
The Brilliance of Borrelia: Mechanisms of Host Immune Evasion by Lyme Disease-Causing Spirochetes


Department of Biomedical Sciences, University of North Dakota, Grand Forks, ND 58202, USA
*
Author to whom correspondence should be addressed.
Pathogens 2021, 10(3), 281; https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10030281
Submission received: 25 January 2021
/
Revised: 23 February 2021
/
Accepted: 24 February 2021
/
Published: 2 March 2021
(This article belongs to the Special Issue The Twists and Turns of Pathogenic Spirochetes: Novel Insights for Prevention, Diagnosis and Treatment)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Lyme disease (LD) has become the most common vector-borne illness in the northern hemisphere. The causative agent, Borrelia burgdorferi sensu lato, is capable of establishing a persistent infection within the host. This is despite the activation of both the innate and adaptive immune responses. B. burgdorferi utilizes several immune evasion tactics ranging from the regulation of surface proteins, tick saliva, antimicrobial peptide resistance, and the disabling of the germinal center. This review aims to cover the various methods by which B. burgdorferi evades detection and destruction by the host immune response, examining both the innate and adaptive responses. By understanding the methods employed by B. burgdorferi to evade the host immune response, we gain a deeper knowledge of B. burgdorferi pathogenesis and Lyme disease, and gain insight into how to create novel, effective treatments.
1. Introduction
Since the first investigations conducted by Steere and Malawista in 1975 [1,2], Lyme borreliosis, otherwise known as Lyme disease (LD), has become the most common vector-borne illness in the northern hemisphere [3]. LD is caused by infection with a member of the Borrelia burgdorferi sensu lato (s.l.) complex. Within the complex, three species cause the majority of LD in humans: Borrelia burgdorferi sensu stricto (s.s.), Borrelia afzelii, and Borrelia garinii [4]. Lyme borrelia are transferred to the vertebrate host by Ixodes ticks. In the northeastern and upper midwest of the US, the main vector is Ixodes scapularis, while Ixodes pacificus is the primary vector in western US [5]. Ixodes ricinus and Ixodes persulcatus are the tick vectors for the Borrelia burgdorferi sensu lato (s.l.) complex in Europe and Asia, respectively [5].
The most common clinical sign of Lyme disease in the United States is the formation of an erythema migrans skin lesion, which is often accompanied by flu-like symptoms. However, Lyme spirochetes are capable of disseminating to other tissues and causing other manifestations, such as Lyme neuroborreliosis, Lyme carditis, or Lyme arthritis [5]. Symptoms of LD can persist following treatment with antibiotics, resulting in a condition known as post-treatment Lyme disease syndrome (PTLDS). PTLDS is often functionally disabling and leaves patients with fatigue, cognitive complaints, or musculoskeletal pain [3].
For B. burgdorferi to persist in the host, the pathogen must employ a variety of tactics to evade the immune response. This review aims to provide a current overview of a majority of the immune evasion tactics employed by B. burgdorferi.
2. Innate Response
2.1. Complement Cascade
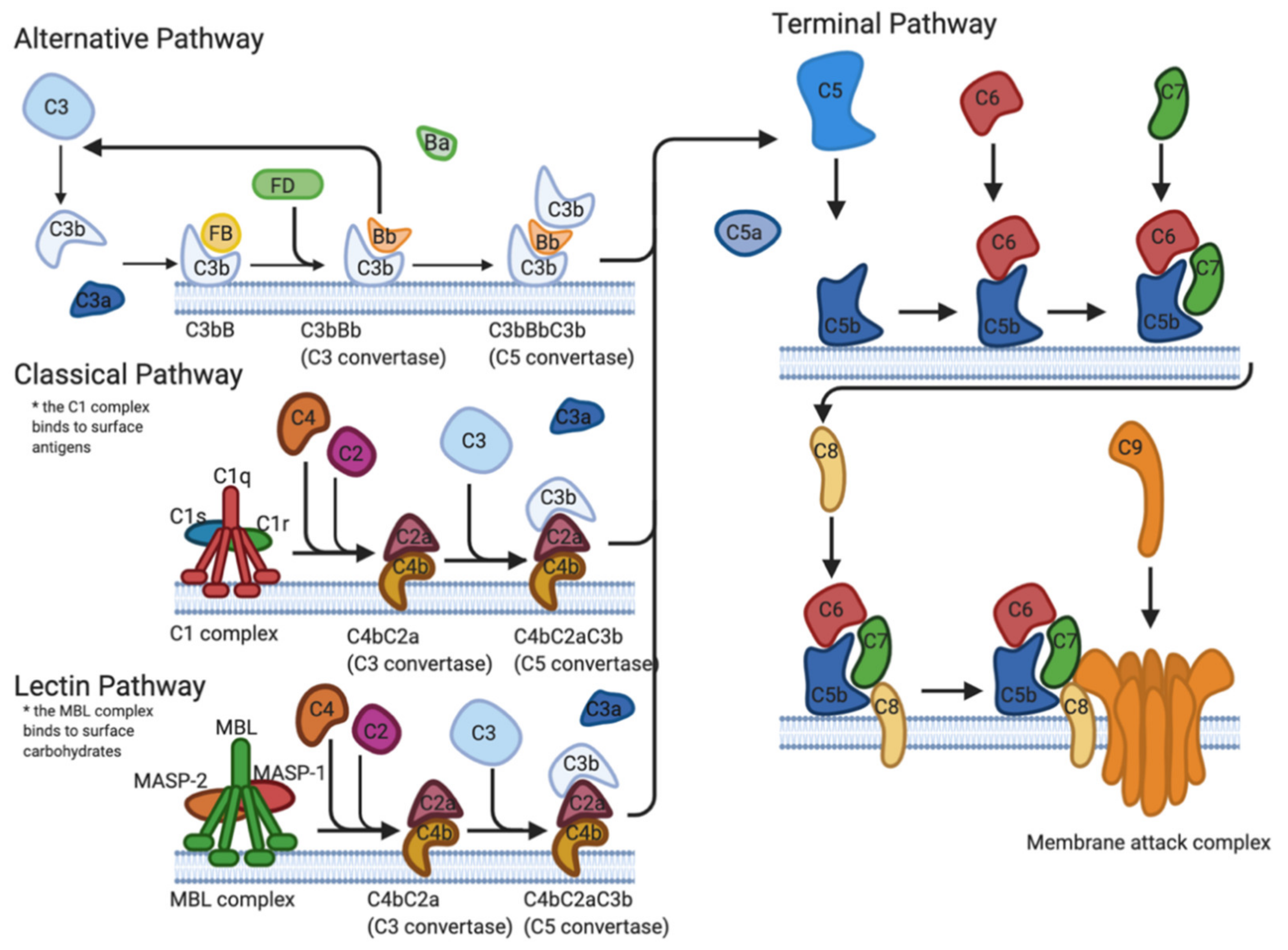
The first line of defense implemented by the immune system to protect the host against pathogens is known as the complement system. The complement system is a tightly regulated cascade of enzymatic proteins responsible for the opsonization of pathogens, phagocytosis, cell lysis, and the establishment of the membrane attack complex (MAC) [6,7]. There are three main activation pathways: the classical pathway (CP), the alternative pathway (AP), and the lectin pathway (LP) (Figure 1). All pathways converge at the complement protein C3 and the formation of activation products C3a, C3b, C5a, and the MAC (C5b-9) [6].
2.1.1. Direct Interference
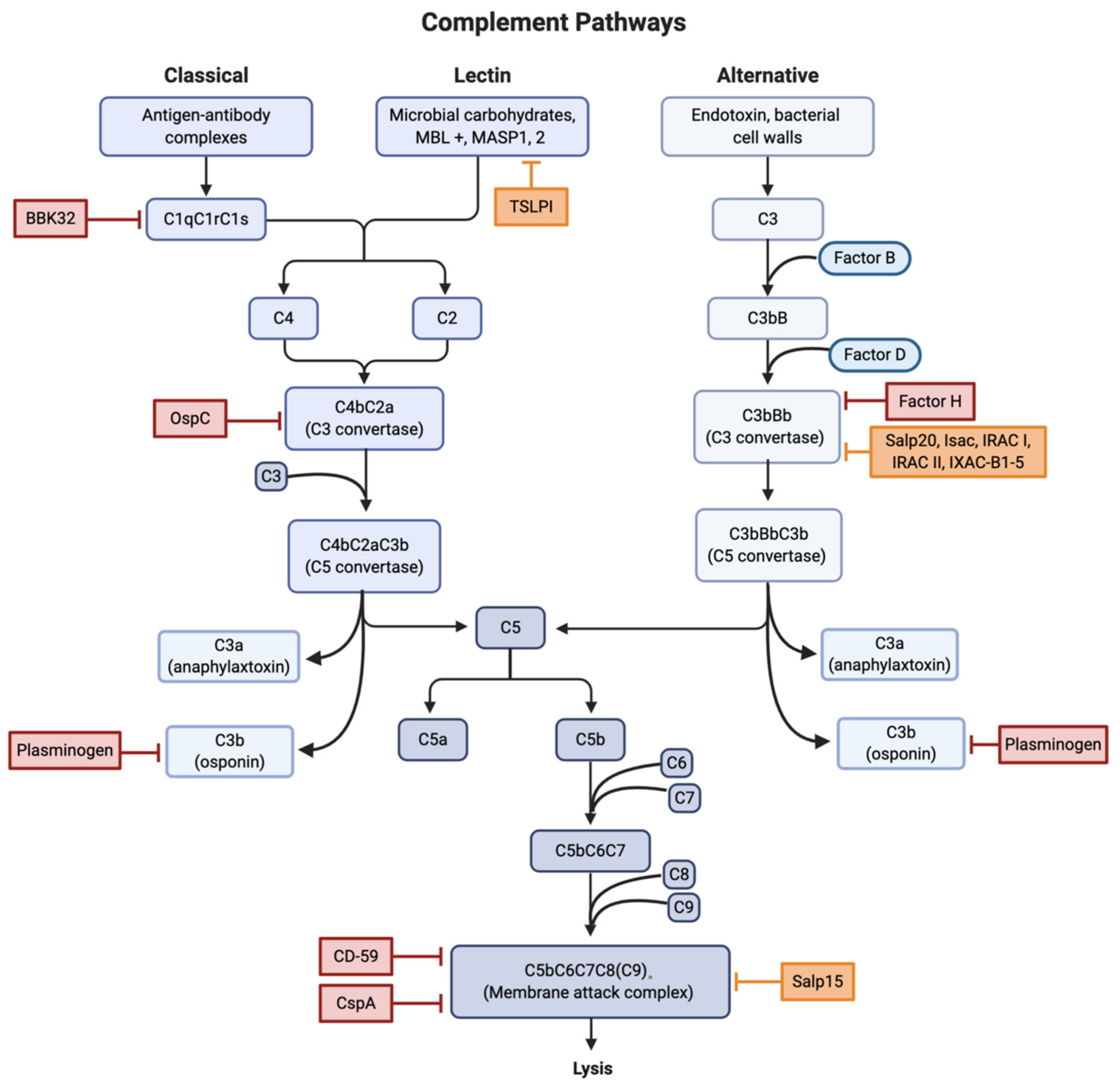
B. burgdorferi evades the complement system in two main methods. The first method involves a direct inference with the components of the complement cascade pathways. Direct interference is accomplished through the use of outer surface proteins, the most notable of which include BBK32, OspA, OspC, and BBA70.
