
Bacterial Cyclic Dinucleotides and the cGAS–cGAMP–STING Pathway: A Role in Periodontitis?

 , , ,
, , , 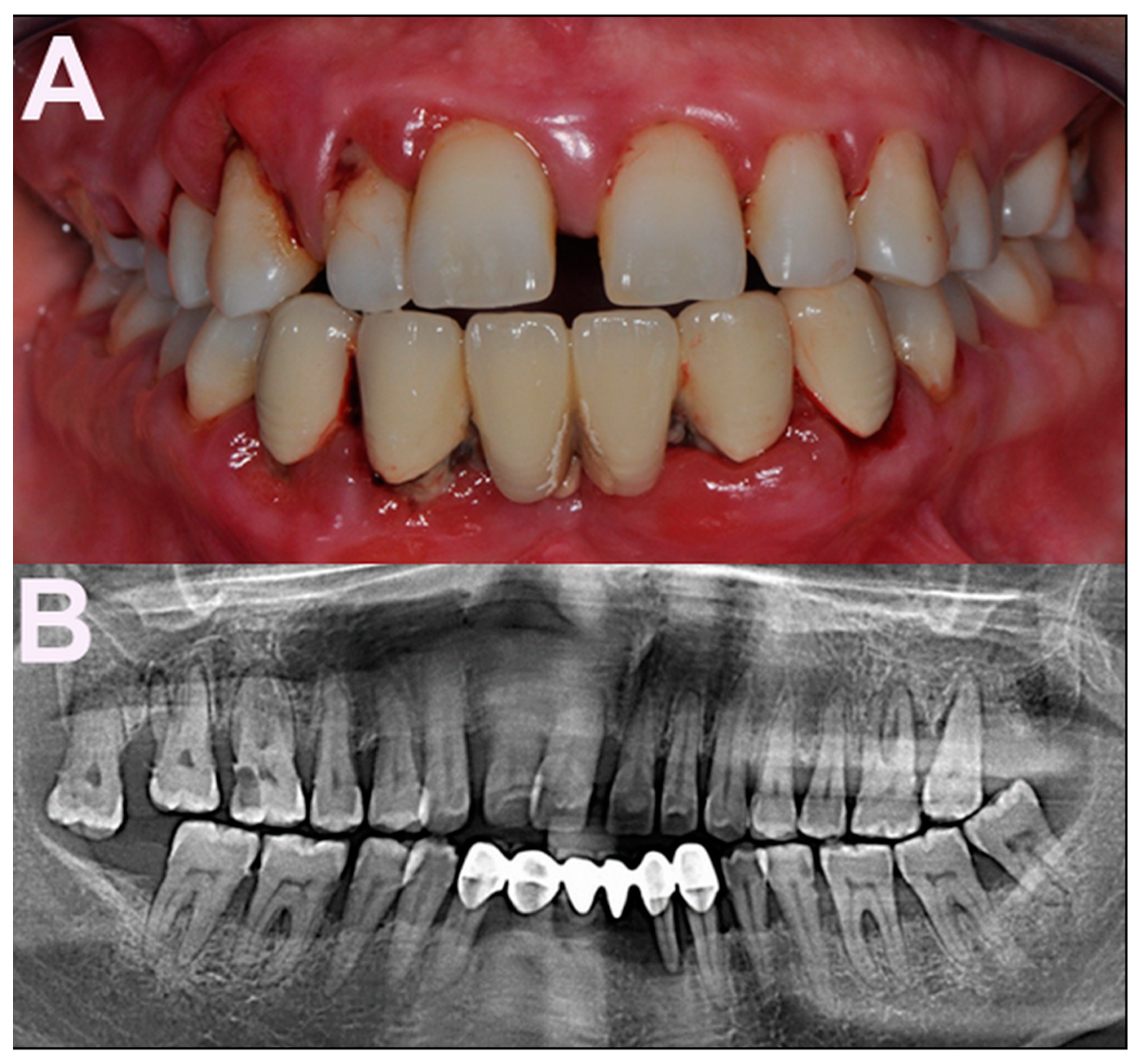{kind=link}
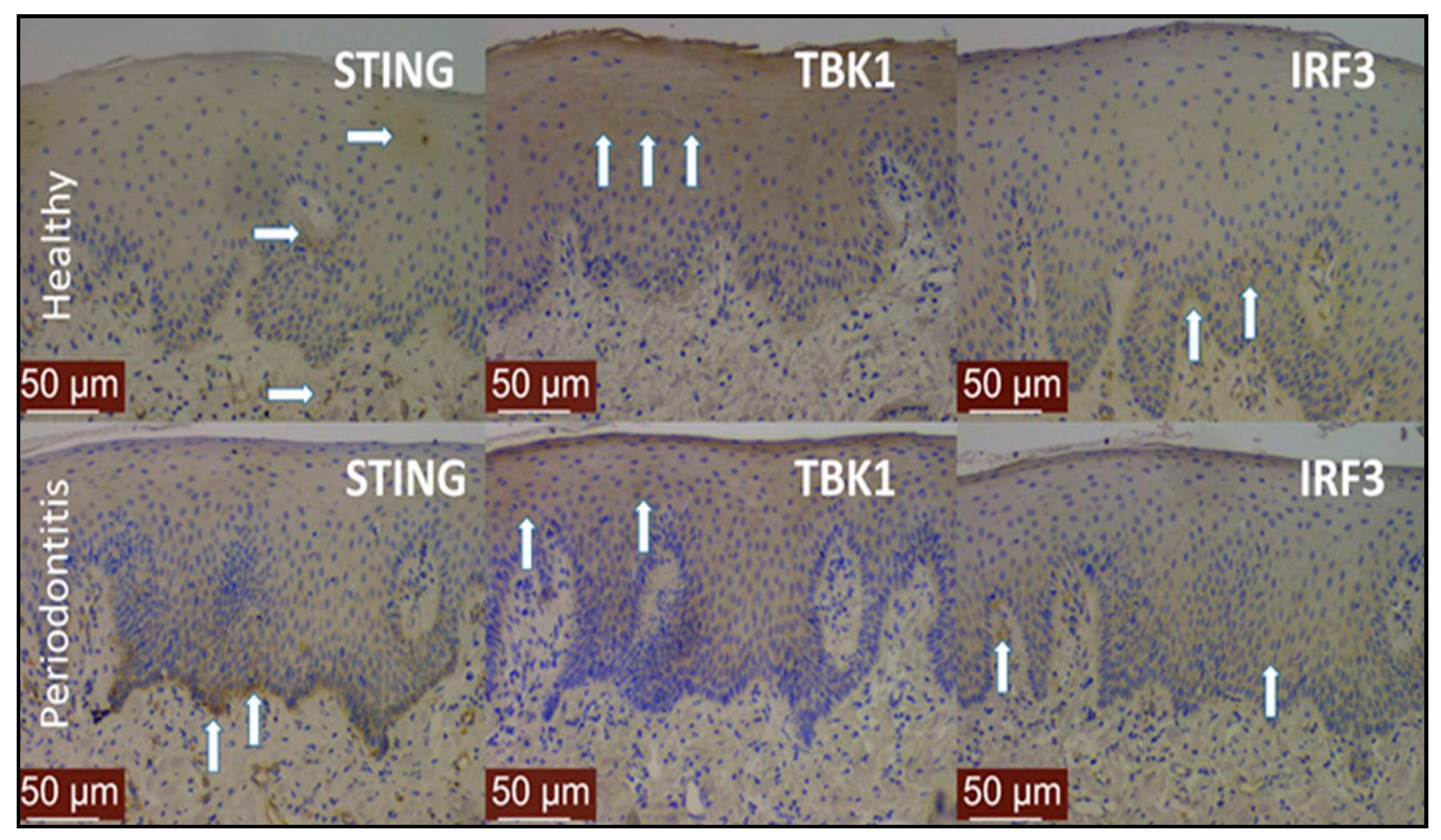{kind=link}
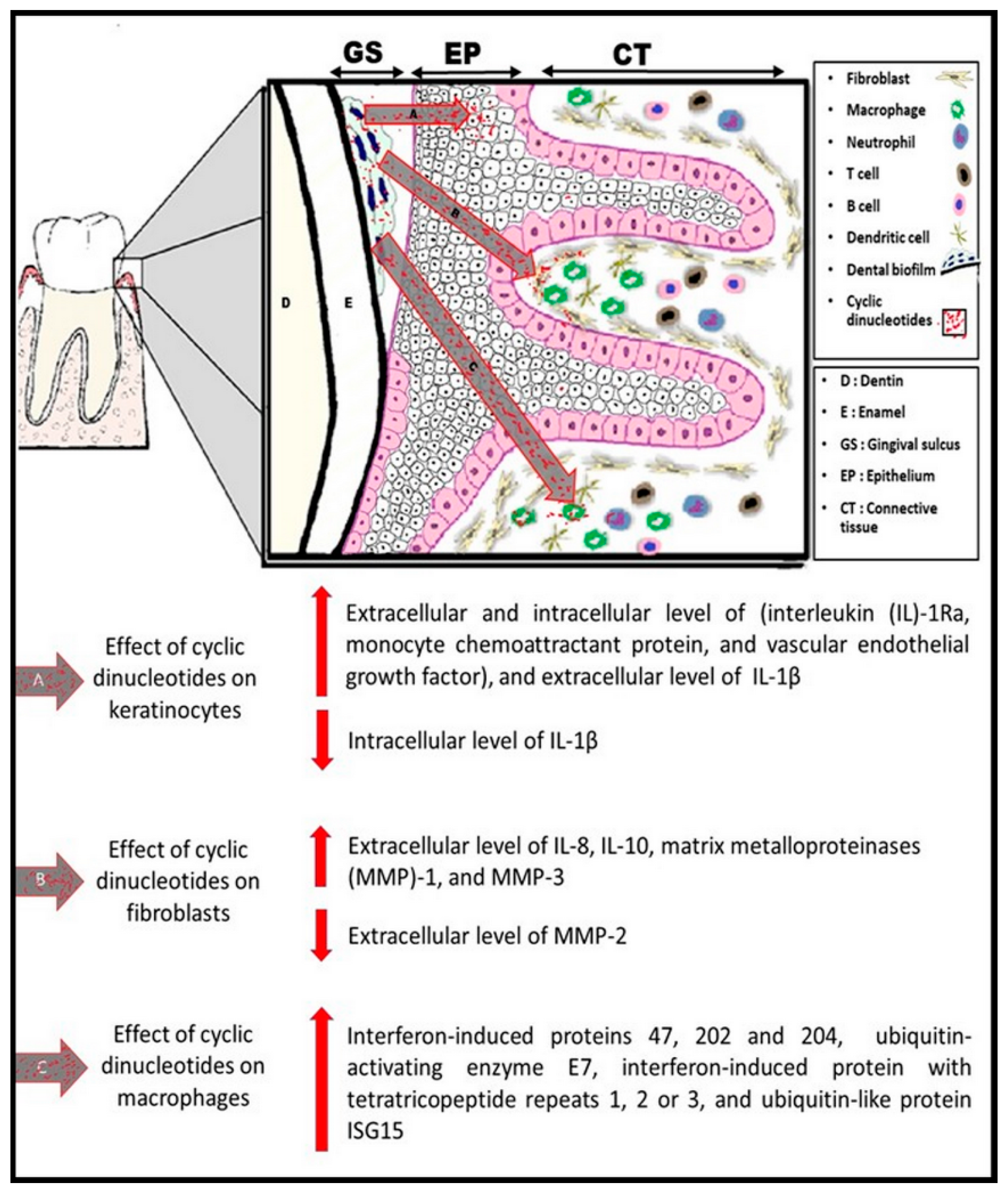{kind=link}
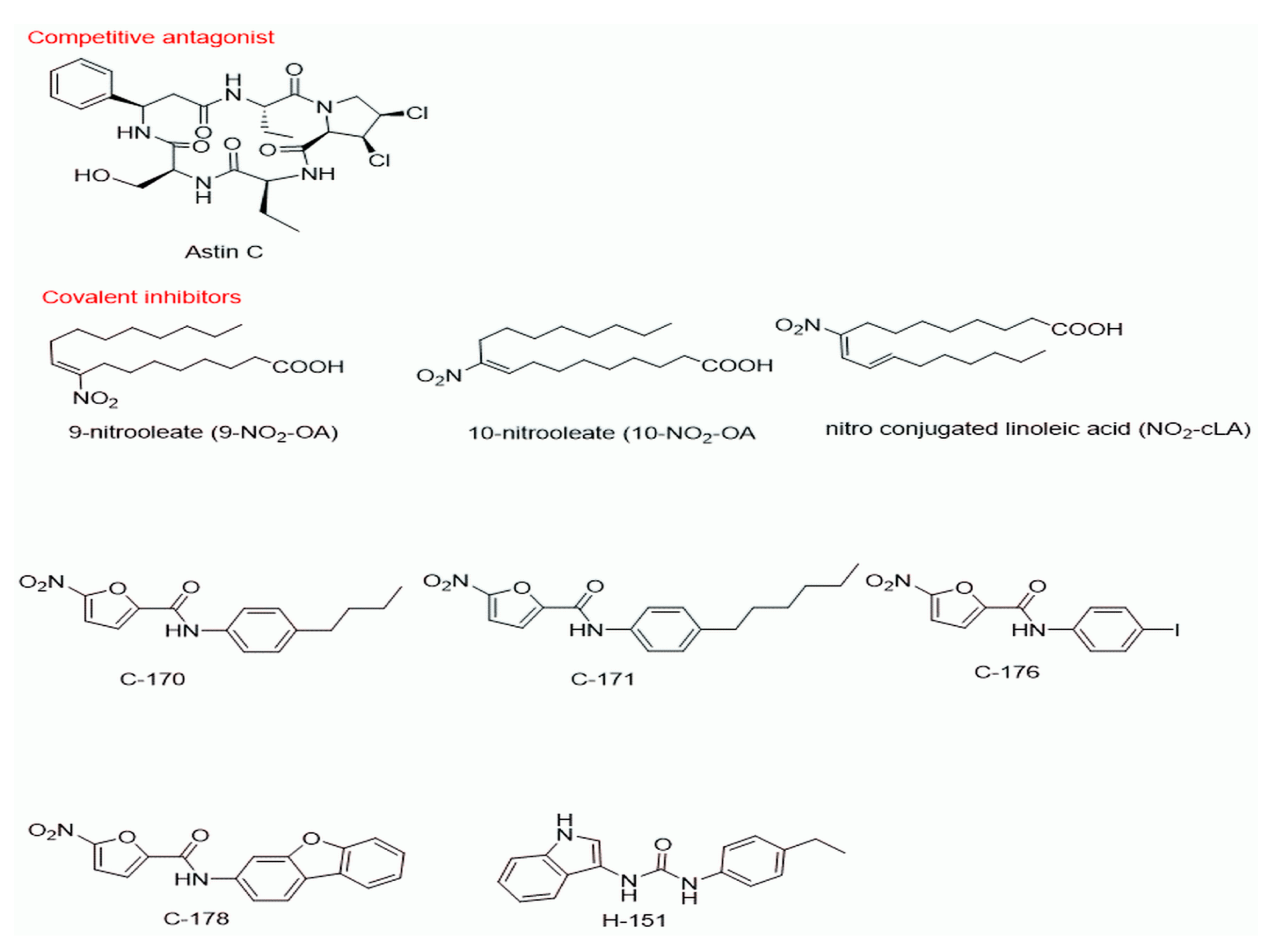{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Intracellular Nucleic Acid Receptors in Mammalian Cells and the STING/TBK1/IRF3 Pathway
3. Bacterial Cyclic Dinucleotides, the cGAS–cGAMP–STING Pathway, and Periodontitis
4. Small Molecule Modulators of the STING Pathway
5. Inhibitors of STING Activation
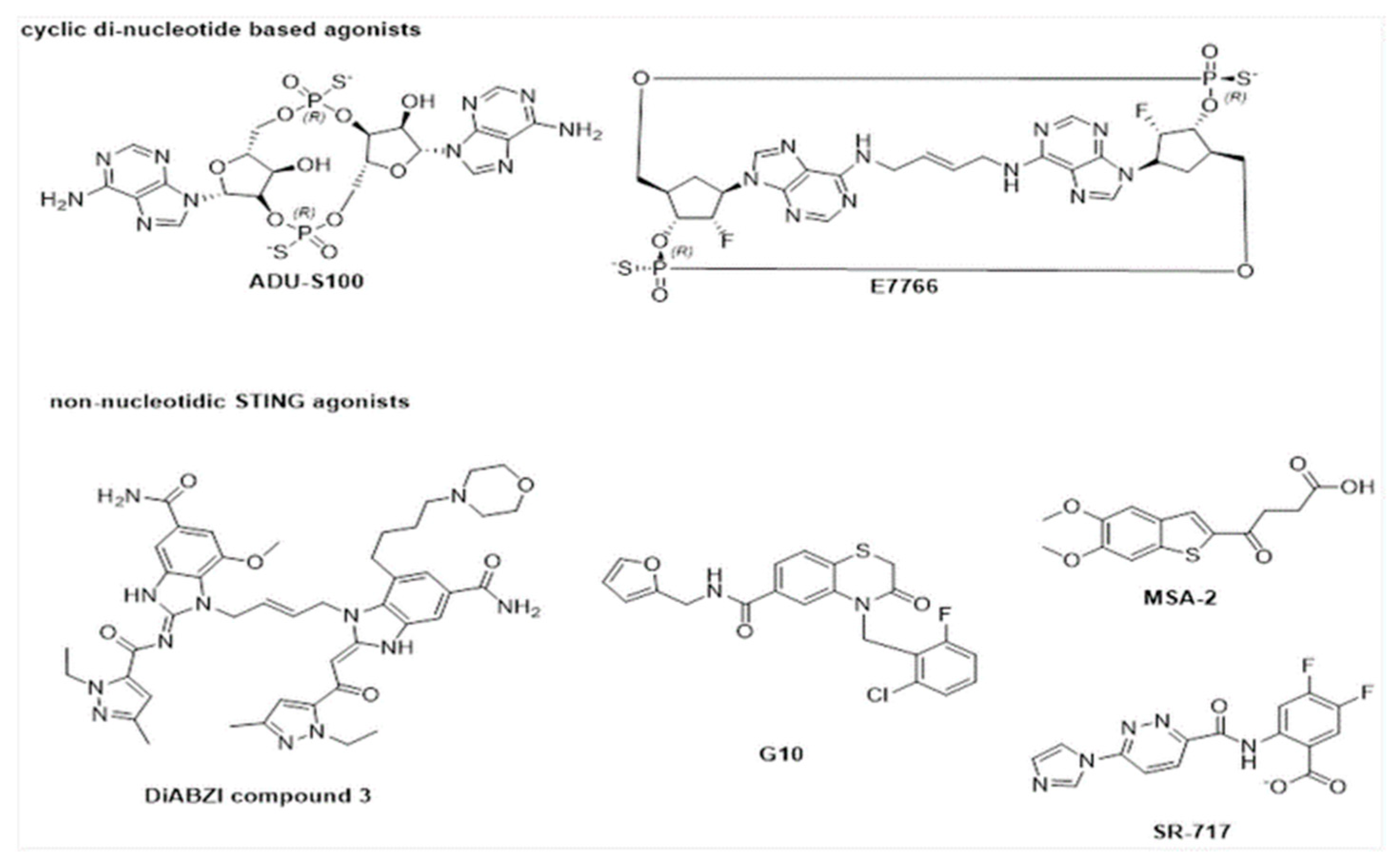
6. Activators of STING Pathway
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Könönen, E.; Gursoy, M.; Gursoy, U.K. Periodontitis: A Multifaceted Disease of Tooth-Supporting Tissues. J. Clin. Med. 2019, 31, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, B.; Zhang, Y.L.; Chen, L.J.; Zhou, T.; Huang, W.K.; Zhou, X.; Shao, L.Q. The role of Toll-like receptors in periodontitis. Oral Dis. 2017, 23, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Belibasakis, G.N.; Bostanci, N. The RANKL-OPG system in clinical periodontology. J. Clin. Periodontol. 2012, 39, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Buduneli, N.; Kinane, D.F. Host-derived diagnostic markers related to soft tissue destruction and bone degradation in periodontitis. J. Clin. Periodontol. 2011, 38, 85–105. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, T.E.; van Winkelhoff, A.J. Infection and inflammatory mechanisms. J. Clin. Periodontol. 2013, 40, S1–S7. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Sahingur, S.E. Novel inflammatory pathways in periodontitis. Adv. Dent. Res. 2014, 26, 23–29. [Google Scholar] [CrossRef]
- Tan, X.; Sun, L.; Chen, J.; Chen, Z.J. Detection of microbial infections through innate immune sensing of nucleic acids. Annu. Rev. Microbiol. 2018, 72, 447–478. [Google Scholar] [CrossRef]
