates from nine southern states of the U.S. that were collected from commercial cucurbit fields. Our study showed that all the 57 U.S. WMV isolates belong to the G3 isolates and none to G1 or G2 isolates. However, within G3, the U.S. isolates divided into three subgroups. The two subgroups (EM1 and EM2) were reported previously in other countries [9,21,22]; none were related to EM3 and EM4, while EM5 (named in this study) was a new distinct subgroup within the G3 that was never reported before within WMV isolates from other countries.
In 1990, only 1 isolate (Accession no. D13913) reported from Florida [25] was classified as G2 [21], but none of the 57 isolates, including 4 from Florida, belong to G1 or G2 and all of them belong to G3. It is evident that the WMV isolates in the U.S. predominantly belong to G3 isolates and are constantly emerging and expanding the genetic variability, particularly with the existence of the new distinct subgroup EM5 and its isolates. This shows that classical isolates are replaced by G3, which are emerging isolates (EM). Similar observations were also reported in France where EM isolates have replaced the classical WMV isolates [20,22,29]. However, the identification of this new subgroups EM5 in G3 suggest that selection and frequent recombination are the main driving forces for the evolution of G3 isolates which has now possible five subgroups. It is important to note that the new subgroup EM5 isolates have specific aa of G2 isolates, while OK11 and OK 12 isolates have specific aa of both G1 and G2 isolates (Figure 2), which confirms that the above forces are responsible for the emergence of new isolates in G3. Another possible reason could be the wide host range of WMV and its transmission by more than 23 vectors that contribute to the selection and genetic bottleneck of G3 isolates of WMV. Genetic bottlenecks, selection, and founder effects were determined during the horizontal transmission of cucumber mosaic virus (CMV) by two different aphid vectors [30].
RNA viruses could accumulate heterogeneous population (termed as quasispecies) in the host due to its error-prone mechanism [31] that provides a pool of genetic variants to be picked by aphid’s vectors during horizontal transmission of the virus. The resulting infection produces a founding population which further increase the pool of genetic variants and could be easily adopted to a particular host. Based on this assumption, the 57 isolates in this study have been collected from various fields and hosts in 9 different states and probably upon introduction in various locations by aphid vectors constituted a founding population that provides a conducive environment for the emergence of new isolates of WMV.
The 57 WMV isolates were collected from various cucurbit hosts, including cucumber, cantaloupe, pumpkin, squash, watermelon, and pigweed (Table 1) in nine different major cucurbit growing states, but interestingly, none of the isolates clustered according to the location or hosts where they were originally isolated, except FL isolates, which clustered together (Figure 1) irrespective of the hosts (cucumber, squash, pumpkin, and watermelon) (Table 1) where they were collected. The possible reason is that the diversity among the FL isolates was very low (<0.5% nt and 0.8% aa) compared with other WMV isolates from other states. However, only four WMV isolates from Florida were analyzed, and meaningful conclusions could be obtained once a large number of isolates from different hosts have been analyzed. Nevertheless, the variability among the U.S. WMV isolates may be due to their frequent introduction from one location to another location through the reservoir hosts, or WMV-infected material during the season or off-season. Similar observations were also noticed previously [24] when 56 WMV isolates from China did not cluster with their geographical locations. It is also possible that WMV is transmitted in the fields located in different states by various vectors and differences in selection, or genetic bottlenecks could be responsible for the evolution of these newly emerged variants, such as subgroup EM5 identified in this work.
Our study showed that the U.S. WMV isolates have a high degree of variation, up to 10.8% at nt level and 8.5% at aa. In comparison, this variation was higher among the WMV isolates reported from other countries including China (6.6% nt and 3.9% aa) [24] or Slovakian and Iranian WMV isolates (5% nt) [23]. This shows that U.S. WMV isolates are more diverse than those reported in other countries. Natural selection and genetic bottlenecks are the two evolutionary forces that affect the population of plant viruses in nature. In this study, negative selection was detected in the CP gene of the U.S. WMV isolates, which could remove the deleterious mutations and stabilize the genetic structure of the virus populations. The majority of the codons in the CP gene were under negative selection but some were also under positive selection, as detected by REL in the Data monkey software. Positive selection in the CP gene of WMV has also been reported in a recent study [32]. These results show that positive selection is also occurring on individual codons, but to a lower extent compared with negative selection. However, some of the codons in the CP genes that differentiate the three groups of WMV (KET, KEA, or KEKET) or in other genes may play an important role in the virus life cycle, such as encapsidation of the virion; interactions with the RNA genomes of the virus and host specificity for infection may not be under the selection pressure because the virus cannot afford any mutations in such conserved regions to be able to survive.
Gene flow and genetic differentiation analysis among WMV isolates from different states indicated that WMV isolates from Florida were unique and has infrequent gene flow with isolates from other states and were well supported statistically with significant p value ((Table 3). This could be due to the long distance between Florida and other states. In contrast, WMV isolates from other states which are in close proximity showed frequent gene flow (Table 3), indicating no significant differences. This notion was well supported by phylogenetic analysis (Figure 1), where WMV isolates from Florida clustered together while WMV isolates from other states did not cluster based on their geographical locations, but instead clustered together in various subgroups including the newly EM5 cluster in G3 (Figure 1).
While comparing the phylogenetic trees based on CP gene nucleotide sequences (Figure 1) and complete genome sequences (Figure 4), it was noted that few isolates available in both analyses clustered mostly in their respective three major groups (G1, G2, and G3) with some exceptions. For example, the Chinese isolate (Accession no. KF274031) did not cluster in the three major groups (Figure 1 and Figure 3). The complete genome sequences of most of the isolates used in the CP gene analysis were not available in the GenBank database. More than 50% of the 37 isolates used in the complete genome analysis were from France while the remaining isolates came from the rest of the countries. To have more comprehensive phylogenetic analysis, future study shall focus to acquire large number of WMV isolates from other countries and shall be sequenced completely in order to get a detailed comparison of the WMV isolates based on both the CP as well as complete genome sequences.