BBK32 is a surface lipoprotein found on Lyme spirochetes that acts as a vascular adhesin and binds both glycosaminoglycans and fibronectin [8,9]. A study by Garcia et al. found that BBK32 is capable of inhibiting the complement CP through high-affinity binding of the C1r subunit of C1 (Figure 2). After C1q binds to the surface of the Borrelia, BBK32 recognizes and binds to C1r, blocking the autocatalysis of the C1r proenzymes and the proteolysis of the C1s proenzymes. Since the C1 complex is the initiating step of the CP, the binding of BBK32 to C1r leaves the pathway in an inactive state. BBK32 knockout mutants were found to have reduced infectivity, indicating that mutants could have a deficiency in either adhesion or complement evasion [10].
The enzyme plasmin is a known inhibitor of the complement system due to its binding and cleaving of Cb3 and C5 [11]. The outer surface protein OspA has been shown to bind plasminogen; however, its expression is downregulated once the spirochete enters the vertebrate host [12]. OspC, another outer surface protein found in Lyme spirochetes, has been shown to bind to plasminogen and its expression is upregulated in the vertebrate host [13]. OspC has also been shown to bind C4b in vivo in both B. burgdorferi and B. garinii [14]. The protein BBA70 is capable of binding plasminogen with a high affinity as well, though it is not able to bind other complement regulators [15].
The complement-regulator acquiring surface protein (CRASP) CspA has also been shown to bind plasminogen [16]. In addition, CspA is capable of interacting with C7, C8, C9, and the MAC [17,18]. By binding C7 and C9, CspA interferes with the MAC formation through the inhibition of C9 polymerization [17,19,20]. The expression of CspA is upregulated during the tick blood meal, but downregulated in the vertebrate host, indicating that it is important in avoiding the immune response during transmission, but is not essential for continued infection [21]. Other CRASP genes cspZ, erpA, erpC, and erpP express proteins that will bind plasminogen [22]. Another species of Borrelia, B. bavariensis, expresses surface proteins BGA66 and BGA71 that share a similarity in sequence with cspA and are capable of inhibiting MAC. BGA66 is capable of inhibiting all three complement pathways, while BGA71 inhibits AP and CP [23].
Pausa et al. found a human-like CD59 protein on the surface of B. burgdorferi, which binds C9 and to a lesser extent the beta subunit of C8 and therefore inhibits the MAC [24]. Currently, there has been only one report on the function of CD59, and further assessment of the function and overall role of this protein in the evasion of the immune response is needed.
2.1.2. Binding of Regulators
In addition to direct interference with the complement system, CRASPs are capable of binding regulators of the complement system. Factor H (FH) is one of the main regulators of the AP (Figure 2). The bunding of FH to C3b accelerates the decay of the AP C3 convertase and promotes Factor I (FI) mediated C3b cleavage [25].
The CRASP protein CspA is essential for the survival of Lyme spirochetes. However, it is expressed by spirochetes only in feeding ticks or at the site of feeding, and not in disseminated spirochetes [26,27]. CspZ is upregulated in the vertebrate host, though it is not essential for the acquisition of spirochetes to the mammalian host [28,29]. When exposed to the spirochetal outer surface, the proteins CspA and CspZ bind complement regulators FH and factor H-like protein-1 (FHL1) [26]. Introducing CspA or CspZ to a serum sensitive strain of spirochete allows survival in vitro in various vertebrate serums, demonstrating the importance of complement evasion [29].
The outer surface protein OspE is expressed throughout the different stages of infection [30,31,32,33]. OspE binds to FH as well as different isotopes of complement factor H-related (CFHR) proteins [33,34]. The complement regulatory domain is on the N terminus, while the C terminal region contains binding sites for C3b, heparin, and microbial surface proteins and is central for FH function, target recognition, discrimination between self and non-self, and anti-inflammatory activities [34].
The OspE-related proteins ErpA and ErpP bind the complement factor H-related proteins CFHR1, CFHR2, and CFHR5, while ErpC binds only CFHR1 and CFHR2 [26]. Studies have shown that in the absence of FH and in the presence of CFHR1, CFHR2, and CFHR5 the complement attack is not inhibited, indicating that FH works in conjunction with CFHR1, CFHR2, and CFHR to support complement evasion [35]. However, the overall role that Erps play in immune evasion or the importance of CFHR binding still remains unclear [29].
2.1.3. Tick Salivary Proteins
The proteins found in tick saliva have been shown to inhibit the complement system (Figure 2). The tick salivary protein, Salp15, binds to OspC both in vitro and in vivo to protect Lyme spirochetes from antibody-mediated killing [36,37,38,39]. This is accomplished by Salp15 preventing the formation of the MAC through the inhibition of the deposition of terminal C5b-9 [40]. The expression of Salp15 was selectively enhanced in the salivary glands during transmission [41].
Salp15 homologs have been identified in other Ixodes species that are known vectors for Lyme disease [40,41]. Salp15 from I. persulcatus is capable of binding to OspC to protect spirochetes from antibody-mediated killing and phagocytosis. The Salp15 found in I. ricinus offers significantly stronger protection from complement-mediated killing from human serum compared with I. scapularis [41].
I. scapularis anti-complement protein (Isac), Salp20, and I. ricinus anti-complement proteins I and II are all part of a homologous protein family called the Isac-like protein (ILP) family, that work to inhibit the complement AP through the dissociation of the C3 convertase components, C3b, and cleaved factor B [42].
Salp20 inhibits the AP by binding and dissociating C3BbP, the active C3 convertase [43,44]. Inhibition of the complement system by Salp20 in murine models was accomplished at concentrations as low as 5 μg [44]; this is significant as the upper limit for any given protein in tick saliva is estimated to be 10 μg [45]. Salp20 prevents the cleavage of C3 into C3a and C3b, thus preventing the deposition of C3b to pathogen surfaces for opsonization and dissociates factor B (fB) from the covalently bound C3b, disrupting the C3 convertase [44]. This method of complement inhibition is also accomplished by I. scapularis anti-complement protein (Isac), as well as the closely related I. ricinus proteins IRAC I, IRAC II, and IXAC-B1-5 [44,46].
The protein properdin (factor P) binds and stabilizes the complement factor C3bBb and suppresses the activity of FH [47]. Properdin that has bound C3b inhibits the FH cofactor ability, impeding the FH-dependent decay acceleration of C3bBb [47,48,49]. Salp20 displaces properdin from C3bP, which leaves C3 vulnerable to FH-mediated cleavage. Salp20 can also displace properdin from C3bBbP, leaving C3bBb vulnerable to FH-mediated decay [44,49].
The Tick Salivary Lectin Pathway Inhibitor (TSLPI) is a dominant complement inhibitor in tick saliva [50]. TSLPI reduces complement-mediated killing and interferes with the complement LP cascade by interfering with the mannose-binding lectin (MBL)-dependent C4 activation [51]. This interference results in impaired neutrophil phagocytosis and chemotaxis and diminished lysis of Borrelia [52].
The infection of I. scapularis nymphs with B. burgdorferi s.s. resulted in a higher expression level of TSLPI mRNA after 24 h of tick attachment compared to uninfected ticks [51]. In I. ricinus ticks, an ortholog of TSLPI is upregulated during tick feeding but was not present in unfed ticks, similar to the ortholog found in I. scapularis ticks [50,51].
2.2. Antimicrobial Protein and Peptide Resistance
The host innate immune system produces antimicrobial proteins and peptides in response to pathogens. B. burgdorferi has demonstrated resistance to antimicrobial proteins lactoferrin, azurocidin, and proteinase 3, as well as a limited susceptibility to lysosomes [53]. The resistance to lactoferrin, an iron-binding and transport protein, is due, in part, to the fact that B. burgdorferi does not require iron [54].
B. burgdorferi is also highly resistant to the antimicrobial peptide cathelicidin [55]. The antimicrobial resistance of B. burgdorferi is thought to be due to the lack of lipopolysaccharide (LPS) in the outer membrane. LPS are typically found in Gram-negative bacteria, where cationic peptides, such as cathelicidins, can bind to the molecule [55,56]. Salp15 also works to inhibit cathelicidin, as well as human defensins (hBD-2 and hBD-3), and psoriasin [56].
In addition to resistance to antimicrobial peptides, B. burgdorferi expresses the surface protein BBA57, which was found to decrease the transcription of antimicrobial peptides. A recent study found that BBA57 decreased the expression of antimicrobial peptides (AMP), bactericidal/permeability-increasing protein (Bpi), lactotransferrin, and secretory leukocyte proteinase inhibitor (Slpi). This protein is conserved within the B. burgdorferi s.l. and lacks homology with other proteins of known function. BBA57 was found to be critical for the early stages of infection, but not for later-stage persistence. The exact mechanism of the suppression of AMP is unknown, but it is thought to be mediated by OspC [57].
2.3. Macrophage Interference
Borrelia can cause an increase in the production of IL-10, an anti-inflammatory interleukin [58,59,60]. Macrophages and dendritic cells are major producers of IL-10 and are known to downregulate immune mechanisms in the presence of IL-10 [58,61]. IL-10 suppresses the secretion of proinflammatory cytokines TNF, IL-6, and IL-12 produced by macrophages and dendritic cells in the presence of B. burgdorferi [58]. IL-10 production also leads to suppression of phagocytosis by macrophages and a decrease in the production of proinflammatory mediators and co-stimulatory molecules in antigen-presenting cells (APCs) [58]. Studies performed on IL-10-/- mice found that a lack of IL-10 resulted in a 10-fold greater clearance of B. burgdorferi, indicating the importance of IL-10 production to the evasion of immune clearance [59].