- Mankan, A.K.; Schmidt, T.; Chauhan, D.; Goldeck, M.; Höning, K.; Gaidt, M.; Kubarenko, A.V.; Andreeva, L.; Hopfner, K.P.; Hornung, V. Cytosolic RNA: DNA hybrids activate the cGAS–STING axis. EMBO. J. 2014, 33, 2937–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Fitzgerald, K.A.; Cancro, M.P.; Marshak-Rothstein, A. Nucleic Acid–Sensing Receptors: Rheostats of Autoimmunity and Autoinflammation. J. Immunol. 2015, 195, 3507–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabeta, K.; Hoebe, K.; Janssen, E.M.; Du, X.; Georgel, P.; Crozat, K.; Mudd, S.; Mann, N.; Sovath, S.; Goode, J. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Barbalat, R.; Ewald, S.E.; Mouchess, M.L.; Barton, G.M. Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol. 2011, 29, 185–214. [Google Scholar] [CrossRef]
- Krug, A.; Towarowski, A.; Britsch, S.; Rothenfusser, S.; Hornung, V.; Bals, R.; Giese, T.; Engelmann, H.; Endres, S.; Krieg, A.M.; et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur. J. Immunol. 2001, 31, 3026–3037. [Google Scholar] [CrossRef]
- Rojo-Botello, N.R.; García-Hernández, A.L.; Moreno-Fierros, L. Expression of toll-like receptors 2, 4 and 9 is increased in gingival tissue from patients with type 2 diabetes and chronic periodontitis. J. Periodontal Res. 2012, 47, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Sahingur, S.E.; Xia, X.J.; Gunsolley, J.; Schenkein, H.A.; Genco, R.J.; De Nardin, E. Single nucleotide polymorphisms of pattern recognition receptors and chronic periodontitis. J. Periodontal Res. 2011, 46, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Lambris, J.D. Microbial manipulation of receptor crosstalk in innate immunity. Nat. Rev. Immunol. 2011, 11, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Kim, Y.G.; McDonald, C.; Kanneganti, T.D.; Hasegawa, M.; Body-Malapel, M.; Inohara, N.; Nunez, G. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J. Immunol. 2007, 178, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly (dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 2016, 16, 566. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Sahingur, S.E.; Xia, X.J.; Voth, S.C.; Yeudall, W.A.; Gunsolley, J.C. Increased nucleic acid receptor expression in chronic periodontitis. J. Periodontol. 2013, 84, e48–e57. [Google Scholar] [CrossRef]
- Xue, F.; Shu, R.; Xie, Y. The expression of NLRP3, NLRP1 and AIM2 in the gingival tissue of periodontitis patients: RT-PCR study and immunohistochemistry. Arch. Oral Biol. 2015, 60, 948–958. [Google Scholar] [CrossRef]
- Bostanci, N.; Meier, A.; Guggenheim, B.; Belibasakis, G.N. Regulation of NLRP3 and AIM2 inflammasome gene expression levels in gingival fibroblasts by oral biofilms. Cell. Immunol. 2011, 270, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Bhan, U.; Ballinger, M.N.; Zeng, X.; Newstead, M.J.; Cornicelli, M.D.; Standiford, T.J. Cooperative interactions between TLR4 and TLR9 regulate interleukin 23 and 17 production in a murine model of gram negative bacterial pneumonia. PLoS ONE 2010, 5, e9896. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Ahn, J.; Xia, T.; Rabasa Capote, A.; Betancourt, D.; Barber, G.N. Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer Cell 2018, 33, 862–873. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Ortiz Serrano, T.P.; Davis, J.; Prigge, A.D.; Ridge, K.M. The cGAS-STING pathway: The role of self-DNA sensing in inflammatory lung disease. FASEB J. 2020, 34, 13156–13170. [Google Scholar] [CrossRef]
- Marinho, F.V.; Benmerzoug, S.; Oliveira, S.C.; Ryffel, B.; Quesniaux, V.F.J. The Emerging Roles of STING in Bacterial Infections. Trends Microbiol. 2017, 25, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Barber, G.N. STING signaling and host defense against microbial infection. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Civril, F.; Deimling, T.; De Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; et al. Cyclic [G (2′, 5′) pA (3′, 5′) p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef] [Green Version]