Recombination is one of the driving forces of plant virus evolution [33] and has been commonly detected in Potyviruses [34,35]. Recombination plays an important role in the evolution of WMV and has been reported previously in WMV populations from other countries [9,20,22] In our analysis, strong recombination events were detected among the U.S. WMV isolates that confirms that genetic recombination is very important in the evolution of global WMV populations. It is also noteworthy to mention that OK-11 isolate was collected from an Amaranthus palmeri (Palmer amaranth) and was a possible recombinant (Table 3) where three algorithms had detected the recombination event in the OK11 WMV isolate. It is clear that WMV isolates not only move through their natural cucurbit hosts but also through various weeds as an infected material.
In conclusion, we showed that the population structure of WMV isolates in the U.S. is highly variable and predominantly belongs to G3 isolates. However, a new subgroup has been identified which includes isolates from Mississippi, Oklahoma, and Texas that is driven by different evolutionary forces, including strong purifying selection and frequent recombination events in nature. We hypothesized that WMV moves from one state to another state through virus-infected material, mostly by human interference. The current information obtained in this study has considerably expanded our knowledge about WMV variability in the U.S. and provides a comprehensive analysis of the population structure of WMV, which could be beneficial in epidemiological studies, as well as in the management of WMV in the U.S. In future, more isolates shall be sequenced from various southern states, including other states where cucurbits are grown, to completely analyze the diversity of WMV and the expansion of the newly detected distinct subgroup (EM5) isolates as well as biological characterization.
4. Materials and Methods
4.1. Sources of WMV Isolates
During our previous study [11], more than 700 cucurbit leaf samples were collected from 10 different southern states and were tested by dot-immunobinding assay (DIBA) against 17 viruses, including WMV. A total of 57 DIBA-positive WMV samples (Table 1) were randomly selected, representing nine southern states, that included 22 WMV isolates from Texas (TX), 15 from Oklahoma (OK), 7 from Arkansas (AR), 4 each from Florida (FL) and Mississippi (MS), 2 from Georgia (GA), and 1 each from Alabama (AL), Kentucky (KY), and Louisiana (LA).
4.2. RNA Extraction, Amplification, and Sequencing
Total RNA was extracted from the DIBA-positive samples as reported before [35]. Two-step reverse transcription-polymerase chain reaction (RT-PCR) was performed to amplify the CP gene (849 nt) of WMV using WMV specific primers: forward 5′-AACACACAACCAAGT-3′ and reverse 5′-TAACGACCCGAAATGCTAACT-3′, as described previously [36]. The PCR products were analyzed on a 1% agarose gel, and purified using a QIAquick PCR Purification Kit (QIAGEN, Germantown, MD, USA). Purified PCR products were cloned using pGEM®-T vector (Promega, Madison, WI, USA), and transformed into Escherichia coli DH5α competent cells (New England Biolabs, Ipswish, MA, USA). Three to five positive recombinant clones were sequenced in both directions using dye terminator cycle sequencing (Applied Biosystems 3130 genetic analyzer) at the core facility lab, Department of Biological Science, the University of Tulsa, Oklahoma.
4.3. Consensus Sequences and Phylogenetic Analysis of the CP Gene
Sequences of the complete CP gene of all 57 WMV isolates were submitted to GenBank with the accession numbers from MG021247 to MG021303 (Table 1). For comparison, complete CP sequences of 89 WMV isolates, reported from 13 different countries and including representative isolates of all the three reported groups (Group 1, 2, and 3), were obtained from GenBank database (Table S1) and used in phylogenetic analysis.
Consensus sequences were obtained from the alignment of three-five clones for each individual WMV isolate using EditSeq ™ and MegAlign ™ within the DNASTAR suite of programs (Madison, WI, USA). Multiple sequence alignment was performed using the Clustal X program [37] and BioEdit [38]. A phylogenetic tree was constructed using maximum likelihood (ML) in Mega 7 with 1000 bootstrap replications [38]. The Tamura-Nei model (TN93+G+I) [39,40] was used for making a ML tree of the complete CP sequences as a result of model tests in Mega7 for determining the best model of nucleotide substitution. The CP sequence of soybean mosaic virus (SMV) was used as an outgroup. Trees were visualized in Figtree v.1.3.1 [41].
4.4. Selection Pressure Analysis
Selection pressure on the CP gene was determined using SNAP [42], as well as for each codon using the Datamonkey online positive selection interface [43]. The ratio of dN/dS (ω) was estimated for the CP gene, where dN means the average number of non-synonymous substitutions per non-synonymous site and dS represents the average number of synonymous substitutions per synonymous site [44]. Selection pressure was considered negative, or purifying, when the ratio of ω < 1, or positive, or diversifying, when ω > 1, and neutral when ω = 1. To detect positive selection on each codon, three different methods were used: single likelihood ancestor counting (SLAC), random effects likelihood (REL), and fixed effects likelihood (FEL), with default parameters including the significance level [43]. The HKY85 nucleotide substitution biased model available on the Datamonkey server was used for all analyses (SLAC, FEL and REL) by selecting the automatic model selection [43]. To measure the synonymous codon usage biases in the CP gene, the effective number of codons (ENC) was calculated using DnaSPv6 [45]. ENC values range from 20 to 61 for a gene [46], where the value of 20 indicates that one codon is used for each amino acid (extreme biased), while a value of 61 shows that all codons are used equally (no bias).
4.5. Genetic Differentiating and Gene Flow Analysis
Genetic differentiation, gen flow among, and permutation-based statistical tests Ks, Ks*, Z*, and Snn among WMV isolates collected from different states were measured in DnaSP6v6 using F statistics (FST) [45]
4.6. Co-Evolution Analysis of CP Amino Acids
Evidence of coevolution sites in the CP gene sequences of WMV isolates was evaluated using the Bayesian Graphical Models (BGM) method implemented in Spidermoneky through the Datamonkey web-based interface [47].