Macrophages and dendric cells act as APCs and activate the T cell response to pathogens through antigen presentation on MHC II molecules, providing a bridge between innate and adaptive immunity [58,62]. During Borrelia infection, dendritic cells downregulate co-stimulatory receptors, except for CD86, which was found to be upregulated. It is thought that the reduced response of co-stimulatory receptors is due to IL-10 [58].
2.4. Disabling of Chemokines and Alarmin Molecules
In addition to disabling the complement system, tick saliva also contains a chemokine-inhibitory evasin protein. Chemokine proteins are responsible for the recruitment of leukocytes to the site of infection, where they bind to the chemokine receptors and direct leukocytes to the site of the tick bite [63]. Evasins are widely expressed within the Ixodidae family, are found in tick saliva, and reduce the migration of immune cells to the site of infection by inhibiting the recruitment of leukocytes [63]. Evasins inhibit the binding of chemokines to glycosaminoglycans, inhibiting chemokine activity [63,64]. Tick saliva proteins inhibit chemokines CCL2, CCL3, CCL5, and CCL11 [64]. (For a comprehensive review of evasin classification and activity, see Bhusal et al. 2020 [65].
In addition to their antimicrobial, enzymatic, or chromatin-binding functions, AMPs can act as “alarmin molecules” to initiate migration and activation of APCs [66]. Specific motifs from conserved bacterial structures, known as pathogen-associated molecular patterns (PAMPs), stimulate antigen-presenting cells (APCs) through Toll-like receptors (TLR) [67,68]. Activation of TLRs results in the differential expression of chemokines and cytokines [67,68].
Salp15 was found to inhibit the mRNA expression of chemokines, monocyte chemoattractant protein 1 (MCP-1), and IL-8, as well as the expression of the alarmin molecules hBD-2, hBD-3, RNase 7, and psoriasin [56]. The inhibition of chemokines and alarmin molecules and the subsequent migration of leukocytes and APCs to the site of infection allows for B. burgdorferi transmission, multiplication, and dissemination.
2.5. Neutralize Reactive Oxygen Species
At the site of infection, neutrophils release what is known as an “oxidative burst” of reactive oxygen species (ROS) to combat infection. The production of ROS and reactive nitrogen species (RNS) is essential to the destruction of bacterial pathogens. ROS consist of superoxide radicals (O−2), hydrogen peroxide (H2O2), and hydroxyl radicals (OH−), and RNS consists of nitric oxide (NO), dinitrogen trioxide (N2O3), nitrogen dioxide (NO2), and peroxynitrite (NO3−) ROS can oxidize cysteinyl residues, iron–sulfur clusters, DNA, polyunsaturated lipids, proteins, and cellular membranes [69].
ROS are most damaging due to the Fenton reaction, where H2O2 and Fe2+ interact to produce OH. Fe2+ is found along the phosphodiester backbone of DNA, and the OH produced from the Fenton reaction can react with deoxyribose and damage DNA [69]. B. burgdorferi was found to be resistant to ROS-mediated DNA damage when compared to other bacterial pathogens. B. burgdorferi is iron independent, encoding few genes of known iron-containing proteins, and does not require iron for growth [54]. A study by Chung et al. found that IL-10, which is increased in production during B. burgdorferi infection, significantly reduces the production of both ROS and NO by macrophages, providing further protection for the pathogen [58].
The lipid membrane of B. burgdorferi was found to be a target of damage by ROS. Free radicals attack the polyunsaturated fatty acids in the cell membranes and initiate lipid peroxidation. This results in a decrease in membrane fluidity, altering the physical properties of the membrane. Oxidation only occurs with certain lipids, such as linoleic and linolenic acid, which B. burgdorferi scavenges from its surroundings and incorporates into its membrane. Around 10% of the total lipid concentration in B. burgdorferi membranes was found to be made up of linoleic acid, while linolenic acid had a concentration of around 1% of the membrane [70].
In order to mitigate cellular damage caused by ROS, bacteria require antioxidant defenses. One of the most important are enzymes known as superoxide dismutases (SODs) that are used to break down superoxides. Borrelia utilizes the manganese-dependent (Mn) SOD SodA, an essential virulence factor with clear contributions to mammalian infection [71]. It is unclear if the role of SodA is to detoxify ROS produced by the innate immune response or endogenous ROS produced by the organism itself. High levels of manganese are required to activate SodA. The limitation of the bioavailability of manganese by macrophages and neutrophils to, B. burgdorferi is also ineffective, as B. burgdorferi are still capable of accumulating enough manganese to activate SodA [72]. The metal transporter A (BmtA) is responsible for B. burgdorferi’s uptake of manganese and is essential for the infection of mammals [73]. B. burgdorferi Mn-SOD protects intracellular targets and not the membrane from ROS damage [54].
While B. burgdorferi is more resistant to ROS and RNSs than other pathogenic bacteria, it is still susceptible to damage by H2O2 [70].
2.6. Pleomorphic Forms
B. burgdorferi s.l. is a pleomorphic bacterium, and therefore is capable of changing its morphology based upon varying environmental conditions. Beyond the most common spirochete conformation, B. burgdorferi can exist as round bodies (RBs) or in a biofilm-like (BFL) aggregation [74].
B. burgdorferi have been seen to change conformation from spirochetes to the spherical RBs during harsh conditions in vitro [74]. In vivo observations of RBs have been noted in few clinical studies, as well [75,76]. A study by Meriläinen et al., found that Borrelia which had entered the RBs conformation were capable of reverting back to viable and motile spirochetes. RBs Borrelia were found to have reduced levels of metabolic activity, though reverted spirochetes were noted to have normal metabolic levels [74].
RBs formation has been thought to enhance the survival of the bacteria in poor environmental conditions and the evasion of the immune system [77,78,79,80]. The lower metabolic activity of RBs may aid in the survival of the bacteria during antibiotic treatments, though RBs could only withstand exposure to harsh environments for short spans of time [74]. B. burgdorferi may have additional strategies to evade antibiotic treatment, including antibiotic tolerance, although the mechanisms remain undefined [80,81,82,83,84].
Biofilms are a complex aggregate of microorganisms that bacteria and other microorganisms use to protect themselves from the hostile host environment [85]. In response to the extreme environment, the bacteria secrete extracellular polymeric substances (EPS) to act as a shield against stressors [85,86]. Biofilm production may help Borrelia to survive in extreme environmental conditions, such as non-physiologic pH, extreme temperature, high concentration of metals, the addition of xenobiotics, or antimicrobials [86,87,88]. It has been hypothesized that the aggregation of B. burgdorferi may aid in the binding of the bacteria to host tissues and may allow the bacteria to avoid phagocytosis [89].
B. burgdorferi biofilm-like growth in suspension and on surfaces been observed in many studies [88,89,90]. Though the spirochete conformation is the most commonly observed, BFL aggregations are observed at relatively low concentrations [74]. Meriläinen et al. found that BFL colonies were a part of B. burgdorferi’s normal in vitro growth and that the colonies formed before the bacteria reached the exponential growth phase [74]. The aggregation of Borrelia occurs preferentially in conditions with high temperatures, low pH, and high cell density in vitro [89].
Borrelia aggregates are comprised of extracellular polysaccharides, similar to that of other microorganism biofilms [85,86]. Borrelia biofilms also have channels, similar to that produced by Leptospira spp., that provide oxygen and nutrients to the aggregates, as well as the removal of waste [86].
Sapi et al. 2016 was the first study to show the presence of a Borrelia biofilm in human skin tissue. The study found that B. burgdorferi s.s. and s.l. have specific surface biofilm markers, like alginate, a biofilm marker found in other pathogenic bacteria [90]. The extracellular polysaccharides found in biofilms play an essential role in the protection of pathogens, immune evasion, and antibiotic resistance [90]. Borrelia exhibit a preference for collagen and fibronectin surfaces for biofilm production [90].
Suspension biofilms found in normal cultures were found by Meriläinen et al. to produce proteins in an extracellular polymeric substance (EPS) matrix, the most notable of which was collagen. It was reasoned that the presence of collagen in the EPS may promote binding of the suspended biofilm to the host tissue [74].
However, the presence of B. burgdorferi biofilms or pleomorphic forms in vivo is controversial. There is still much more research that needs to be performed in order to elucidate the potential roles of RBs and biofilm formation in B. burgdorferi persistence and the evasion of the immune system.
2.7. Intracellular Localization
Borrelia burgdorferi is capable of hiding from the immune system in vitro by invaginating itself through binding to fibrocytic cells [91]. Borrelia can also enter endothelial cells and macrophages. While intracellular localization of B. burgdorferi appears to be a rare occurrence, the ability of Borrelia to be internalized and survive within host macrophages could represent a potential reservoir for chronic or reoccurring Lyme disease [92].
Studies have shown that Borrelia spirochetes are capable of intracellular localization. It was found that actin-containing microfilaments were required for intracellular localization and that the host cell is a participant in the process [93].
A study conducted by Wu et al. 2011 found that intracellular localization of B. burgdorferi into mammalian cells led to brief protection from antibiotic killing. Borrelia were shown to invade several non-phagocytic cells, such as endothelial cells, fibroblasts, neuronal and neuroglial cells. B. burgdorferi that did not synthesize the 1 integrin subunit had a reduced capability of invading fibroblasts, indicating a necessity for 1 integrin for borrelial invasion [94]. The ability for B. burgdorferi to form long-term co-cultures with primary human fibroblasts supports the concept of intracellular localization aiding Borrelia in immune evasion.
3. Adaptive Response
3.1. The Humoral Response
The humoral response is one of the main mechanisms by which the adaptive immune response operates. The humoral response works to protect the extracellular spaces of the host, through the production of antigens from activated B cells in the lymph nodes [95]. During early infection, Borrelia target the lymph nodes where they continue to occupy the lymphoid tissue for the duration of infection. Once there, B. burgdorferi causes rapid B cell proliferation, leading to the enlargement of the lymph nodes, structural damage, and the deterioration of the T and B cell zones in mice [96,97,98].
B cells are the primary adaptive response for the clearing of B. burgdorferi infections in mice, as B cell deficiencies were found to lead to a more severe illness [99]. The increased cell accumulation in the lymph nodes was due to CD19+ B cells, of which some produced antibodies specific for B. burgdorferi. There was a lack of CD4+ T cell accumulation in the lymph nodes [97]. A study by Elsner et al. showed that despite their induction, the CD4+ T cells and T follicular helper cells did not function properly and led to rapid B cell proliferation but not differentiation in vivo [100]. The B cells packed in the lymph nodes lacked the typical follicular arrangement and the CD4+ T helper cells were scattered and did not preside inside discernable T cell zones [97].