- Kranzusch, P.J.; Lee, A.S.; Berger, J.M.; Doudna, J.A. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013, 3, 1362–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, M.; Chen, Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lässig, C.; De Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Gulen, M.F. The role of cGAS in innate immunity and beyond. J. Mol. Med. 2016, 94, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Liu, W.; Zhang, Q.; Huang, Y.; Li, W.; Wang, W.; Zhao, P.; Huang, S.; Song, Y.; Shereen, M.A.; et al. MYSM1 Represses Innate Immunity and Autoimmunity through Suppressing the cGAS-STING Pathway. Cell Rep. 2020, 33, 108297. [Google Scholar]
- Kasnak, G.; Firatli, E.; Könönen, E.; Olgac, V.; Zeidán-Chuliá, F.; Gursoy, U.K. Elevated levels of 8-OHdG and PARK7/DJ-1 in peri-implantitis mucosa. Clin. Implant. Dent. Relat. Res. 2018, 20, 574–582. [Google Scholar] [CrossRef]
- Nakayama, S.; Zhou, J.; Zheng, Y.; Szmacinski, H.; Sintim, H.O. Supramolecular polymer formation by cyclic dinucleotides and intercalators affects dinucleotide enzymatic processing. Future Sci. 2016, 2, FSO93. [Google Scholar] [CrossRef] [Green Version]
- Zaver, S.A.; Woodward, J.J. Cyclic dinucleotides at the forefront of innate immunity. Curr. Opin. Cell Biol. 2020, 63, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, U.K.; Gursoy, M.; Kononen, E.; Sintim, H.O. Cyclic dinucleotides in oral bacteria and in oral biofilms. Front. Cell Infect. Microbiol. 2017, 7, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danilchanka, O.; Mekalanos, J.J. Cyclic dinucleotides and the innate immune response. Cell 2013, 154, 962–970. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morehouse, B.R.; Govande, A.A.; Millman, A.; Keszei, A.F.A.; Lowey, B.; Ofir, G.; Shao, S.; Sorek, R.; Kranzusch, P.J. STING cyclic dinucleotide sensing originated in bacteria. Nature 2020, 586, 429–433. [Google Scholar] [CrossRef]