4.7. Phylogenetic Analysis of the Complete Genome Sequences
In order to confirm the WMV grouping (G1, G2, and G3) based on the CP gene sequence, complete genome sequences of selected 37 WMV isolates including 2 U.S. isolates and 35 from 12 different countries representing all the three groups (G1, G2, and G3) were downloaded from GenBank NCBI database and used in the phylogenetic analysis (Table S3). One of the U.S. WMV isolates (Accession no. HQ384216) collected from Dendrobium anosmum (orchid) and directly submitted to GenBank while the other U.S. WMV isolate (Accession no. KU246036) was collected from watermelon in Texas during our previous study [11]. Later, it was sequenced in our lab [48] and used for comparison in the complete genome analysis of WMV isolates.
A maximum likelihood (ML) tree with 1000 bootstrap replications [39] was constructed based on the best-fit model of nucleotide substitution in Mega 7. The general time-reversible substitution model with a gamma distribution and invariant sites (GTR+G+I) was used for making a ML tree of the complete genome sequences while soybean mosaic virus (SMV) (Accession number AJ628750) was used as an outgroup. Trees were visualized in Figtree v.1.3.1 [41].
The pairwise nucleotide sequence identities scores among the 37 WMV isolates were determined using SDT software version 1.2 [49].
4.8. Detection of Recombination Events
The recombination events were investigated in the complete CP gene nucleotide sequences as well as complete genome sequences using RDR4 Beta 4.69, implementing all seven different recombination detection algorithms with default parameters [50]. Only potential recombination events supported by at least four RDR4-implemnted algorithms coupled with phylogenetic evidence of recombination were included in the analysis of recombination events. Furthermore, the potential recombination was also confirmed by Simplot software version 3.5 [51] using a window of 200 nt and step size of 20 nt.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/pathogens10101245/s1, Table S1: Nucleotide sequences of the coat protein gene of watermelon mosaic virus isolates downloaded from GenBank [9,24,25,52,53,54,55,56,57]. Table S2: Selection pressures in the coat protein (CP) gene of watermelon mosaic virus isolates from the U.S. Table S3: Complete genome sequences of watermelon mosaic virus isolates downloaded from GenBank [10,48,52,53,57,58]. Table S4: Potentially co-evolving sites in CP gene sequences of watermelon mosaic virus isolates identified using Bayesian Graphical Models (BGM).
Author Contributions
A.A. conceived the idea and obtained the funding. A.A. and O.A.A. designed and performed the experiments. O.A.A. analyzed the data: O.A.A. and A.A. wrote the paper and A.A. made several revisions. Both authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Watermelon Association (NWA), and partial support from Oklahoma EPSCoR and the Office of Research and Sponsored Programs, The University of Tulsa, Oklahoma grant number 2021211474.
Institutional Review Board Statement
Not applicable as no humans or animals are involved in this study.
Informed Consent Statement
Ethical approval: This article does not contain any studies with human or animals studies performed by any of the authors.
Data Availability Statement
All 57 coat protein gene sequences of WMV isolates from the US presented in this study (Table 1) were submitted to NCBI database. The accession numbers from (MG021270- MG0212303) can be found online https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/genbank/ (accessed on 30 August 2021).
Acknowledgments
We are thankful to Peggy Hill, the University of Tulsa, for careful review of the manuscript and valuable suggestions. The authors are also thankful to David Langston, University of Georgia; Gary Vallad, University of Florida; Edward Sikora, Auburn University; Kenneth Seebold, University of Kentucky; Sarah R Denkler, University of Missouri; Sead Sabanadzovicjo, Mississippi State University; Rodrigo Valverde, Louisiana State University; and Terry Kirkpatrick, University of Arkansas, for the collection of cucurbit samples from their respective states.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Khanal, V.; Wells, H.; Ali, A. High Prevalence of Three Potyviruses Infecting Cucurbits in Oklahoma and Phylogenetic Analysis of Cucurbit Aphid-Borne Yellows Virus Isolated from Pumpkins. Pathogens 2021, 10, 53. [Google Scholar] [CrossRef]
- Purcifull, D.; Hiebert, E.; Edwardson, J. Watermelon Mosaic Virus 2. Descriptions of Plant Viruses. 1984. no. 293. Available online: https://www.dpvweb.net/dpv/showdpv/?dpvno=293 (accessed on 30 August 2021).