A study by Hastey et al. indicates that B. burgdorferi infection induces type I IFN signaling that increases the proliferation of B cells in draining lymph nodes and aids in the disruption of the lymph node architecture [101].
The invasion of the lymphoid tissue disrupts the ability of the immune response to form functioning germinal centers (GC). GC form in the secondary lymphoid tissue during infection and are required for the formation of long-lived plasma cells to aid the immune response by continuously secreting antibodies and inducing memory B cells [102].
During infection with B. burgdorferi, GC formed in the first two weeks of infection, but they were short-lived and rapidly dissipated over the following two weeks [96,97]. The GC demonstrated changes in structure and was incapable of inducing memory B cells and long-living plasma cells for several months post-infection. Mice that were co-administered a vaccine at the time of infection with B. burgdorferi failed to produce antibodies to the vaccine antigen. The temporary immunosuppression leaves the host open for reinfection with the same strain of B. burgdorferi, especially if the infection was treated with antibiotics. This is also seen in endemic areas, where reinfection with Lyme disease is common [96].
Tick saliva also inhibits the production of antibodies by plasma cells, though this occurs only at the site of infection and has no effect on the formation of memory B cells [103,104]. Salp15 from I. scapularis was found to have an immunomodulatory effect through the inhibition of CD4+ T cell activation and the production of IL-2 in a dose-dependent manner in vivo [105]. The binding of Salp15 to CD4+ T cells is persistent and produces a long-lasting immunomodulatory effect, resulting in a reduction in the production of cross-antigenic antibodies [106].
T follicular helper cells (THD) and follicular dendritic cells (FDC) are also crucial to the function of GC [107]. TFH cells were found to rise in numbers in the GC during B. burgdorferi infection, but rapidly declined to pre-infection numbers after 45 days. The FDC of some GC failed to correctly position themselves opposite of the T cell zone. This disruption of the FDC network could inhibit the ability of the GC to properly function [96].
The presence of Borrelia may result in a lower deposition of the complement component C4 onto FDC. As mentioned earlier, the protein BBK32 on the surface of the spirochete inhibits the classical pathway and prevents the formation of C4. It is possible that the lack of antigen-C4 deposition reduces the antigen presentation by the FDC to the GC B cells, which would cause a premature collapse of the GC [96].
B. burgdorferi induces a T cell-independent B cell response [99]. A study by Elsner et al. found that there was a failure to produce long-lived antibodies to T cell-dependent Borrelial antigens. T cell-independent antigens produce short-lived antibodies that last only as long as the infection, while T cell-dependent antibodies, which are generated by the GC, produce a long-lived response [100].
Borrelia infection results in a failure of the B cells to undergo a class switch recombination of IgM to IgG antibodies [97]. Several studies have shown that the number of IgM-secreting cells exceeds that of IgG-secreting cells in both mouse models and human patients [96,108,109]. The B cell response produces unusually strong and persistent IgM antibodies that are both T cell dependent and T cell independent. The failure to produce a strong T cell-dependent response or long-lived germinal centers leads to a humoral response that is dominated by IgM-secreting B cells in both the lymph nodes and the bone marrow [97]. IgG-secreting cells did accumulate slowly in the bone marrow, but it was insufficient to clear the infection, and production ceased around the same time as the collapse of the GC [97,100]. The animal data are bolstered by clinical evidence for elevated and long-lasting IgM responses in human patients with persistent symptoms and/or late manifestations of Lyme borreliosis [109,110,111,112,113].
IgM may work to clear Borrelia from the blood, but due to its large size, it could fail to reach and clear an infection in the skin. The large and sustained IgM production is indicative of a failure of the B cells to undergo a class switch recombination to an IgG antibody production. This could affect the ability of the host immune response to clear infection in tissues unreachable by IgM antibodies. It is thought to be most likely that the cause for the high IgM production is a failure of the B cell response to undergo a class switch due to interference from B. burgdorferi [97].
3.2. Antigenic Variation
Antigenic variation in Lyme Borrelia is an extensively researched immune evasion tactic. This section aims to provide only a brief overview of the subject; for a more thorough review, please refer to [114,115,116].
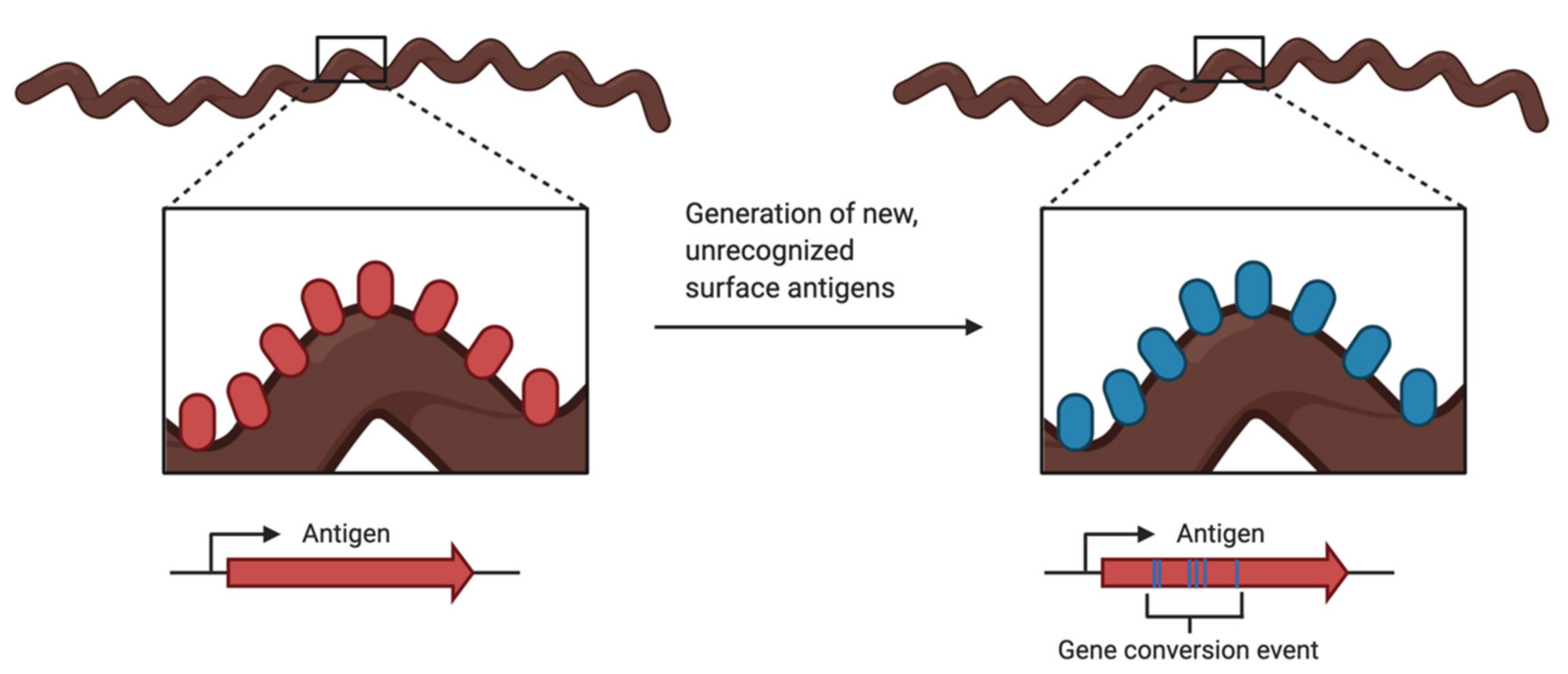
Antigenic variation (Figure 3) is a common evasion tactic employed by pathogens such as bacteria, protozoans, and fungi. While the host’s adaptive immune response works to produce the antigen-specific antibodies to clear infections, the pathogen has created a new variation of the antigen and is now unrecognizable by the antibodies being produced by the host. This variation of the surface antigen is generated by recombination events that produce altered versions of the proteins, by changes in the allele expression, or both [115].
In Lyme Borrelia, the vls locus is the site of recombination for antigenic variation. The vls locus is located on linear plasmid 28-1 (lp28-1) for the B. burgdorferi strain B31. The expression locus, vlsE, encodes for the outer surface lipoprotein. The vlsE gene is located near a hairpin telomere and has 15 silent cassettes located adjacent to and upstream of vlsE going in the opposite orientation [116]. The expressed protein, VlsE, is a surface-bound lipoprotein that is continuously modified as the gene goes through segmental gene conversion events with the silent cassettes [114]. Please see Chaconas et al. 2020, Figure 2, for a detailed visual of the vls locus and recombination process [114].
The locus is the most evolutionary diverse gene in the pathogen [114]. Lyme Borrelia genomes typically show a high degree of conservation, for example, the RecA protein from nine different Borrelia species show 95% sequence identity. However, comparisons of VlsE in B. burgdorferi strains B31 and 297 showed only 46% identity, while B31 and three other Lyme species share only 35–49% [116].
The vlsE gene is essential for the initial and persistent infection of Lyme, as the absence of lp28-1 resulted in low infectivity of B. burgdorferi [117,118,119]. The variation of vlsE has also been found to be required for reinfection and is an advantage for the enzootic cycle [120,121,122]. Three-dimensional modeling was used to determine that amino acid changes that resulting from recombination events with the silent cassettes to the vlsE are exposed and accessible to antibodies [123].
B31 has an inverted repeat (IR) upstream of vlsE, in the prompter region, that is 100 bp in length [123]. Under conditions that cause negative supercoiling, such as replication or transcription, the IR can take on a cruciform structure [114,124]. In this event, the cruciform could act as a flag for recognition of proteins involved in the recombination events, as it is a distinct marker not found in the silent cassettes [114]. There are proteins that use cruciform as recognition sites for replication, recombination, and repair. In addition, cruciform can be sites for the introduction of double or single-stranded breaks [125,126]. The exact role of the IR has yet to be demonstrated, but it is thought that it could play a role in vlsE transcription or recombination [114].
The locus has several conserved regions, despite its ability to constantly change the genetic code at this location. These conserved regions include the telomeric location of the locus, the inverse orientation of vlsE and the silent cassettes, the inverted repeat consisting of 100 bp near the 5′ end of vlsE, and the high concentrations of G runs in vlsE and the silent cassettes [114]. There may also be well conserved direct repeats (DR) present that are 17 bp in length and flank the cassettes and variable region of vlsE [114,126].
