
Evaluation of Inhibitory Activity In Silico of In-House Thiomorpholine Compounds between the ACE2 Receptor and S1 Subunit of SARS-CoV-2 Spike

 , ,
, , 
Abstract
:
1. Introduction
2. Results and Discussion
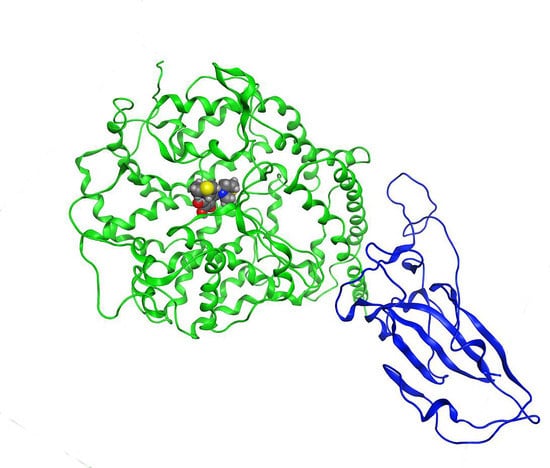
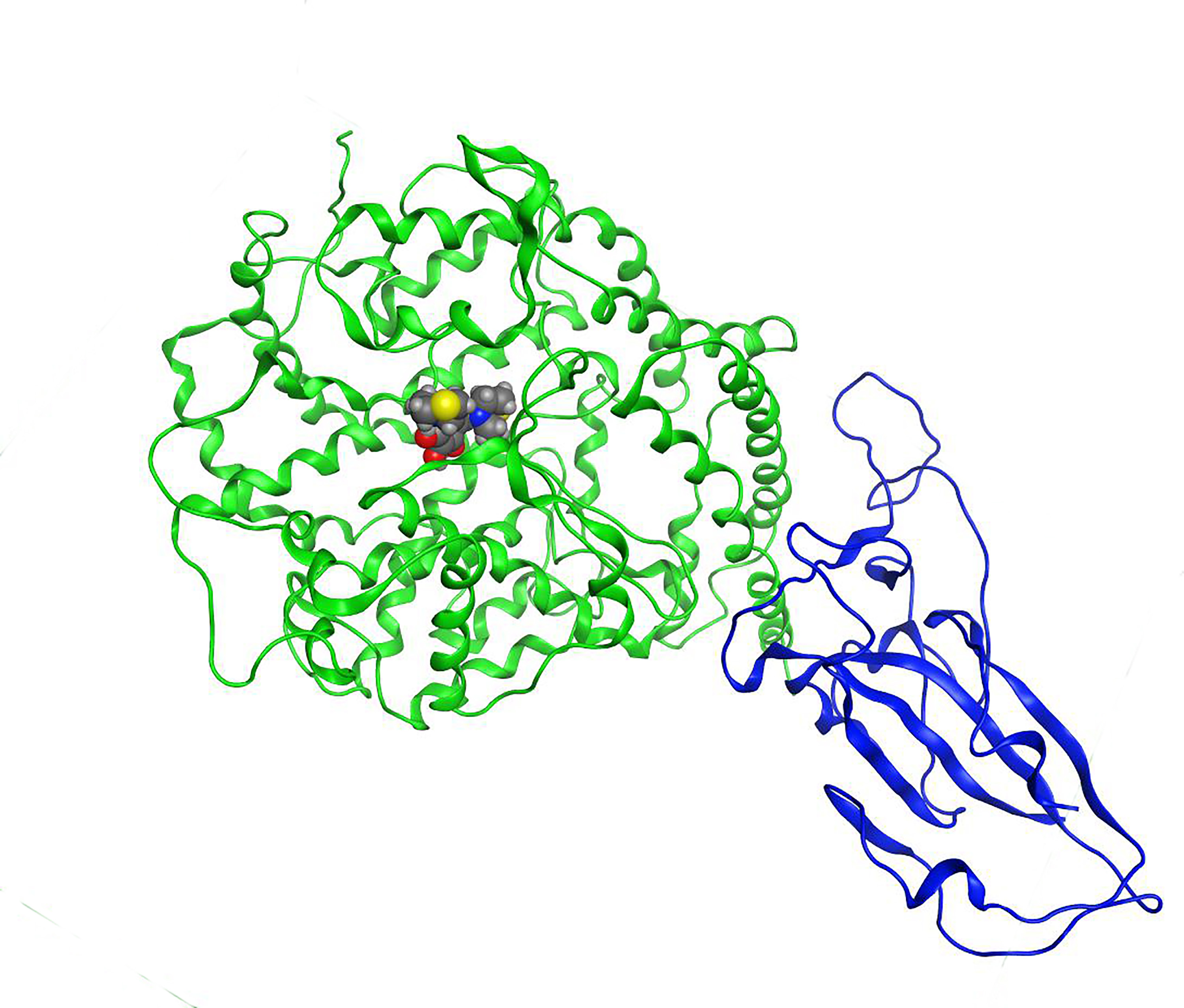