- Grafton-Cardwell, E.E.; Perring, T.M.; Smith, R.F.; Valencia, J.; Farrar, C.A. Occurrence of mosaic viruses in melons in the central valley of California. Plant Dis. 1996, 80, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Lecoq, H.; Desbiez, C. Watermelon mosaic virus and Zucchini yellow mosaic virus. In Encyclopedia of Virology, 3rd ed.; Mahy, B.W.J., van Regenmortel, M.H.V., Eds.; Academic Press: Waltham, MA, USA, 2008; pp. 433–440. [Google Scholar]
- Ali, A. Epidemiology and evolution of poytviruses infecting cucurbits. In Applied Plant Virology; Awasthi, L.P., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 405–417. [Google Scholar]
- Riechmann, J.I.; Lain, S.; Garcia, J.A. Highlights and prospects of Potyvirus molecular biology. J. Gen. Virol. 1992, 73, 1–16. [Google Scholar] [CrossRef]
- Sharifi, M.; Massumi, H.; Heydarnejad, J.; Pour, A.H.; Shaabanian, M.; Rahimian, H. Analysis of the biological and molecular variability of Watermelon mosaic virus isolates from Iran. Virus Genes 2008, 37, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, M.J.; Ward, C.W.; Shukla, D.D. The use of 3 non-codig nucleotide sequences in the taxonomy of potyviruses: Application to Watermelon mosaic virus 2 and Soybean mosaic virus-N. J. Gen. Virol. 1989, 70, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Desbiez, C.; Lecoq, H. The nucleotide sequence of Watermelon mosaic virus (WMV, Potyvirus) reveals interspecific recombination between two related potyviruses in the 5 part of the genome. Arch Virol. 2004, 149, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Natsuaki, T.; Okuda, S. The complete nucleotide sequence of a Pakistani isolate of Watermelon mosaic virus provides further insight into the taxonomic status in the Bean common mosaic virus subgroup. Virus Genes 2006, 32, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Abdalla, O.; Bruton, B.; Fish, W.; Sikora, E.; Zhang, S.; Taylor, M. Occurrence of viruses infecting watermelon, other cucurbits, and weeds in the parts of southern United States. Plant Health Prog. 2012, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Luis-Arteaga, M.; Alvarez, J.M.; Alonso-Prados, J.L.; Bernal, J.J.; Garcia-Arenal, F.; Lavina, A.; Batlle, A.; Moriones, E. Occurrence, distribution, and relative incidence of mosaic viruses infecting field-grown melon in Spain. Plant Dis. 1998, 82, 979–982. [Google Scholar] [CrossRef] [Green Version]
- Shoeibi, S.; Masumi, M.; Nasrollanezhad, D.; Heydari, S.; Izadpanah, K.; Ahmadikhah, S. Sequencing of six Iranian isolates of Watermelon mosaic virus and phylogenetic comparison of Iranian isolates with other isolates of the world. Iran J. Plant Path. 2009, 45, 34–37. [Google Scholar]
- Yakoubi, S.; Lecoq, H.; Desbiez, C. Algerian watermelon mosaic virus (AWMV): A new Potyvirus species in the PRSV cluster. Virus Genes 2008, 37, 103–109. [Google Scholar] [CrossRef]
- Webb, R.E.; Scott, H.A. Isolation and identification of watermelon mosaic viruses 1 and 2. Phytopathology 1965, 55, 895–900. [Google Scholar]
- Chala, V.H.; Harrison, C.W.; Halliwell, R.S. Identification of two distinct strains of Watermelon mosaic virus 2 affecting cucurbits in Texas. Plant Dis. 1987, 71, 750–752. [Google Scholar] [CrossRef]
- Davis, R.E.; Mizuki, M.K. Detection of cucurbit viruses in New Jersey. Plant Dis. 1987, 71, 40–44. [Google Scholar] [CrossRef]
- Jossey, S.; Babadoost, M. Occurrence and distribution of pumpkin and squash viruses in Illinois. Plant Dis. 2008, 92, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Mohammad, O.; Khattab, A. Distribution of viruses infecting cucurbit crops and isolation of potential new virus-like sequences from weeds on Oklahoma. Plant Dis. 2012, 96, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desbiez, C.; Joannon, B.; Wipf-Scheibel, C.; Chandeysson, C.; Lecoq, H. Emergence of new strains of Watermelon mosaic virus in south-eastern France: Evidence for limited spread but rapid local population shift. Virus Res. 2009, 14, 201–228. [Google Scholar] [CrossRef]
- Desbiez, C.; Costa, C.; Wipf-Scheibel, C.; Girard, M.; Lecoq, H. Serological and molecular variability of Watermelon mosaic virus (genus Potyvirus). Arch Virol. 2007, 152, 775–781. [Google Scholar] [CrossRef]
- Lecoq, H.; Fabre, F.; Joannon, B.; Wipf-Scheiberl, C.; Chandeysson, C.; Schoeny, A.; Desbiez, C. Search for factors involved in the rapid shift of Watermelon mosaic virus (WMV) populations in South-eastern France. Virus Res. 2011, 159, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Glasa, M.; Bananej, K.; Predajna, L.; Vahdat, A. Genetic diversity of Watermelon mosaic virus is Slovakia and Iran shows distinct pattern. Plant Dis. 2011, 95, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Li, G.; Shan, S.D. Occurrence of viruses infecting melon in Xinjiang of China and molecular characterization of Watermelon mosaic virus isolates. Eur. J. Plant Pathol. 2017, 147, 919–931. [Google Scholar] [CrossRef]
- Quemada, H.; Sieu, L.C.; Siemieniak, D.R.; Gonsalves, D.; Slightom, J.L. Watermelon mosaic virus II and Zucchini yellow mosaic virus: Cloning of 3-terminal regions, nucleotide sequences and phylogenetic comparisons. J. Gen. Virol. 1990, 71, 203–210. [Google Scholar] [CrossRef]
- Gal-On, A.; Antignus, Y.; Rosner, A.; Raccah, B. A zucchini yellow mosaic virus coat protein gene mutation restores aphid transmissibility but has no effect on multiplication. J. Gen. Virol. 1992, 73, 2183–2187. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, M.A.; Coults, B.A.; Buirchell, B.J.; Jones, R.A.C. Split Personality of a Potyvirus: To Specialize or Not to Specialize? PLoS ONE 2014, 9, e105770. [Google Scholar] [CrossRef] [PubMed]
- Wylie, S.J.; Jones, R.A.C. Role of recombination in the evolution of host specialization within Bean yellow mosaic virus. Phytopathology 2009, 99, 512–518. [Google Scholar] [CrossRef] [Green Version]
- Lecoq, H.; Wisler, G.; Pitrat, M. Cucurbit viruses: The classics and the emerging. In Cucurbitacea, Evaluation and Enhancement of Cucurbit Germplasm; McCreight, J.D., Ed.; Alexandria ASHS: Alexandria, LA, USA, 1998; pp. 126–142. [Google Scholar]
- Ali, A.; Li, H.; Schneider, W.L.; Sherman, D.J.; Gray, S.; Smith, D.; Roossinck, M.J. Analysis of genetic bottlenecks during horizontal transmission of Cucumber mosaic virus. J. Virol. 2006, 80, 8345–8350. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Arenal, F.; Fraile, A.; Malpica, J.M. Variation and evolution of plant virus populations. Int. Microbiol. 2003, 6, 225–232. [Google Scholar] [CrossRef]
- Nigam, D.; LaTourrette, K.; Souza, P.F.N.; Garcia-Ruiz, H. Genome- Wide Variation in Potyviruses. Front. Plant Sci. 2019, 10, 1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roossinck, M.J.; Ali, A. Mechanisms of plant virus evolution and identification of geneticbottlenecks: Impact on disease management. In Biotechnology and Plant Disease Management; Punja, Z.K., DeBoer, S., Sanfacon, H., Eds.; CAB International: Wallingford, UK, 2007; pp. 109–124. [Google Scholar]
- Chare, E.R.; Holmes, E.C. A phylogenetic survey of recombination frequency in plant RNAviruses. Arch. Virol. 2006, 151, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Sztuba-Solińska, J.; Urbanowicz, A.; Figlerowicz, M.; Bujarski, J.J. RNA-RNA recombination in plant virus replication and evolution. Ann. Rev. Phytopathol. 2011, 49, 415–443. [Google Scholar] [CrossRef]
- Abdalla, O.A.; Ali, A. Genetic diversity in the 3’-terminal region of Papaya ringspot virus (PRSV-W) isolates from watermelon in Oklahoma. Arch. Virol. 2011, 157, 405–412. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Hall, T. BioEdit: A user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Nei, M.; Kumar, S. Prospects of inferring very large phylogenies by using the neighbor-joining method. Pro. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7, Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2015, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree 1.3.1. Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2010. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 30 August 2021).