The DR found in B31 contains G-runs that form intermolecular G-quadruplex structures in vitro, though it is not known whether the G-quadruplex can be formed in vivo. A G-quadruplex, also known as G4 or 4-stranded DNA, is stabilized by Hoogsteen hydrogen bonding of the bases. The G-quadruplex could play a role in the gene conversion events that occur at the vlsE. The vls locus is rich in G-C nucleotides, tacking up 48% of the locus. This is in comparison to a very A-T-rich genome, with only 29.75% of the genome comprised of G-C pairs. The frequency of G-runs is also much higher than to be expected [116]. This was observed in several different B. burgdorferi strains. The G-runs are not frequently found on either the coding or non-coding strands of the non-vls DNA on the plasmid that carries the vls locus. The G-runs are found in high numbers on the coding strand. It is believed that there is an essential function that the G-C-rich content and the G-runs serve due to its preservation [114].
G4 DNA is an inhibitor of DNA replication and can show specificity for either the leading or the lagging strand [127,128]. G4 may facilitate interactions and synapsis from distant locations, or the non-specific formation of G4 could provide sites of stalled DNA replication, where recombination is prevalent [129,130,131].
The mechanism for the recombination events is still largely unknown, but it has been shown that the RuvAB Holliday junction branch migrase is required. The cis location of the vlsE and the silent cassettes, as well as the high G-C content and the GC skew, may be another requirement for recombination [116].
4. Conclusions
B. burgdorferi utilizes several different methods to evade the host immune response. Borrelia disable the complement system through the regulation of outer surface proteins, the binding of complement regulators, and the use of tick salivary proteins. The innate immune response signaling through chemokines and alarmin molecules is disrupted through the use of tick salivary proteins, preventing the migration of immune cells to the site of infection, allowing B. burgdorferi to establish infection. Resistance to antimicrobial proteins and ROS-mediated killing, as well as the disabling of macrophages, prevents the removal of B. burgdorferi spirochetes from the host. Pleomorphism and intracellular localization may also play a role.
The adaptive immune response is disabled through the invasion of the lymph nodes by Borrelia, and the resulting collapse of the germinal center, the lack of memory cell production, and antibody class switching. B. burgdorferi also evades detection through the antigenic variation of the outer surface protein VlsE.
Though research in recent decades has shed light on the mechanisms by which B. burgdorferi evades the immune response, there is still much to be learned. More research is needed to elucidate the exact mechanisms that B. burgdorferi uses to interact with and evade the different parts of the immune response. A better understanding of how B. burgdorferi subverts the host immune response is important to the development of novel treatments and preventative measures.
Author Contributions
Conceptualization, C.A. and C.A.B.; writing—original draft preparation, C.A.; writing—review and editing, C.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institutes of Health, grant number P20GM113123-01.
Acknowledgments
The authors thank Yvonne Tourand for reading and critical review of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Steere, A.C.; Malawista, S.E.; Snydman, D.R.; Shope, R.E.; Andiman, W.A.; Ross, M.R.; Steele, F.M. An epidemic of oligoarticular arthritis in children and adults in three connecticut communities. Arthritis Rheum. 1977, 20, 7–17. [Google Scholar] [CrossRef]
- Elbaum-Garfinkle, S. Close to home: A history of yale and lyme disease. Yale J. Biol. Med. 2011, 84, 103–108. [Google Scholar] [PubMed]
- Steere, A.C.; Strle, F.; Wormser, G.P.; Hu, L.T.; Branda, J.A.; Hovius, J.W.R.; Li, X.; Mead, P.S. Lyme borreliosis. Nat. Rev. Dis. Prim. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Cerar, T.; Strle, F.; Stupica, D.; Ruzic-Sabljic, E.; McHugh, G.; Steere, A.C.; Strle, K. Differences in genotype, clinical features, and inflammatory potential of borrelia burgdorferi sensu stricto strains from Europe and the United States. Emerg. Infect. Dis. 2016, 22, 818–827. [Google Scholar] [CrossRef] [Green Version]
- Stanek, G.; Wormser, G.P.; Gray, J.; Strle, F. Lyme borreliosis. Lancet 2012, 379, 461–473. [Google Scholar] [CrossRef]
- Sarma, J.V.; Ward, P.A. The complement system. Cell Tissue Res. 2011, 343, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.W. Complement evasion in Borrelia spirochetes: Mechanisms and opportunities for intervention. Antibiotics 2019, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Ebady, R.; Niddam, A.F.; Boczula, A.E.; Kim, Y.R.; Gupta, N.; Tang, T.T.; Odisho, T.; Zhi, H.; Simmons, C.A.; Skare, J.T.; et al. Biomechanics of Borrelia burgdorferi Vascular Interactions. Cell Rep. 2016, 16, 2593–2604. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.R.; LeBlanc, K.T.; Leong, J.M. Fibronectin binding protein BBK32 of the Lyme disease spirochete promotes bacterial attachment to glycosaminoglycans. Infect. Immun. 2006, 74, 435–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, B.L.; Zhi, H.; Wager, B.; Höök, M.; Skare, J.T. Borrelia burgdorferi BBK32 Inhibits the Classical Pathway by Blocking Activation of the C1 Complement Complex. PLoS Pathog. 2016, 12, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Barthel, D.; Schindler, S.; Zipfel, P.F. Plasminogen is a complement inhibitor. J. Biol. Chem. 2012, 287, 18831–18842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, H.; Wallich, R.; Simon, M.M.; Kramer, M.D. The outer surface protein A of the spirochete Borrelia burgdorferi is a plasmin(ogen) receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 12594–12598. [Google Scholar] [CrossRef] [Green Version]
- Önder, Ö.; Humphrey, P.T.; McOmber, B.; Korobova, F.; Francella, N.; Greenbaum, D.C.; Brisson, D. OspC is potent plasminogen receptor on surface of borrelia burgdorferi. J. Biol. Chem. 2012, 287, 16860–16868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caine, J.A.; Lin, Y.P.; Kessler, J.R.; Sato, H.; Leong, J.M.; Coburn, J. Borrelia burgdorferi outer surface protein C (OspC) binds complement component C4b and confers bloodstream survival. Cell. Microbiol. 2017, 19, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Koenigs, A.; Hammerschmidt, C.; Jutras, B.L.; Pogoryelov, D.; Barthel, D.; Skerka, C.; Kugelstadt, D.; Wallich, R.; Stevenson, B.; Zipfel, P.F.; et al. BBA70 of borrelia burgdorferi is a novel plasminogen-binding protein. J. Biol. Chem. 2013, 288, 25229–25243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallström, T.; Haupt, K.; Kraiczy, P.; Hortschansky, P.; Wallich, R.; Skerka, C.; Zipfel, P.F. Complement Regulator–Acquiring Surface Protein 1 of Borrelia burgdorferi Binds to Human Bone Morphogenic Protein 2, Several Extracellular Matrix Proteins, and Plasminogen. J. Infect. Dis. 2010, 202, 490–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraiczy, P.; Hellwage, J.; Skerka, C.; Becker, H.; Kirschfink, M.; Simon, M.M.; Brade, V.; Zipfel, P.F.; Wallich, R. Complement Resistance of Borrelia burgdorferi Correlates with the Expression of BbCRASP-1, a Novel Linear Plasmid-encoded Surface Protein That Interacts with Human Factor H and FHL-1 and Is Unrelated to Erp Proteins. J. Biol. Chem. 2004, 279, 2421–2429. [Google Scholar] [CrossRef] [Green Version]
- Kraiczy, P.; Skerka, C.; Kirschfink, M.; Brade, V. Further Characterization of Complement Regulator-Acquiring Surface Proteins of. Society 2001, 69, 7800–7809. [Google Scholar] [CrossRef]
- Hallström, T.; Siegel, C.; Mörgelin, M.; Kraiczy, P.; Skerka, C.; Zipfel, P.F. CspA from Borrelia burgdorferi inhibits the terminal complement pathway. MBio 2013, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hart, T.; Nguyen, N.T.T.; Nowak, N.A.; Zhang, F.; Linhardt, R.J.; Diuk-Wasser, M.; Ram, S.; Kraiczy, P.; Lin, Y.P. Polymorphic Factor H-Binding Activity of CspA Protects Lyme Borreliae from the Host Complement in Feeding Ticks to Facilitate Tick-to-Host Transmission. PLoS Pathog. 2018, 14, 21007106. [Google Scholar] [CrossRef] [Green Version]
- Kenedy, M.R.; Vuppala, S.R.; Siegel, C.; Kraiczy, P.; Akins, D.R. CspA-mediated binding of human factor H inhibits complement deposition and confers serum resistance in Borrelia burgdorferi. Infect. Immun. 2009, 77, 2773–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brissette, C.A.; Haupt, K.; Barthel, D.; Cooley, A.E.; Bowman, A.; Skerka, C.; Wallich, R.; Zipfel, P.F.; Kraiczy, P.; Stevenson, B. Borrelia burgdorferi infection-associated surface proteins ErpP, ErpA, and ErpC bind human plasminogen. Infect. Immun. 2009, 77, 300–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerschmidt, C.; Klevenhaus, Y.; Koenigs, A.; Hallström, T.; Fingerle, V.; Skerka, C.; Pos, K.M.; Zipfel, P.F.; Wallich, R.; Kraiczy, P. BGA66 and BGA71 facilitate complement resistance of Borrelia bavariensis by inhibiting assembly of the membrane attack complex. Mol. Microbiol. 2016, 99, 407–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pausa, M.; Pellis, V.; Cinco, M.; Giulianini, P.G.; Presani, G.; Perticarari, S.; Murgia, R.; Tedesco, F. Serum-Resistant Strains of Borrelia burgdorferi Evade Complement-Mediated Killing by Expressing a CD59-Like Complement Inhibitory Molecule. J. Immunol. 2003, 170, 3214–3222. [Google Scholar] [CrossRef] [Green Version]