- Millman, A.; Melamed, S.; Amitai, G.; Sorek, R. Diversity and classification of cyclic-oligonucleotide-based anti-phage signalling systems. Nat. Microbiol. 2020, 5, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.S.; Fitzgerald, K.A. The cGAS-STING pathway for DNA sensing. Mol. Cell 2013, 51, 135–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmanfi, S.; Zhou, J.; Sintim, H.O.; Könönen, E.; Gürsoy, M.; Gürsoy, U.K. Regulation of gingival epithelial cytokine response by bacterial cyclic dinucleotides. J. Oral Microbiol. 2018, 11, 1538927. [Google Scholar] [CrossRef] [Green Version]
- Elmanfi, S.; Sintim, H.O.; Zhou, J.; Gürsoy, M.; Könönen, E.; Gürsoy, U.K. Activation of gingival fibroblasts by bacterial cyclic dinucleotides and lipopolysaccharide. Pathogens 2020, 9, 792. [Google Scholar] [CrossRef] [PubMed]
- Sooreshjani, M.A.; Gursoy, U.K.; Aryal, U.K.; Sintim, H.O. Proteomic analysis of RAW macrophages treated with cGAMP or c-di-GMP reveals differentially activated cellular pathways. RSC Adv. 2018, 8, 36840–36851. [Google Scholar] [CrossRef] [Green Version]
- Aryal, U.K.; Hedrick, V.; Onyedibe, K.I.; Sobreira, T.J.P.; Sooreshjani, M.A.; Wang, M.; Gürsoy, U.K.; Sintim, H.O. Global proteomic analyses of STING-positive and -negative macrophages reveal STING and Non-STING differentially regulated cellular and molecular pathways. Proteom. Clin. Appl. 2020, 14, e1900109. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.R.; Garlet, G.P.; Moreira, A.P.; Júnior, W.M.; Rossi, M.A.; Silva, J.S. Characterization of CD4+CD25+ natural regulatory T cells in the inflammatory infiltrate of human chronic periodontitis. J. Leukoc. Biol. 2008, 84, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Wilensky, A.; Segev, H.; Mizraji, G.; Shaul, Y.; Capucha, T.; Shacham, M.; Hovav, A.H. Dendritic cells and their role in periodontal disease. Oral Dis. 2014, 20, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Swiggard, W.J.; Heufler, C.; Peng, M.; Mirza, A.; Steinman, R.M.; Nussenzweig, M.C. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature 1995, 375, 151–155. [Google Scholar] [CrossRef]