- Korber, B. HIV Signature and Sequence Variation Analysis. In Computational Analysis of HIV Molecular Sequences; Rodrigo, A.G., Learn, G.H., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000; pp. 55–72. [Google Scholar]
- Kosakovsky, P.S.L.; Frost, S.D.W. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar]
- Nei, M.; Gojobori, T. Simple methods for estimating the number of synonymous and non-synonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-Delbarrio, J.S.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.; Sanchez-Garcia, A. DnaSP 6, D Sequence Polymorphism Analysis of Large Datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Fuglsang, A. Estimating the Effective Number of Codons: The Wright way of Determining Codon Homozygosity Leads to Superior Estimates. Genetics 2006, 172, 1301–1307. [Google Scholar] [CrossRef]
- Poon, A.F.; Lewis, F.I.; Frost, S.D.W.; Pond, S.L. Spidermonkey: Rapid Detection of Co-Evolving Sites Using Bayesian Graphical Models. Bioinformatics 2008, 24, 1949–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajbanshi, N.; Ali, A. First complete genome sequence of a Watermelon mosaic virus isolated from watermelon in the United States. Genome Announc. 2016, 4, e00299-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4, Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Garkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Desbiez, C.; Lecoq, H. Evidence for multiple interspecific recombination in natural population of Watermelon mosaic virus (WMV, Potyvirus). Arch. Virol. 2008, 153, 1749–1754. [Google Scholar] [CrossRef]
- Desbiez, C.; Joannon, B.; Wipf-Scheibel, C.; Chandeysson, C.; Lecoq, H. Recombination in natural populations of watermelon mosaic virus: New agronomic threat or damp squib? J. Gen. Virol. 2011, 92, 1939–1948. [Google Scholar] [CrossRef]
- Moreno, M.; Malpice, J.M.; Diaz-Pendon, J.A.; Moriones, E.; Fraile, A.; Garcia-Arenal, F. Variability and genetic structure of the population of Watermelon mosaic virus infecting melon in Spain. Virology 2004, 318, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Bau, H.; Yeh, S. Comparison of the nuclear inclusion b protein and coat protein genes of five Papaya ringspot virus strains distinct in geographic origin and pathogenicity. Phytopathology 1994, 84, 1205–1210. [Google Scholar] [CrossRef]
- Salvaudon, L.; De Moraes, C.M.; Mescher, M.C. Outcomes of co-infection by two potyviruses: Implications for the evolution of manipulative strategies. Proc. R. Soc. B 2013, 280, 20122959. [Google Scholar] [CrossRef] [Green Version]
- Romay, G.; Lecoq, H.; Desbiez, C. First report of watermelon mosaic virus naturally infecting Cucumis anguria. Plant Dis. 2013, 97, 1515. [Google Scholar] [CrossRef]
- Finetti-Sialer, M.M.; Mascia, T.; Cillo, F.; Vovlas, C.; Gallitelli, D. Biological and molecular characterization of a recombinant isolate of Watermelon mosaic virus associated with a watermelon necrotic disease in Italy. Eur. J. Plant Pathol. 2012, 132, 317–322. [Google Scholar] [CrossRef]
Figure 1.
Maximum likelihood (ML) tree showing the phylogenetic relationships based on the nucleotide sequences of complete coat protein (CP) genes among 57 U.S. WMV isolates obtained in this study and 89 published WMV isolates reported worldwide previously (available from GenBank). In the ML trees, the names of U.S. WMV isolates obtained in this study are without accession number and denoted by the abbreviation of various states and number of isolates. Detailed information for the U.S. WMV isolates is listed in Table 1, while sequences of the 89 isolates from other countries that were downloaded from the GenBank are listed in Table S1. ML trees were generated using MEGA7 with the Tamura-Nei model (TN93+G+I) model. Bootstrap values (1000 replicates) greater than 50% are indicated at the tree nodes. soybean mosaic virus (SMV) was used as the outgroup.
Figure 1.
Maximum likelihood (ML) tree showing the phylogenetic relationships based on the nucleotide sequences of complete coat protein (CP) genes among 57 U.S. WMV isolates obtained in this study and 89 published WMV isolates reported worldwide previously (available from GenBank). In the ML trees, the names of U.S. WMV isolates obtained in this study are without accession number and denoted by the abbreviation of various states and number of isolates. Detailed information for the U.S. WMV isolates is listed in Table 1, while sequences of the 89 isolates from other countries that were downloaded from the GenBank are listed in Table S1. ML trees were generated using MEGA7 with the Tamura-Nei model (TN93+G+I) model. Bootstrap values (1000 replicates) greater than 50% are indicated at the tree nodes. soybean mosaic virus (SMV) was used as the outgroup.