- Józsi, M.; Tortajada, A.; Uzonyi, B.; Goicoechea de Jorge, E.; Rodríguez de Córdoba, S. Factor H-related proteins determine complement-activating surfaces. Trends Immunol. 2015, 36, 374–384. [Google Scholar] [CrossRef] [Green Version]
- Kraiczy, P.; Stevenson, B. Complement regulator-acquiring surface proteins of Borrelia burgdorferi: Structure, function and regulation of gene expression. Ticks Tick. Borne Dis. 2013, 4, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, A.S.; Yang, X.; Kumar, M.; Zhang, X.; Promnares, K.; Shroder, D.; Kenedy, M.R.; Anderson, J.F.; Akins, D.R.; Pal, U. Borrelia burgdorferi Complement Regulator-Acquiring Surface Protein 2 does not contribute to complement resistance or host infectivity. PLoS ONE 2008, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bykowski, T.; Woodman, M.E.; Cooley, A.E.; Brissette, C.A.; Brade, V.; Wallich, R.; Kraiczy, P.; Stevenson, B. Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the lyme disease spirochete’s mammal-tick infection cycle. Infect. Immun. 2007, 75, 4227–4236. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-P.; Frye, A.M.; Nowak, T.A.; Kraiczy, P. New Insights Into CRASP-Mediated Complement Evasion in the Lyme Disease Enzootic Cycle. Front. Cell. Infect. Microbiol. 2020, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Hefty, P.S.; Jolliff, S.E.; Caimano, M.J.; Wikel, S.K.; Radolf, J.D.; Akins, D.R. Regulation of OspE-related, OspF-related, and Elp lipoproteins of Borrelia burgdorferi strain 297 by mammalian host-specific signals. Infect. Immun. 2001, 69, 3618–3627. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.C.; Von Lackum, K.; Babb, K.; McAlister, J.D.; Stevenson, B. Temporal Analysis of Borrelia burgdorferi Erp Protein Expression throughout the Mammal-Tick Infectious Cycle. Infect. Immun. 2003, 71, 6943–6952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.C.; Narayan, K.; Stevenson, B.; Pachner, A.R. Expression of Borrelia burgdorferi erp genes during infection of non-human primates. Microb. Pathog. 2005, 39, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Hellwage, J.; Meri, T.; Heikkilä, T.; Alitalo, A.; Panelius, J.; Lahdenne, P.; Seppälä, I.J.T.; Meri, S. The Complement Regulator Factor H Binds to the Surface Protein OspE of Borrelia burgdorferi. J. Biol. Chem. 2001, 276, 8427–8435. [Google Scholar] [CrossRef] [Green Version]
- Zipfel, P.F.; Skerka, C.; Hellwage, J.; Jokiranta, S.T.; Meri, S.; Brade, V.; Kraiczy, P.; Noris, M.; Remuzzi, G. Structure-function studies of the complement system. Biochem. Soc. Trans. 2002, 30, 971–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, C.; Hallström, T.; Skerka, C.; Eberhardt, H.; Uzonyi, B.; Beckhaus, T.; Karas, M.; Wallich, R.; Stevenson, B.; Zipfel, P.F.; et al. Complement factor H-related proteins CFHR2 and CFHR5 represent novel ligands for the infection-associated CRASP proteins of Borrelia burgdorferi. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Nuttall, P.A. Tick saliva and its role in pathogen transmission. Wien. Klin. Wochenschr. 2019. [Google Scholar] [CrossRef] [Green Version]
- Schwan, T.G.; Piesman, J.; Golde, W.T.; Dolan, M.C.; Rosa, P.A. Induction of an outer surface protein on Borrelia burgdorferi during tick feeding. Proc. Natl. Acad. Sci. USA 1995, 92, 2909–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamoorthi, N.; Narasimhan, S.; Pal, U.; Bao, F.; Yang, X.F.; Fish, D.; Anguita, J.; Norgard, M.V.; Kantor, F.S.; Anderson, J.F.; et al. The Lyme disease agent exploits a tick protein to infect the mammalian host. Nature 2005, 436, 573–577. [Google Scholar] [CrossRef] [Green Version]
- Pal, U.; Yang, X.; Chen, M.; Bockenstedt, L.K.; Anderson, J.F.; Flavell, R.A.; Norgard, M.V.; Fikrig, E. OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J. Clin. Investig. 2004, 113, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Schuijt, T.J.; Hovius, J.W.R.; Van Burgel, N.D.; Ramamoorthi, N.; Fikrig, E.; Van Dam, A.P. The tick salivary protein Salp15 inhibits the killing of serum-sensitive Borrelia burgdorferi sensu lato isolates. Infect. Immun. 2008, 76, 2888–2894. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.; Wang, F.; Ji, Z.; Pan, Y.Y.; Jian, M.; Bi, Y.F.; Zhou, G.; Luo, L.; Chen, T.; Li, L.; et al. Salp15, a Multifunctional Protein From Tick Saliva With Potential Pharmaceutical Effects. Front. Immunol. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Tyson, K.R.; Elkins, C.; de Silva, A.M. A Novel Mechanism of Complement Inhibition Unmasked by a Tick Salivary Protein That Binds to Properdin. J. Immunol. 2008, 180, 3964–3968. [Google Scholar] [CrossRef] [Green Version]
- Tyson, K.; Elkins, C.; Patterson, H.; Fikrig, E.; De Silva, A. Biochemical and functional characterization of Salp20, an Ixodes scapularis tick salivary protein that inhibits the complement pathway. Insect Mol. Biol. 2007, 16, 469–479. [Google Scholar] [CrossRef]
- Hourcade, D.E.; Akk, A.M.; Mitchell, L.M.; Zhou, H.; Hauhart, R.; Pham, C.T.N. Anti-complement activity of the Ixodes scapularis salivary protein Salp20. Mol. Immunol. 2016, 69, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Mans, B.J. Chemical equilibrium at the tick-host feeding interface: A critical examination of biological relevance in hematophagous behavior. Front Physiol. 2019, 10, 530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvreur, B.; Beaufays, J.; Charon, C.; Lahaye, K.; Gensale, F.; Denis, V.; Charloteaux, B.; Decrem, Y.; Prévôt, P.P.; Brossard, M.; et al. Variability and action mechanism of a family of anticomplement proteins in ixodes ricinus. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [Green Version]
- Medicus, R.G.; Gotze, O.; Muller-Eberhard, H.J. Alternative pathway of complement: Recruitment of precursor properdin by the labile C3/ C5 convertsade and the potentiation of the pathway. J. Exp. Med. 1976, 144, 1076–1093. [Google Scholar] [CrossRef]
- Farries, T.C.; Lachmann, P.J.; Harrison, R.A. Analysis of the interactions between properdin, the third component of complement (C3), and its physiological activation products. Biochem. J. 1988, 252, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcorlo, M.; Tortajada, A.; De Córdoba, S.R.; Llorca, O. Structural basis for the stabilization of the complement alternative pathway C3 convertase by properdin. Proc. Natl. Acad. Sci. USA 2013, 110, 13504–13509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagemakers, A.; Coumou, J.; Schuijt, T.J.; Oei, A.; Nijhof, A.M.; Van ’T Veer, C.; Van Der Poll, T.; Bins, A.D.; Hovius, J.W.R. An Ixodes ricinus Tick Salivary Lectin Pathway Inhibitor Protects Borrelia burgdorferi sensu lato from Human Complement. Vector-Borne Zoonotic Dis. 2016, 16, 223–228. [Google Scholar] [CrossRef]
- Schuijt, T.J.; Coumou, J.; Narasimhan, S.; Dai, J.; Deponte, K.; Wouters, D.; Brouwer, M.; Oei, A.; Roelofs, J.J.T.H.; Van Dam, A.P.; et al. A tick mannose-binding lectin inhibitor interferes with the vertebrate complement cascade to enhance transmission of the Lyme disease agent. Cell Host Microbe 2011, 10, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Lusitani, D.; Malawista, S.E.; Montgomery, R.R. Borrelia burgdorferi Are Susceptible to Killing by a Variety of Human Polymorphonuclear Leukocyte Components. J. Infect. Dis. 2002, 185, 797–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, A.; Tilly, K.; Stewart, P.; Bestor, A.; Battisti, J.M.; Rosa, P.A. Borrelia burgdorferi resistance to a major skin antimicrobial peptide is independent of outer surface lipoprotein content. Antimicrob. Agents Chemother. 2009, 53, 4490–4494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troxell, B.; Xu, H.; Yang, X.F. Borrelia burgdorferi, a pathogen that lacks iron, encodes manganese-dependent superoxide dismutase essential for resistance to streptonigrin. J. Biol. Chem. 2012, 287, 19284–19293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambri, V.; Marangoni, A.; Giacani, L.; Gennaro, R.; Murgia, R.; Cevenini, R.; Cinco, M. Comparative in vitro activity of five cathelicidin-derived sythetic peptides against Leptospira, Borrelia and Treponema pallidum. J. Antimicrob. Chemother. 2002, 50, 895–902. [Google Scholar] [CrossRef] [Green Version]
- Marchal, C.; Schramm, F.; Kern, A.; Luft, B.J.; Yang, X.; Schuijt, T.; Hovius, J.; Jaulhac, B.; Boulanger, N. Antialarmin effect of tick saliva during the transmission of lyme disease. Infect. Immun. 2011, 79, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Bernard, Q.; Smith, A.A.; Yang, X.; Koci, J.; Foor, S.D.; Cramer, S.D.; Zhuang, X.; Dwyer, J.E.; Lin, Y.P.; Mongodin, E.F.; et al. Plasticity in early immune evasion strategies of a bacterial pathogen. Proc. Natl. Acad. Sci. USA 2018, 115, E3788–E3797. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.; Zhang, N.; Wooten, R.M. Borrelia burgdorferi elicited-IL-10 suppresses the production of inflammatory mediators, phagocytosis, and expression of co-stimulatory receptors by murine macrophages and/or dendritic cells. PLoS ONE 2013, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, J.J.; Meadows, M.J.; Lintner, R.E.; Wooten, R.M. IL-10 Deficiency Promotes Increased Borrelia burgdorferi Clearance Predominantly through Enhanced Innate Immune Responses. J. Immunol. 2006, 177, 7076–7085. [Google Scholar] [CrossRef]
- Brown, J.P.; Zachary, J.F.; Teuscher, C.; Weis, J.J.; Wooten, R.M. Dual role of interleukin-10 in murine lyme disease: Regulation of arthritis severity and host defense. Infect. Immun. 1999, 67, 5142–5150. [Google Scholar] [CrossRef] [Green Version]
- Sabat, R.; Grütz, G.; Warszawska, K.; Kirsch, S.; Witte, E.; Wolk, K.; Geginat, J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010, 21, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Hume, D.A. Macrophages as APC and the Dendritic Cell Myth. J. Immunol. 2008, 181, 5829–5835. [Google Scholar] [CrossRef] [Green Version]