- Hans, M.; Hans, V.M. Toll-like receptors and their dual role in periodontitis: A review. J. Oral Sci. 2011, 53, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Santegoets, K.C.; Wenink, M.H.; Braga, F.A.; Cossu, M.; Lamers-Karnebeek, F.B.; van Riel, P.L.; Sturm, P.D.; van den Berg, W.B.; Radstake, T.R. Impaired Porphyromonas gingivalis-Induced Tumor Necrosis Factor Production by Dendritic Cells Typifies Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Kanaya, S.; Nemoto, E.; Ogawa, T.; Shimauchi, H. Porphyromonas gingivalis lipopolysaccharides induce maturation of dendritic cells with CD14+CD16+ phenotype. Eur. J. Immunol. 2004, 34, 1451–1460. [Google Scholar] [CrossRef]
- De Oliveira, S.; Reyes-Aldasoro, C.C.; Candel, S.; Renshaw, S.A.; Mulero, V.; Calado, A. Cxcl8 (IL-8) mediates neutrophil recruitment and behavior in the zebrafish inflammatory response. J. Immunol. 2013, 190, 4349–4359. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Hasturk, H.; Kantarci, A.; Van Dyke, T.E. Oral inflammatory diseases and systemic inflammation: Role of the macrophage. Front. Immunol. 2012, 3, 118. [Google Scholar] [CrossRef] [Green Version]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Hong, Z.; Wang, Z.; Li, F.; Mei, J.; Huang, L.; Lou, X.; Zhao, S.; Song, L.; Chen, W.; et al. The cyclopeptide astin C specifically inhibits the innate immune CDN sensor STING. Cell Rep. 2018, 25, 3405–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; You, Q.D.; Xu, X.L. Targeting Stimulator of Interferon Genes (STING): A Medicinal Chemistry Perspective. J. Med. Chem. 2020, 63, 3785–3816. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef]
- Hansen, A.L.; Buchan, G.J.; Rühl, M.; Mukai, K.; Salvatore, S.R.; Ogawa, E.; Andersen, S.D.; Iversen, M.B.; Thielke, A.L.; Gunderstofte, C.; et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc. Natl. Acad. Sci. USA. 2018, 115, 7768–7775. [Google Scholar] [CrossRef] [Green Version]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef]