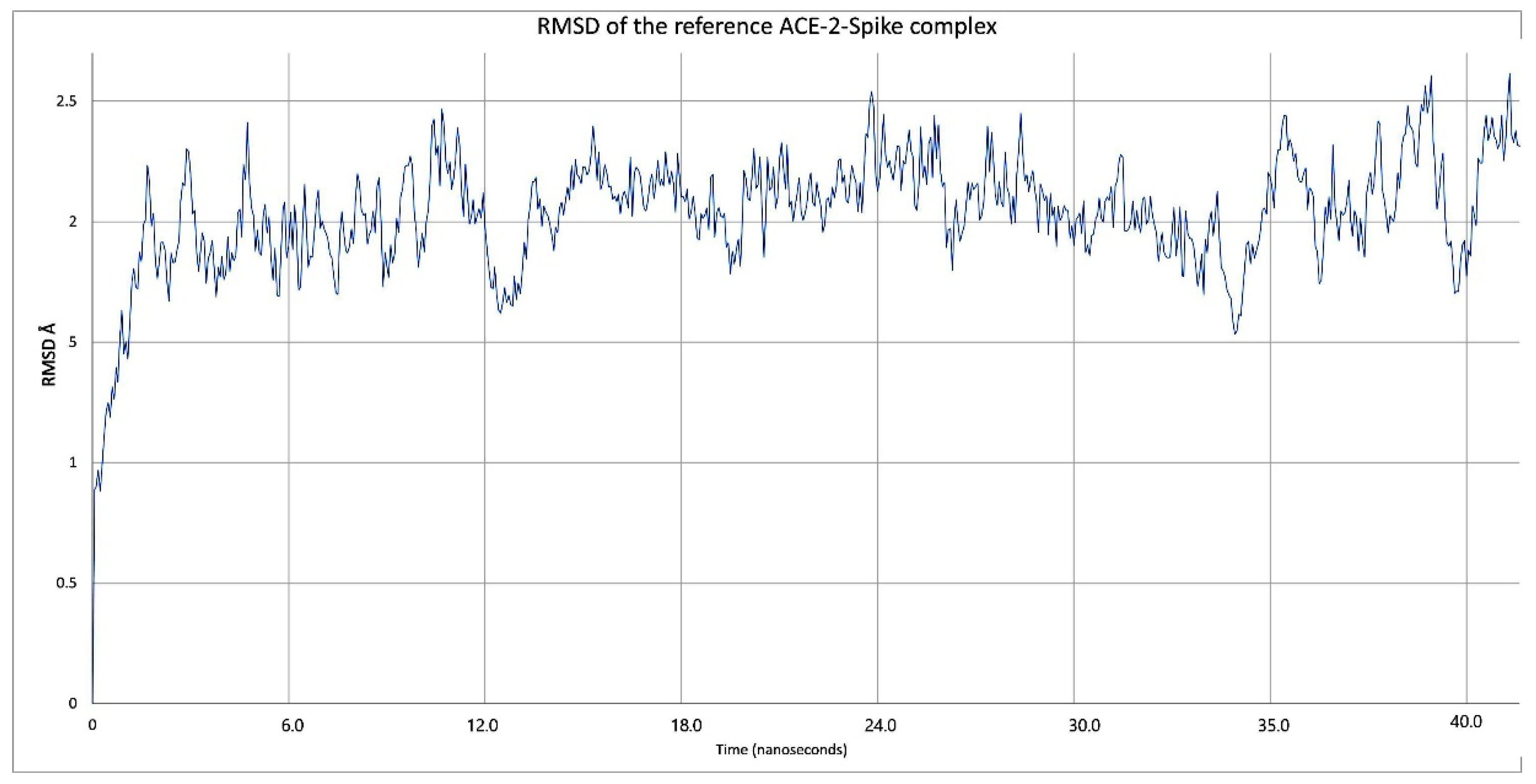
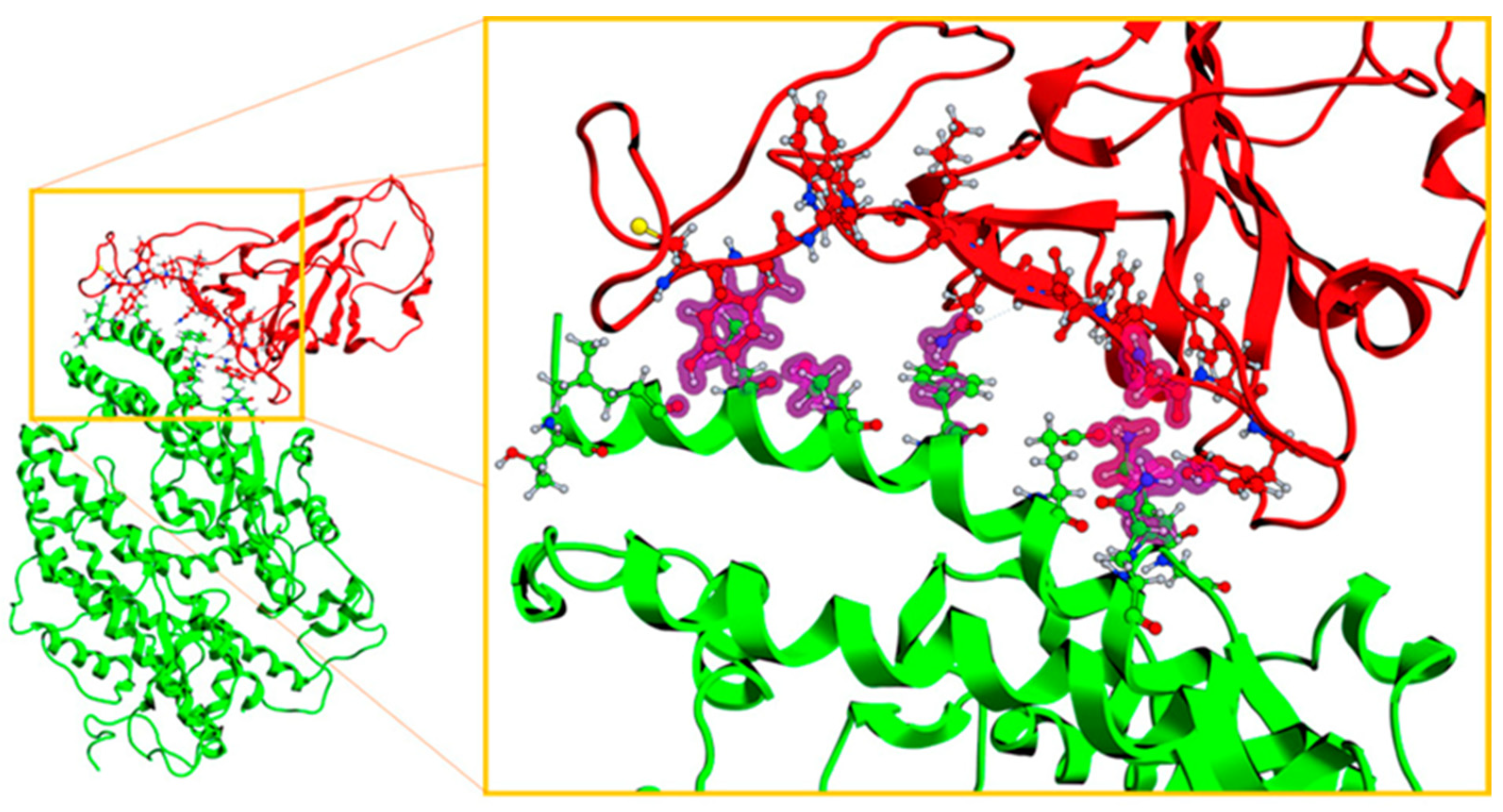