- Hayward, J.; Sanchez, J.; Perry, A.; Huang, C.; Rodriguez Valle, M.; Canals, M.; Payne, R.J.; Stone, M.J. Ticks from diverse genera encode chemokine-inhibitory evasin proteins. J. Biol. Chem. 2017, 292, 15670–15680. [Google Scholar] [CrossRef] [Green Version]
- Crijns, H.; Vanheule, V.; Proost, P. Targeting Chemokine—Glycosaminoglycan Interactions to Inhibit Inflammation. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Bhusal, R.P.; Eaton, J.R.O.; Chowdhury, S.T.; Power, C.A.; Proudfoot, A.E.I.; Stone, M.J.; Bhattacharya, S. Evasins: Tick Salivary Proteins that Inhibit Mammalian Chemokines. Trends Biochem. Sci. 2020, 45, 108–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppenheim, J.J.; Tewary, P.; de la Rosa, G.; Yang, D. Alarmins initiate host defense. Adv. Exp. Med. Biol. 2007, 601, 185–194. [Google Scholar]
- Guerau-De-Arellano, M.; Huber, B.T. Chemokines and Toll-like receptors in Lyme disease pathogenesis. Trends Mol. Med. 2005, 11, 114–120. [Google Scholar] [CrossRef]
- Hirschfeld, M.; Kirschning, C.J.; Schwandner, R.; Wesche, H.; Weis, J.H.; Wooten, R.M.; Weis, J.J. Cutting edge: Inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by toll-like receptor. J. Immunol. 1999, 163, 2382–2386. [Google Scholar]
- Thannickal, V.J.; Fanburg, B.L. Reactive Oxygen Species in Cell Walls. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, L1005–L1028. [Google Scholar] [CrossRef]
- Boylan, J.A.; Lawrence, K.A.; Downey, J.S.; Gherardini, F.C. Borrelia burgdorferi membranes are the primary targets of reactive oxygen species. Mol. Microbiol. 2008, 68, 786–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteve-Gassent, M.D.; Elliot, N.L.; Seshu, J. sodA is essential for virulence of Borrelia burgdorferi in the murine model of Lyme disease. Mol. Microbiol. 2009, 71, 594–612. [Google Scholar] [CrossRef]
- Aguirre, J.D.; Clark, H.M.; McIlvin, M.; Vazquez, C.; Palmere, S.L.; Grab, D.J.; Seshu, J.; Hart, P.J.; Saito, M.; Culotta, V.C. A manganese-rich environment supports superoxide dismutase activity in a lyme disease pathogen, borrelia burgdorferi. J. Biol. Chem. 2013, 288, 8468–8478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, Z.; He, M.; Oman, T.; Yang, X.F.; Norgard, M.V. A manganese transporter, BB0219 (BmtA), is required for virulence by the Lyme disease spirochete, Borrelia burgdorferi. Proc. Natl. Acad. Sci. USA 2009, 106, 3449–3454. [Google Scholar] [CrossRef] [Green Version]
- Meriläinen, L.; Herranen, A.; Schwarzbach, A.; Gilbert, L. Morphological and biochemical features of Borrelia burgdorferi pleomorphic forms. Microbiology 2015, 161, 516–527. [Google Scholar] [CrossRef]
- Aberer, E.; Kersten, A.; Klade, H.; Poitschek, C.; Jurecka, W. Heterogeneity of Borrelia burgdorferi in the skin. Am. J. Dermatopathol. 1996, 18, 571–579. [Google Scholar] [CrossRef]
- Miklossy, J.; Kasas, S.; Zurn, A.D.; McCall, S.; Yu, S.; McGeer, P.L. Persisting atypical and cystic forms of Borrelia burgdorferi and local inflammation in Lyme neuroborreliosis. J. Neuroinflamm. 2008, 5, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Murgia, R.; Cinco, M. Induction of cystic forms by different stress conditions in Borrelia burgdorferi. Apmis 2004, 112, 57–62. [Google Scholar] [CrossRef]
- Al-Robaiy, S.; Dihazi, H.; Kacza, J.; Seeger, J.; Schiller, J.; Huster, D.; Knauer, J.; Straubinger, R.K. Metamorphosis of Borrelia burgdorferi organisms—RNA, lipid and protein composition in context with the spirochetes’ shape. J. Basic Microbiol. 2010, 50, 5–17. [Google Scholar] [CrossRef]
- Brorson, S.H. Transformation of cystic forms of Borrelia burgdorferi to normal, mobile spirochetes. Infection 1997, 25, 240–246. [Google Scholar] [CrossRef]
- Smith, A.J.; Oertle, J.; Prato, D. Chronic Lyme Disease: Persistent Clinical Symptoms Related to Immune Evasion, Antibiotic Resistance and Various Defense Mechanisms of Borrelia burgdorferi. Open J. Med. Microbiol. 2014, 04, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Pothineni, V.R.; Potula, H.S.K.; Ambati, A.; Mallajosyula, V.V.A.; Sridharan, B.; Inayathullah, M.; Ahmed, M.S.; Rajadas, J. Azlocillin can be the potential drug candidate against drug-tolerant Borrelia burgdorferi sensu stricto JLB31. Sci. Rep. 2020, 10, 3798. [Google Scholar] [CrossRef] [Green Version]
- Hodzic, E. Lyme borreliosis: Is there a preexisting (natural) variation in antimicrobial susceptibility among Borrelia burgdorferi strains? Bosn. J. Basic Med. Sci. 2015, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hodzic, E.; Imai, D.M.; Escobar, E. Generality of post-antimicrobial treatment persistence of Borrelia burgdorferi strains N40 and B31 in genetically susceptible and resistant mouse strains. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [Green Version]
- Cabello, F.C.; Godfrey, H.P.; Bugrysheva, J.V.; Newman, S.A. Sleeper cells: The stringent response and persistence in the Borreliella (Borrelia) burgdorferi enzootic cycle. Environ. Microbiol. 2017, 19, 3846–3862. [Google Scholar] [CrossRef] [Green Version]
- Costerton, A.J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial Biofilms: A Common Cause of Persistent Infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [Green Version]
- Sapi, E.; Bastian, S.L.; Mpoy, C.M.; Scott, S.; Rattelle, A.; Pabbati, N.; Poruri, A.; Burugu, D.; Theophilus, P.A.S.; Pham, T.V.; et al. Characterization of Biofilm Formation by Borrelia burgdorferi In Vitro. PLoS ONE 2012, 7, 1–11. [Google Scholar] [CrossRef]
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138. [Google Scholar] [CrossRef]
- Barbour, A.G. Isolation and cultivation of Lyme disease spirochetes. Yale J. Biol. Med. 1984, 57, 521–525. [Google Scholar]
- Srivastava, S.Y.; De Silva, A.M. Characterization of borrelia burgdorferi aggregates. Vector-Borne Zoonotic Dis. 2009, 9, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Sapi, E.; Balasubramanian, K.; Poruri, A.; Maghsoudlou, J.S.; Socarras, K.M.; Timmaraju, A.V.; Filush, K.R.; Gupta, K.; Shaikh, S.; Theophilus, P.A.S.; et al. Evidence of in vivo existence of Borrelia biofilm in borrelial lymphocytomas. Eur. J. Microbiol. Immunol. 2016, 6, 9–24. [Google Scholar] [CrossRef] [Green Version]
- Grab, D.J.; Lanners, H.N.; Martin, L.N.; Chesney, J.; Cai, C.; Adkisson, H.D.; Bucala, R. Interaction of Borrelia burgdorferi with peripheral blood fibrocytes, antigen-presenting cells with the potential for connective tissue targeting. Mol. Med. 1999, 5, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, R.R.; Nathanson, M.H.; Malawista, S.E. The fate of Borrelia burgdorferi, the agent for Lyme disease, in mouse macrophages: Destruction, survival, recovery. J. Immunol. 1993, 150, 909–915. [Google Scholar]
- Ma, Y.; Sturrock, A.; Weis, J.J. Intracellular localization of Borrelia burgdorferi within human endothelial cells. Infect. Immun. 1991, 59, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Weening, E.H.; Faske, J.B.; Höök, M.; Skare, J.T. Invasion of eukaryotic cells by Borrelia burgdorferi requires β1 integrins and Src kinase activity. Infect. Immun. 2011, 79, 1338–1348. [Google Scholar] [CrossRef] [Green Version]
- Janeway, C.J.; Travers, P.; Walport, M.; Shlomchik, M.J. The Humoral Immune Response. In Immunobiology: The Immune System in Healt and Disease; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Elsner, R.A.; Hastey, C.J.; Olsen, K.J.; Baumgarth, N. Suppression of Long-Lived Humoral Immunity Following Borrelia burgdorferi Infection. PLoS Pathog. 2015, 11, 1–19. [Google Scholar] [CrossRef]
- Hastey, C.J.; Elsner, R.A.; Barthold, S.W.; Baumgarth, N. Delays and diversions mark the development of B cell responses to Borrelia burgdoferi infection. J. Immunol. 2012, 188, 5612–5622. [Google Scholar] [CrossRef] [Green Version]
- Tunev, S.S.; Hastey, C.J.; Hodzic, E.; Feng, S.; Barthold, S.W.; Baumgarth, N. Lymphoadenopathy during lyme borreliosis is caused by spirochete migration-induced specific B cell activation. PLoS Pathog. 2011, 7, 20–24. [Google Scholar] [CrossRef] [Green Version]
- McKisic, M.D.; Barthold, S.W. T-cell-independent responses to Borrelia burgdorferi are critical for protective immunity and resolution of lyme disease. Infect. Immun. 2000, 68, 5190–5197. [Google Scholar] [CrossRef] [Green Version]
- Elsner, R.A.; Hastey, C.J.; Baumgarth, N. CD4+ T cells promote antibody production but not sustained affinity maturation during Borrelia burgdorferi infection. Infect. Immun. 2015, 83, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Hastey, C.J.; Ochoa, J.; Olsen, K.J.; Barthold, S.W.; Baumgarth, N. MyD88- and TRIF-independent induction of Type I interferon drives naive B cell accumulation but not loss of lymph node architecture in lyme disease. Infect. Immun. 2014, 82, 1548–1558. [Google Scholar] [CrossRef] [Green Version]
- Good-Jacobson, K.L.; Shlomchik, M.J. Plasticity and Heterogeneity in the Generation of Memory B Cells and Long-Lived Plasma Cells: The Influence of Germinal Center Interactions and Dynamics. J. Immunol. 2010, 185, 3117–3125. [Google Scholar] [CrossRef] [PubMed]
- Menten-Dedoyart, C.; Couvreur, B.; Thellin, O.; Drion, P.V.; Herry, M.; Jolois, O.; Heinen, E. Influence of the Ixodes ricinus tick blood-feeding on the antigen-specific antibody response in vivo. Vaccine 2008, 26, 6956–6964. [Google Scholar] [CrossRef]
- Menten-Dedoyart, C.; Couvreur, B.; Jolois, O.; Van Lerberghe, P.B.; Duwez, L.; Drion, P.; Heinen, E. Kinetic study of the antibody response during the blood meal of Ixodes ricinus: Implication on plasma cell maturation in vivo and for anti-Ixodes vaccination. Vaccine 2011, 29, 2044–2050. [Google Scholar] [CrossRef]