- Hansen, A.L.; Mukai, K.; Schopfer, F.J.; Taguchi, T.; Holm, C.K. STING palmitoylation as a therapeutic target. Cell. Mol. Immunol. 2019, 16, 236–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdette, D.L.; Vence, R.E. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 2013, 14, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Onyedibe, K.I.; Wang, M.; Sintim, H.O. ENPP1, an Old Enzyme with New Functions, and Small Molecule Inhibitors-A STING in the Tale of ENPP1. Molecules 2019, 24, 4192. [Google Scholar] [CrossRef] [Green Version]
- Mikek, C.; Wang, M.; Sooreshjani, M.A.; Sintim, H.O. Interrupting cyclic dinucleotide-cGAS-STING axis with small molecules. Med. Chem. Commun. 2019, 10, 1999–2023. [Google Scholar]
- Chin, E.N.; Yu, C.; Vartabedian, V.F.; Jia, Y.; Kumar, M.; Gamo, A.M.; Vernier, W.; Ali, S.H.; Kissai, M.; Lazar, D.C.; et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 2020, 369, 993–999. [Google Scholar] [CrossRef]
- Pan, B.S.; Perera, S.A.; Piesvaux, J.A.; Presland, J.P.; Schroeder, G.K.; Cumming, J.N.; Wesley Trotter, B.; Altman, M.D.; Buevich, A.V.; Cash, B.; et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 2020, 369, eaba6098. [Google Scholar] [CrossRef]
- Zandberg, D.P.; Ferris, R.; Mehra, R.; Nabell, L.; Kaczmar, J.; Gibson, M.K.; Kim, Y.J.; Neupame, P.; Bauman, J.; Julian, R.; et al. A phase II study of ADU-S100 in combination with Pembrolizumab in adult patients with PD-L1+ recurrent or metastatic HNSCC: Preliminary safety, efficacy and PK/PD results. Ann. Oncol. 2020, 31, S1446–S1447. [Google Scholar] [CrossRef]
- Kim, D.S.; Fang, F.; Endo, A.; Choi, H.; Hao, M.; Bao, X.; Huang, K.C.; Eisai R & D Management Co. Ltd. Compounds for the Treatment of Cancer. U.S. Patent US8633174B2, 21 January 2014. [Google Scholar]