Figure 2.
Multiple sequence alignment of the complete coat protein gene amino acids (283 aa) of EM5 subgroup watermelon mosaic virus (WMV) isolates from the U.S. and their comparison with the representative WMV isolates from G1, G2, and G3 groups reported from other countries of the world. The three WMV isolates reported previously from the U.S. are also included for comparison. In the top panel, the horizontal boxes show the specific conserved amino acid motifs in the N-terminal of the CP gene among the G1, G2, and G3 isolates. Vertical boxes show specific conserved amino acids that only exist in EM5 subgroups isolates.
Figure 2.
Multiple sequence alignment of the complete coat protein gene amino acids (283 aa) of EM5 subgroup watermelon mosaic virus (WMV) isolates from the U.S. and their comparison with the representative WMV isolates from G1, G2, and G3 groups reported from other countries of the world. The three WMV isolates reported previously from the U.S. are also included for comparison. In the top panel, the horizontal boxes show the specific conserved amino acid motifs in the N-terminal of the CP gene among the G1, G2, and G3 isolates. Vertical boxes show specific conserved amino acids that only exist in EM5 subgroups isolates.


Figure 3.
Maximum likelihood (ML) tree showing the phylogenetic relationships based on the nucleotide sequences of complete genome sequences of 37 WMV isolates containing 2 U.S WMV isolates and 35 other WMV isolates reported worldwide previously (available from GenBank). ML trees were generated using MEGA7 with the Tamura-Nei model (TN93+G+I) model. Bootstrap values (1000 replicates) greater than 50% are indicated at the tree nodes. soybean mosaic virus (SMV) was used as an outgroup.
Figure 3.
Maximum likelihood (ML) tree showing the phylogenetic relationships based on the nucleotide sequences of complete genome sequences of 37 WMV isolates containing 2 U.S WMV isolates and 35 other WMV isolates reported worldwide previously (available from GenBank). ML trees were generated using MEGA7 with the Tamura-Nei model (TN93+G+I) model. Bootstrap values (1000 replicates) greater than 50% are indicated at the tree nodes. soybean mosaic virus (SMV) was used as an outgroup.

Figure 4.
Average incidences of synonymous, and non-synonymous and indel mutations in the codons of the coat protein (CP) gene of watermelon mosaic virus isolates subgroups: (A) EM1 isolates, (B) EM2 isolates, (C) EM5 isolates. The X-axis represents the position of the codon while the Y-axis represents the average cumulative number of synonymous, non-synonymous mutations estimated at specific codon positions in the CP gene. No indel was present in the CP of any WMV isolates in the three subgroups.
Figure 4.
Average incidences of synonymous, and non-synonymous and indel mutations in the codons of the coat protein (CP) gene of watermelon mosaic virus isolates subgroups: (A) EM1 isolates, (B) EM2 isolates, (C) EM5 isolates. The X-axis represents the position of the codon while the Y-axis represents the average cumulative number of synonymous, non-synonymous mutations estimated at specific codon positions in the CP gene. No indel was present in the CP of any WMV isolates in the three subgroups.

Figure 5.
The distribution of pairwise identity score of the complete genome sequences of 37 WMV isolates as determined by the MUSCL multiple sequence alignment program in SDT software version 1.2. All 37 isolates have been shown in three molecular groups (G1, G2 and G3).
Figure 5.
The distribution of pairwise identity score of the complete genome sequences of 37 WMV isolates as determined by the MUSCL multiple sequence alignment program in SDT software version 1.2. All 37 isolates have been shown in three molecular groups (G1, G2 and G3).

Figure 6.
Recombination events analysis confirmed by SimPlot (version 3.5) with a sliding window of 200 bases and a step of 20 bases. Recombination breakpoints between the U.S. watermelon mosaic virus isolates are shown: (A) recombination event number 3 (Table 3) in subgroup EM1 isolates (OK15 and OK12), (B) recombination event number 3 (Table 3) in subgroup EM5 isolates (MS-3 and TX-6). The X-axis shows the nucleotide positions of the coat protein gene while the Y-axis shows the percent similarity between the major and minor parent isolates.
Figure 6.
Recombination events analysis confirmed by SimPlot (version 3.5) with a sliding window of 200 bases and a step of 20 bases. Recombination breakpoints between the U.S. watermelon mosaic virus isolates are shown: (A) recombination event number 3 (Table 3) in subgroup EM1 isolates (OK15 and OK12), (B) recombination event number 3 (Table 3) in subgroup EM5 isolates (MS-3 and TX-6). The X-axis shows the nucleotide positions of the coat protein gene while the Y-axis shows the percent similarity between the major and minor parent isolates.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Watermelon mosaic virus isolates collected in nine different states of the U.S. and analyzed in this study.
Table 1.
Watermelon mosaic virus isolates collected in nine different states of the U.S. and analyzed in this study.