2.1. Validation Process of Molecular Protein–Protein Coupling
2.2. Molecular Coupling Protocol and MOE-NAMD (Molecular Operating Environment-Nanoscale Molecular Dynamics)
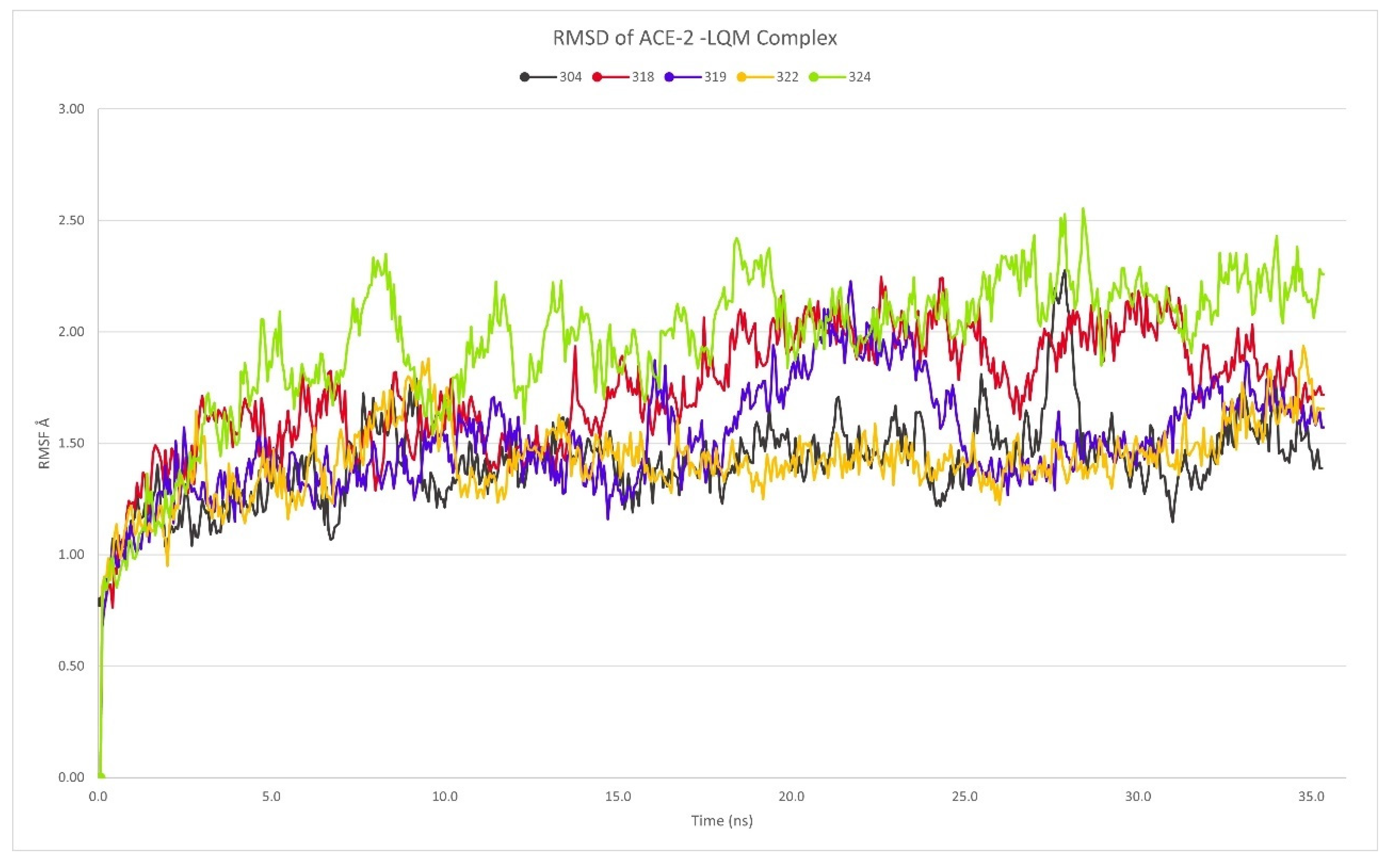
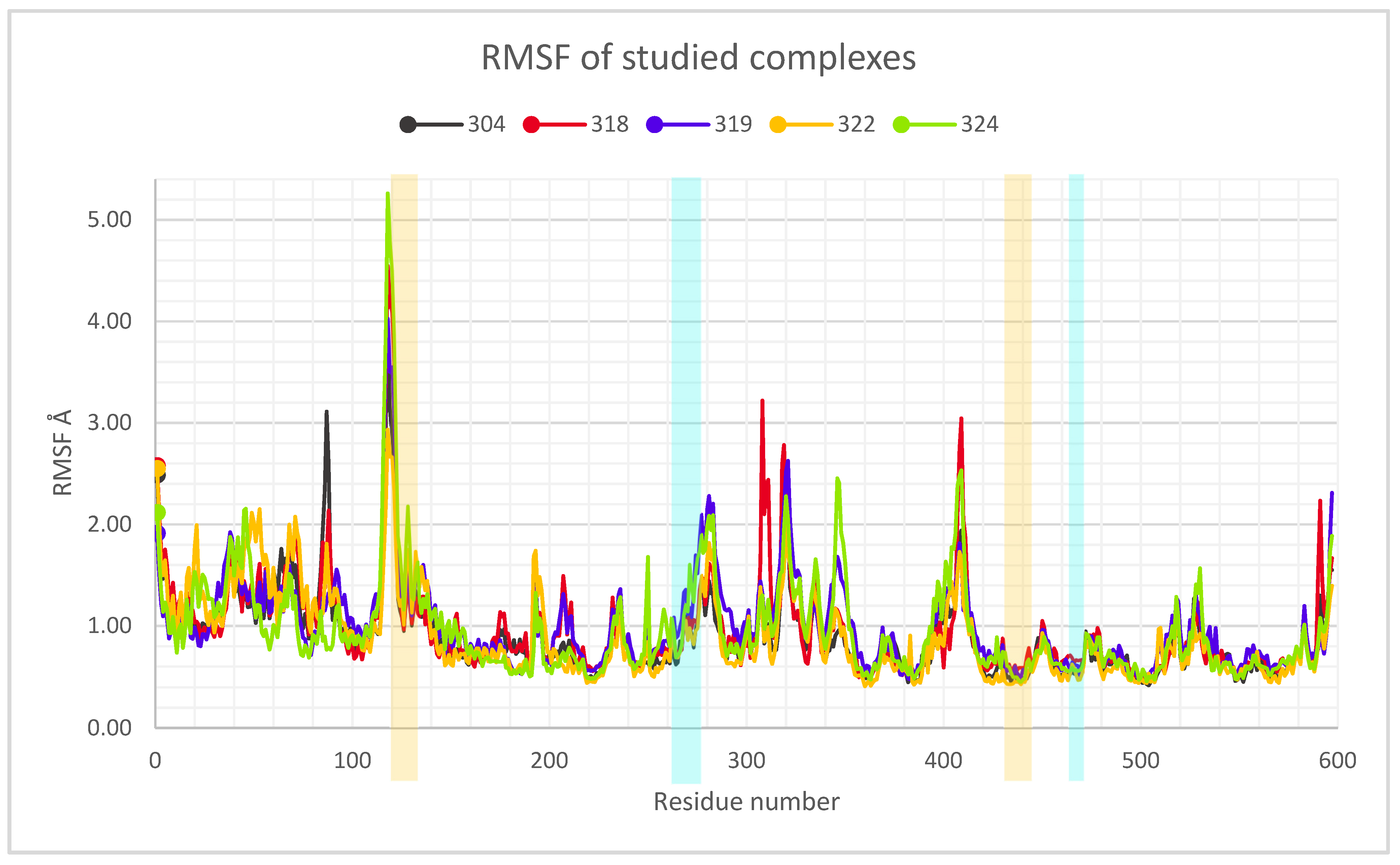
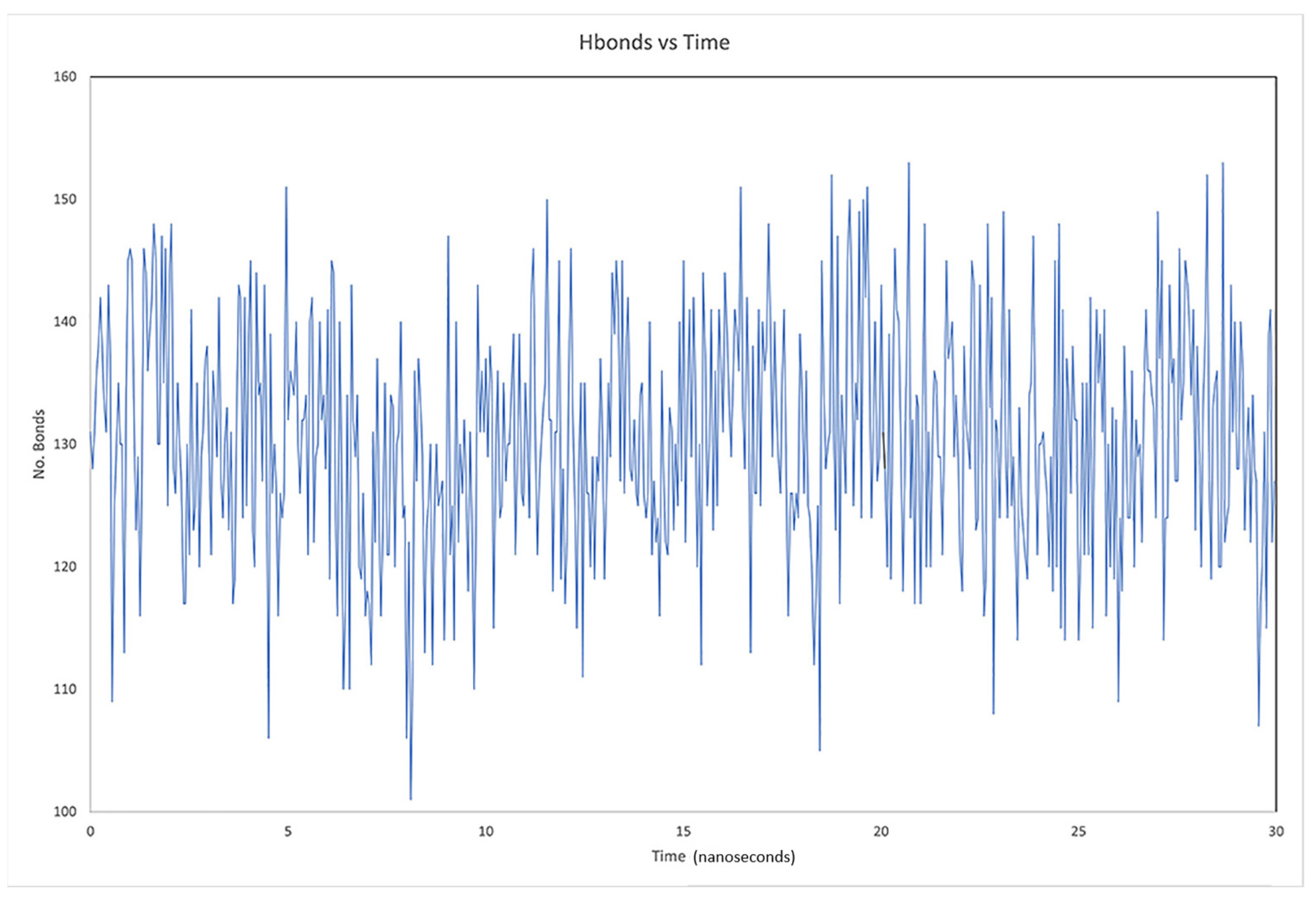