- Sali, T.M.; Pryke, K.M.; Abraham, J.; Liu, A.; Archer, I.; Broeckel, R.; Staverosky, J.A.; Smith, J.L.; Al-Shammari, A.; Amsler, L.; et al. Characterization of a Novel Human-Specific STING Agonist that Elicits Antiviral Activity Against Emerging Alphaviruses. PLoS Pathog. 2015, 11, e1005324. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Middya, S.; Shrivastava, R.; Basu, S.; Ghosh, R.; Pryde, D.C.; Yadav, D.B.; Surya, A. G10 is a direct activator of human STING. PLoS ONE 2020, 15, e0237743. [Google Scholar] [CrossRef] [PubMed]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.Y.; Tran, J.L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Kalia, D.; Merey, G.; Nakayama, S.; Zheng, Y.; Zhou, J.; Luo, Y.; Guo, M.; Roembke, B.T.; Sintim, H.O. Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP,(p) ppGpp signaling in bacteria and implications in pathogenesis. Chem. Soc. Rev. 2013, 42, 305–341. [Google Scholar] [CrossRef] [PubMed]
- Opoku-Temeng, C.; Zhou, J.; Zheng, Y.; Su, J.; Sintim, H.O. Cyclic dinucleotide (c-di-GMP, c-di-AMP, and cGAMP) signalings have come of age to be inhibited by small molecules. Chem. Commun. 2016, 52, 9327–9342. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elmanfi, S.; Yilmaz, M.; Ong, W.W.S.; Yeboah, K.S.; Sintim, H.O.; Gürsoy, M.; Könönen, E.; Gürsoy, U.K. Bacterial Cyclic Dinucleotides and the cGAS–cGAMP–STING Pathway: A Role in Periodontitis? Pathogens 2021, 10, 675. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10060675
Elmanfi S, Yilmaz M, Ong WWS, Yeboah KS, Sintim HO, Gürsoy M, Könönen E, Gürsoy UK. Bacterial Cyclic Dinucleotides and the cGAS–cGAMP–STING Pathway: A Role in Periodontitis? Pathogens. 2021; 10(6):675. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10060675
Chicago/Turabian StyleElmanfi, Samira, Mustafa Yilmaz, Wilson W. S. Ong, Kofi S. Yeboah, Herman O. Sintim, Mervi Gürsoy, Eija Könönen, and Ulvi K. Gürsoy. 2021. "Bacterial Cyclic Dinucleotides and the cGAS–cGAMP–STING Pathway: A Role in Periodontitis?" Pathogens 10, no. 6: 675. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10060675