| No | Name of State | Name of Isolate | Host | Year Collected | Accession Number |
|---|---|---|---|---|---|
| 1 | Alabama | AL-1 | Watermelon | 2010 | MG021270 |
| 2 | Arkansas | AR-1 | Pumpkin | 2010 | MG021275 |
| 3 | AR-2 | Watermelon | 2010 | MG021276 | |
| 4 | AR-3 | Pumpkin | 2010 | MG021277 | |
| 5 | AR-4 | Cantaloupe | 2010 | MG021278 | |
| 6 | AR-5 | Watermelon | 2010 | MG021279 | |
| 7 | AR-6 | Watermelon | 2010 | MG021280 | |
| 8 | AR-7 | Watermelon | 2010 | MG021281 | |
| 9 | Florida | FL-1 | Squash | 2011 | MG021262 |
| 10 | FL-2 | Pumpkin | 2011 | MG021263 | |
| 11 | FL-3 | Cucumber | 2011 | MG021264 | |
| 12 | FL-4 | Watermelon | 2011 | MG021265 | |
| 13 | Georgia | GA-1 | Watermelon | 2010 | MG021271 |
| 14 | GA-2 | Watermelon | 2010 | MG021272 | |
| 15 | Kentucky | KY-1 | Watermelon | 2010 | MG021273 |
| 16 | Louisiana | LA-1 | Watermelon | 2010 | MG021274 |
| 17 | Mississippi | MS-1 | Cantaloupe | 2007 | MG021266 |
| 18 | MS-2 | Squash | 2007 | MG021267 | |
| 19 | MS-3 | Squash | 2008 | MG021268 | |
| 20 | MS-4 | Watermelon | 2010 | MG021269 | |
| 21 | Oklahoma | OK-1 | Squash | 2008 | MG021247 |
| 22 | OK-2 | Watermelon | 2010 | MG021248 | |
| 23 | OK-3 | Watermelon | 2009 | MG021249 | |
| 24 | OK-4 | Watermelon | 2009 | MG021250 | |
| 25 | OK-5 | Watermelon | 2009 | MG021251 | |
| 26 | OK-6 | Cantaloupe | 2009 | MG021252 | |
| 27 | OK-7 | Watermelon | 2010 | MG021253 | |
| 28 | OK-8 | Watermelon | 2010 | MG021254 | |
| 29 | OK-9 | Watermelon | 2010 | MG021255 | |
| 30 | OK-10 | Watermelon | 2010 | MG021256 | |
| 31 | OK-11 | Palmer amaranth | 2010 | MG021257 | |
| 32 | OK-12 | Pumpkin | 2010 | MG021258 | |
| 33 | OK-13 | Watermelon | 2010 | MG021259 | |
| 34 | OK-14 | Watermelon | 2010 | MG021260 | |
| 35 | OK-15 | Pumpkin | 2010 | MG021261 | |
| 36 | Texas | TX-1 | Watermelon | 2010 | MG021282 |
| 37 | TX-2 | Cucumber | 2010 | MG021283 | |
| 38 | TX-3 | Watermelon | 2010 | MG021284 | |
| 39 | TX-4 | Watermelon | 2010 | MG021285 | |
| 40 | TX-5 | Watermelon | 2010 | MG021286 | |
| 41 | TX-6 | Watermelon | 2010 | MG021287 | |
| 42 | TX-7 | Watermelon | 2010 | MG021288 | |
| 43 | TX-8 | Watermelon | 2010 | MG021289 | |
| 44 | TX-9 | Watermelon | 2010 | MG021290 | |
| 45 | TX-10 | Watermelon | 2010 | MG021291 | |
| 46 | TX-11 | Watermelon | 2010 | MG021292 | |
| 47 | TX-12 | Watermelon | 2010 | MG021293 | |
| 48 | TX-13 | Watermelon | 2010 | MG021294 | |
| 49 | TX-14 | Watermelon | 2010 | MG021295 | |
| 50 | TX-15 | Watermelon | 2010 | MG021296 | |
| 51 | TX-16 | Watermelon | 2010 | MG021297 | |
| 52 | TX-17 | Watermelon | 2010 | MG021298 | |
| 53 | TX-18 | Watermelon | 2010 | MG021299 | |
| 54 | TX-19 | Watermelon | 2010 | MG0212300 | |
| 55 | TX-20 | Watermelon | 2010 | MG0212301 | |
| 56 | TX-21 | Watermelon | 2010 | MG0212302 | |
| 57 | TX-22 | Watermelon | 2010 | MG0212303 |
Table 2.
Selection pressure acting on the coat protein (CP) gene of watermelon mosaic virus isolates from the U.S.
Table 2.
Selection pressure acting on the coat protein (CP) gene of watermelon mosaic virus isolates from the U.S.
| Subgroups within G3 (EM Isolates) | No. of Isolates a | ENC b | No. of Nnegatively c Selected Codons | dNSd | Variance (dNS) | Standard Deviation (dNS) | dSe | Variance (dNS) | Standard Deviation (dNS) | dNS/dS | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SLAC | FEL | REL | ||||||||||
| EM1 | 17 | 50.092 | 2 | 29 | 55 | 0.0129 | 0.0000 | 0.0022 | 0.0639 | 0.0000 | 0.0098 | 0.2019 |
| EM2 | 31 | 51.828 | 9 | 25 | 1 | 0.0148 | 0.0000 | 0.0024 | 0.0479 | 0.0001 | 0.0077 | 0.3089 |
| EM5 | 9 | 50.573 | 2 | 17 | 31 | 0.0188 | 0.0001 | 0.0037 | 0.0906 | 0.0002 | 0.0154 | 0.2075 |
a Number of WMV isolates in each subgroup within G3 (emerging isolates). EM-V is the new distinct subgroup identified in this work. b ENC- effective number of codons, were estimated by DnaSPv6 (see materials and methods). c Number of negatively selected codons was calculated suing SLAC, FEL, and REL programs within Datamonkey software d Average number of non-synonymous substitutions per non-synonymous site. e Average number of synonymous substitutions per synonymous site. d,e dNS and dS along with standard variation and variance was calculated using SNAP (non-synonymous analysis program) (see materials and methods).
Table 3.
Estimates of genetic differentiation and gene flow among different population of watermelon mosaic virus isolates from different states.
Table 3.