- Anguita, J.; Ramamoorthi, N.; Hovius, J.W.R.; Das, S.; Thomas, V.; Persinski, R.; Conze, D.; Askenase, P.W.; Rincón, M.; Kantor, F.S.; et al. Salp15, an Ixodes scapularis salivary protein, inhibits CD4+ T cell activation. Immunity 2002, 16, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Tomás-Cortázar, J.; Martín-Ruiz, I.; Barriales, D.; Pascual-Itoiz, M.Á.; De Juan, V.G.; Caro-Maldonado, A.; Merino, N.; Marina, A.; Blanco, F.J.; Flores, J.M.; et al. The immunosuppressive effect of the tick protein, Salp15, is long-lasting and persists in a murine model of hematopoietic transplant. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Vinuesa, C.G.; Linterman, M.A.; Goodnow, C.C.; Randall, K.L. T cells and follicular dendritic cells in germinal center B-cell formation and selection. Immunol. Rev. 2010, 237, 72–89. [Google Scholar] [CrossRef]
- Kalish, R.A.; McHugh, G.; Granquist, J.; Shea, B.; Ruthazer, R.; Steere, A.C. Persistence of immunoglobulin M or immunoglobulin G antibody responses to Borrelia burgdorferi 10-20 years after active Lyme disease. Clin. Infect. Dis. 2001, 33, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Craft, J.E.; Grodzicki, R.L.; Shrestha, M.; Fischer, D.K.; García-Blanco, M.; Steere, A.C. The antibody response in Lyme disease. Yale J. Biol. Med. 1984, 57, 561–565. [Google Scholar]
- Hilton, E.; Tramontano, A.; DeVoti, J.; Sood, S.K. Temoral study of immunglobulin M seroreactivity to Borrelia burgdorferi in patients treated for Lyme borreliosis. J. Clin. Microbiol. 1997, 35, 774–776. [Google Scholar] [CrossRef] [Green Version]
- Kalish, R.A.; Kaplan, R.F.; Taylor, E.; Jones-Woodward, L.; Steere, A.C. Evaluation of study patients with Lyme disease, 10-20-year follow-up. J. Infect. Dis. 2001, 183, 453–460. [Google Scholar] [CrossRef]
- Henriksson, A.; Link, H.; Cruz, M.; Stiernstdedt, G. Immunoglobulin abnormalities in cerebrospinal fluid and blood over the course of lymphocytic meningoradiculitis (Bannwarth’s syndrome). Ann. Neurol. 1986, 20, 337–345. [Google Scholar] [CrossRef]
- Hammers-Berggren, S.; Lecech, A.M.; Karlsson, M.; Andersson, U.; Hansen, K.; Stiernstedt, G. Serological follow-up after treatment of Borrelia arthritis and acrodermatitis chronica atrophicans. Scand. J. Infect. Dis. 1994, 26, 339–347. [Google Scholar] [CrossRef]
- Chaconas, G.; Castellanos, M.; Verhey, T.B. Changing of the guard: How the Lyme disease spirochete subverts the host immune response. J. Biol. Chem. 2020, 295, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-R.; Hardham, J.M.; Barbour, A.G.; Norris, S.J. Antigenic VAriation in Lyme Disease Borreliae by Promiscuous Recombination of VMP-like Sequence Cassettes. Cell 1997, 89, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Norris, S.J. vls Antigenic Variation Systems of Lyme Disease Borrelia: Eluding Host Immunity through both Random, Segmental Gene Conversion and Framework Heterogeneity. Microbiol. Spectr. 2015, 2, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Purser, J.E.; Norris, S.J. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. USA 2000, 97, 13865–13870. [Google Scholar] [CrossRef] [Green Version]
- Labandeira-Rey, M.; Skare, J.T. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect. Immun. 2001, 69, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Bankhead, T.; Chaconas, G. The role of VlsE antigenic variation in the Lyme disease spirochete: Persistence through a mechanism that differs from other pathogens. Mol. Microbiol. 2007, 65, 1547–1558. [Google Scholar] [CrossRef]
- Rogovskyy, A.S.; Casselli, T.; Tourand, Y.; Jones, C.R.; Owen, J.P.; Mason, K.L.; Scoles, G.A.; Bankhead, T. Evaluation of the importance of VlsE antigenic variation for the enzootic cycle of Borrelia burgdorferi. PLoS ONE 2015, 10, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankhead, T. Role of the VlsE lipoprotein in immune avoidance by the lyme disease spirochete borrelia burgdorferi. For. Immunopathol. Dis. Ther. 2016, 7, 191–203. [Google Scholar] [CrossRef]
- Eicken, C.; Sharma, V.; Klabunde, T.; Lawrenz, M.B.; Hardham, J.M.; Norris, S.J.; Sacchettini, J.C. Crystal structure of Lyme disease variable surface antigen VlsE of Borrelia burgdorferi. J. Biol. Chem. 2002, 277, 21691–21696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, C.R.; Frye, J.G.; Quinn, F.D.; Gherardini, F.C. Increased expression of Borrelia burgdorferi vlsE in response to human endothelial cell membranes. Mol. Microbiol. 2001, 41, 229–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brázda, V.; Coufal, J.; Liao, J.C.C.; Arrowsmith, C.H. Preferential binding of IFI16 protein to cruciform structure and superhelical DNA. Biochem. Biophys. Res. Commun. 2012, 422, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Bikard, D.; Loot, C.; Baharoglu, Z.; Mazel, D. Folded DNA in Action: Hairpin Formation and Biological Functions in Prokaryotes. Microbiol. Mol. Biol. Rev. 2010, 74, 570–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brázda, V.; Laister, R.C.; Jagelská, E.B.; Arrowsmith, C. Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Walia, R.; Chaconas, G. Suggested Role for G4 DNA in Recombinational Switching at the Antigenic Variation Locus of the Lyme Disease Spirochete. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahan, D.; Tsirkas, I.; Dovrat, D.; Sparks, M.A.; Singh, S.P.; Galletto, R.; Aharoni, A. Pif1 is essential for efficient replisome progression through lagging strand G-quadruplex DNA secondary structures. Nucleic Acids Res. 2018, 46, 11847–11857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, J.; Le Piazza, A.; Bermejo, R.; Kriegsman, B.; Colosio, A.; Teulade-Fichou, M.P.; Foiani, M.; Nicolas, A. G-quadruplex-induced instability during leading-strand replication. EMBO J. 2011, 30, 4033–4046. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Sannino, V.; de Antoni, A.; Técher, H.; Baldi, G.; Costanzo, V. Moonlighting at replication forks—A new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017, 591, 1083–1100. [Google Scholar] [CrossRef]
- Polleys, E.J.; House, N.C.M.; Freudenreich, C.H. Role of recombination and replication fork restart in repeat instability. DNA Repair (Amst.) 2017, 56, 156–165. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Formation of the membrane attack complex formation via the alternative, classical, and lectin pathways. The alternative pathway is activated by the binding of C3b directly to the surface of a microbe. The classical pathway activation is triggered by the presence of antigen–antibody complexes. The lectin pathway is activated by the binding of mannose-binding lectins to mannose-containing liposaccharides on the surface of the pathogen. Created with BioRender.com; https://biorender.com/ (accessed on 1 March 2021).
Figure 1.
Formation of the membrane attack complex formation via the alternative, classical, and lectin pathways. The alternative pathway is activated by the binding of C3b directly to the surface of a microbe. The classical pathway activation is triggered by the presence of antigen–antibody complexes. The lectin pathway is activated by the binding of mannose-binding lectins to mannose-containing liposaccharides on the surface of the pathogen. Created with BioRender.com; https://biorender.com/ (accessed on 1 March 2021).

Figure 2.
Locations of interference for all three complement activation pathways. Red boxes were used to indicate sites of both direct and indirect inhibition involved in B. burgdorferi infection. Orange boxes indicate sites of compliment inhibition due to tick salivary proteins. Abbreviations: Osp, outer surface protein; TSLPI, tick salivary lectin pathway inhibitor; Salp, salivary protein; Isac/Irac/Ixac, Ixodes anti-complement proteins. Created with BioRender.com.
Figure 2.
Locations of interference for all three complement activation pathways. Red boxes were used to indicate sites of both direct and indirect inhibition involved in B. burgdorferi infection. Orange boxes indicate sites of compliment inhibition due to tick salivary proteins. Abbreviations: Osp, outer surface protein; TSLPI, tick salivary lectin pathway inhibitor; Salp, salivary protein; Isac/Irac/Ixac, Ixodes anti-complement proteins. Created with BioRender.com.

Figure 3.
Illustration of antigenic variation, a tactic used by pathogens to avoid detection by the hosts immune system. Evasion is accomplished by the continuous change in a prominent surface antigen through gene conversion events or change in allelic expression. Created with BioRender.com.
Figure 3.
Illustration of antigenic variation, a tactic used by pathogens to avoid detection by the hosts immune system. Evasion is accomplished by the continuous change in a prominent surface antigen through gene conversion events or change in allelic expression. Created with BioRender.com.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Anderson, C.; Brissette, C.A. The Brilliance of Borrelia: Mechanisms of Host Immune Evasion by Lyme Disease-Causing Spirochetes. Pathogens 2021, 10, 281. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10030281
AMA Style
Anderson C, Brissette CA. The Brilliance of Borrelia: Mechanisms of Host Immune Evasion by Lyme Disease-Causing Spirochetes. Pathogens. 2021; 10(3):281. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10030281
Chicago/Turabian StyleAnderson, Cassidy, and Catherine A. Brissette. 2021. "The Brilliance of Borrelia: Mechanisms of Host Immune Evasion by Lyme Disease-Causing Spirochetes" Pathogens 10, no. 3: 281. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10030281
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.