Estimates of genetic differentiation and gene flow among different population of watermelon mosaic virus isolates from different states.
| States | Fst | Nm | Ks | Kst | p-Value | Ks* | Kst* | p-Value | Z* | p-Value | Snn | p-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AR vs. FL | 0.439 | 0.32 | 30.54 | 0.246 | 0.008 ** | 3.06 | 0.112 | 0.007 ** | 2.66 | 0.006 ** | 0.95 | 0.008 ** |
| AR vs. MS | 0.083 | 2.73 | 43.03 | 0.043 | 0.180 ns | 3.54 | 0.021 | 0.095 ns | 2.88 | 0.051 ns | 0.95 | 0.007 ** |
| AR vs. OK | 0.004 | 65.87 | 48.56 | 0.001 | 0.364 ns | 3.68 | 0.000 | 0.036 ns | 4.44 | 0.293 ns | 0.772 | 0.013 * |
| AR vs. TX | 0.061 | 3.84 | 44.73 | 0.024 | 0.100 ns | 3.52 | 0.013 | 0.082 ns | 4.94 | 0.051 ns | 0.84 | 0.004 ** |
| AR vs. OS a | 0.019 | 12.93 | 46.84 | 0.010 | 0.025 ns | 3.66 | -0.000 | 0.280 ns | 3.19 | 0.319 ns | 0.83 | 0.028 * |
| Fl vs. MS | 0.620 | 0.15 | 22.0 | 0.483 | 0.024 * | 2.42 | 0.279 | 0.024 * | 8.62 | 0.024 * | 1.00 | 0.024 * |
| FL vs. OK | 0.470 | 0.28 | 40.58 | 0.174 | 0.001 ** | 3.40 | 0.079 | 0.0010 ** | 3.86 | 0.0010 ** | 1.00 | 0.002 ** |
| FL vs. TX | 0.569 | 0.19 | 38.45 | 0.186 | 0.001 ** | 3.31 | 0.076 | 0.000 *** | 4.52 | 0.0010 ** | 1.00 | 0.000 *** |
| FL vs. OS | 0.270 | 0.68 | 29.42 | 0.158 | 0.028 * | 2.83 | 0.120 | 0.036 * | 2.29 | 0.028 * | 0.888 | 0.024 * |
| MS vs. OK | 0.010 | 24.29 | 47.80 | 0.003 | 0.035 ns | 3.63 | 0.009 | 0.192 ns | 4.08 | 0.115 ns | 0.921 | 0.005 ** |
| MS vs. TX | 0.026 | 9.07 | 43.73 | 0.007 | 0.307 ns | 3.46 | 0.012 | 0.125 ns | 4.69 | 0.040 * | 0.961 | 0.003 ** |
| MS vs. OS | 0.128 | 1.69 | 44.68 | 0.074 | 0.130 ns | 3.51 | 0.033 | 0.157 ns | 2.48 | 0.112 ns | 0.833 | 0.044 * |
| OK vs. TX | 0.027 | 8.88 | 46.81 | 0.014 | 0.162 ns | 3.56 | 0.019 | 0.022 * | 5.39 | 0.005 ** | 0.887 | 0.000 *** |
| OK vs. OS | 0.024 | 10.06 | 49.86 | 0.009 | 0.315 ns | 3.68 | 0.006 | 0.278 ns | 4.21 | 0.181 ns | 0.850 | 0.014 * |
| TX vs. OS | 0.122 | 1.79 | 45.41 | 0.043 | 0.097 ns | 3.51 | 0.024 | 0.041 * | 4.75 | 0.020 * | 0.944 | 0.000 *** |
p value estimates are based on probability obtained by the permutation test with 1000 replicates. ns, not significant; *, 0.01 < p < 0.05; **, 0.001 < p < 0.01; ***, p < 0.001. p > 0.001 significantly rejects the null hypothesis that there is no genetic differentiation between two populations. a WMV isolates from other states include: two isolates from Georgia and one isolate each from Alabama, Louisiana, and Kentucky.
Table 4.
Recombination events in the nucleotide sequence of the coat protein (CP) gene of watermelon mosaic virus isolates from the U.S.
Table 4.
Recombination events in the nucleotide sequence of the coat protein (CP) gene of watermelon mosaic virus isolates from the U.S.
| Sub-Groups a | Event No. | Recomb. | Major Parent | Minor Parent | Recombination Sites | Detection Methods | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM1 Isolates | RDP | GENECON V | Bootscan | Maxchi | Chimaera | Sisscan | 3-Seq | |||||
| 1 | AR-3 | TX-13 | Unknown (Al-1) | 69–735 nt | - | - | - | 8.115 × 10−2 | - | - | 2.864 × 10−2 | |
| 2 | OK-2 | TX-13 | Unknown (Al-1) | 680–832 nt | - | - | - | 8.115 × 10−2 | - | - | 2.864 × 10−2 | |
| 3 b | OK-15 | OK-2 | Unknown(Al-1) | 298–810 nt | 1.243 × 10−4 | 4.828 × 10−3 | - | 6.683 × 10−8 | 2.881 × 10−5 | - | 9.765 × 10−9 | |
| EM2 | None | |||||||||||
| EM5 isolates | ||||||||||||
| 1 | OK-11 | MS-3 | OK-12 | 230–824 nt | 8.231 × 10−4 | 4.035 × 10−10 | 1.655 × 10−6 | |||||
| 2 | TX-7 | TX-5 | OK-12 | 627–743 nt | 3.198 × 10−2 | 1.201 × 10−4 | ||||||
| 3 b | MS-3 | TX-6 | Unknown (OK-12) | 155–724 nt | 7.921 × 10−3 | 1.214 × 10−1 | 4.096 × 10−2 | 4.814 × 10−3 | ||||
| 4 | OK-13 | OK-12 | TX-6 | 58–783 nt | 3.987 × 10−3 | 6.198 × 10−3 | ||||||
a All three subgroups are within G3 (Emerging isolates) of WMV; b recombination event detected by at least four algorithms was considered statistically significant.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Abdalla, O.A.; Ali, A. Genetic Variability and Evidence of a New Subgroup in Watermelon Mosaic Virus Isolates. Pathogens 2021, 10, 1245. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10101245
AMA Style
Abdalla OA, Ali A. Genetic Variability and Evidence of a New Subgroup in Watermelon Mosaic Virus Isolates. Pathogens. 2021; 10(10):1245. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10101245
Chicago/Turabian StyleAbdalla, Osama A., and Akhtar Ali. 2021. "Genetic Variability and Evidence of a New Subgroup in Watermelon Mosaic Virus Isolates" Pathogens 10, no. 10: 1245. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10101245
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.