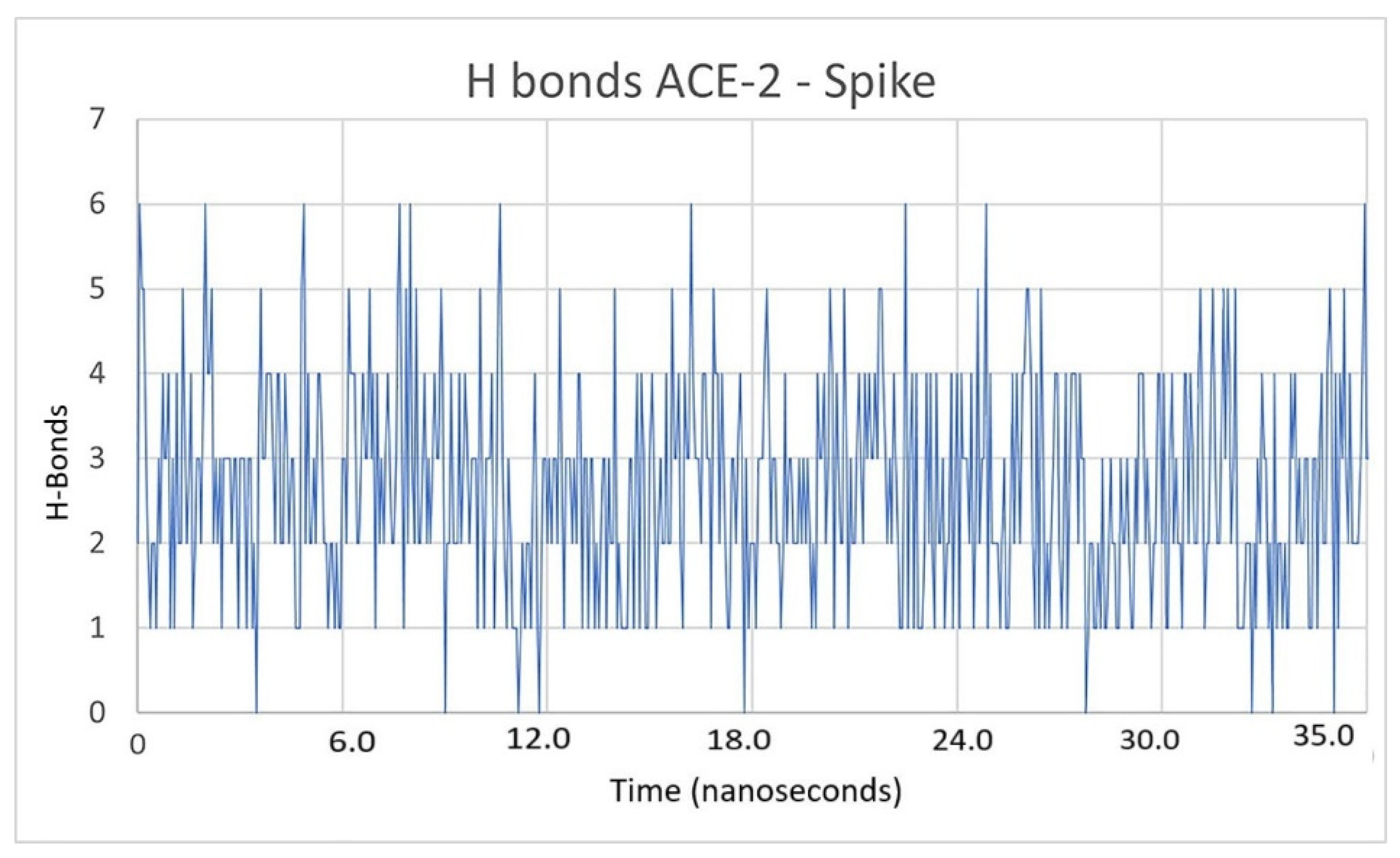
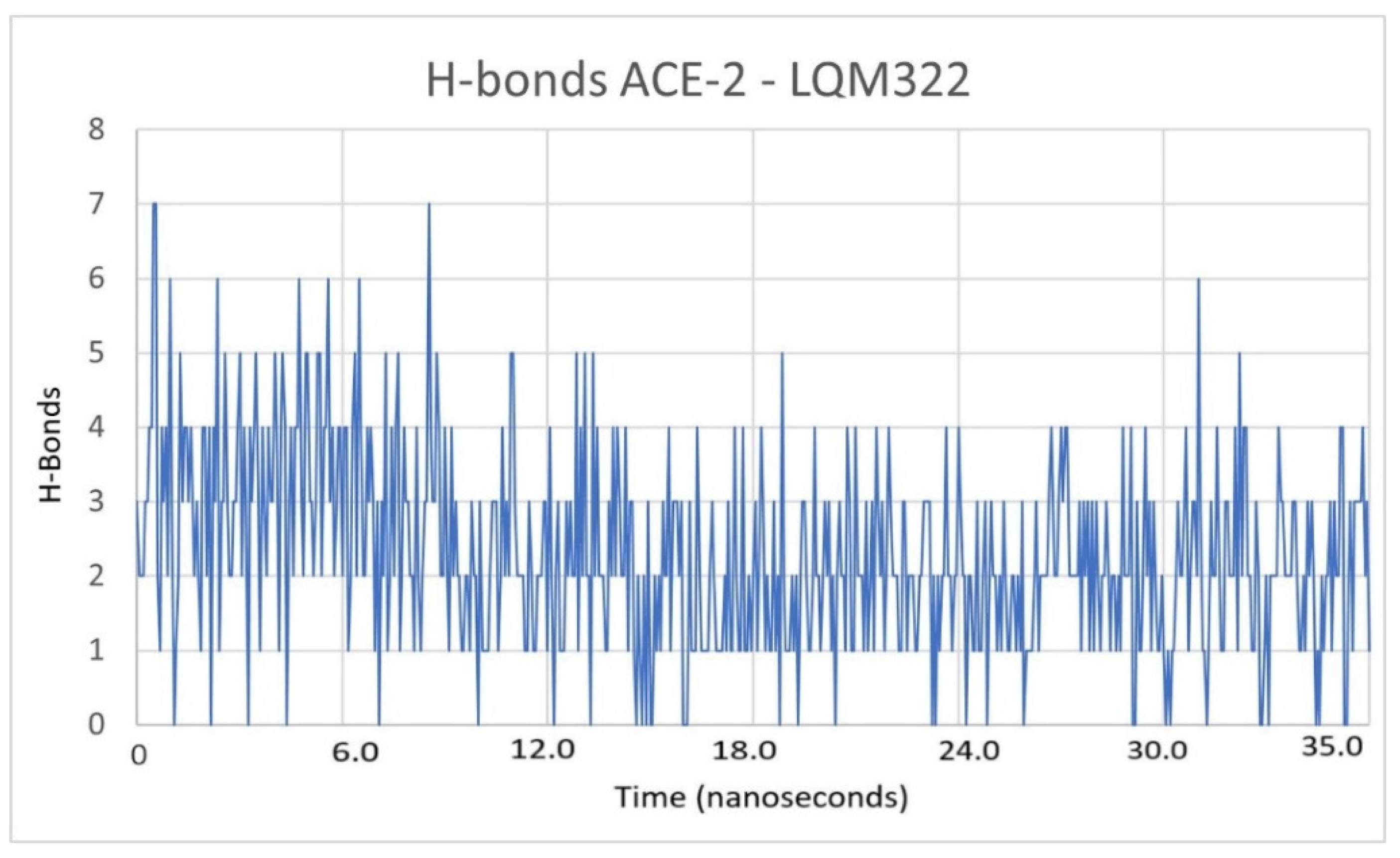
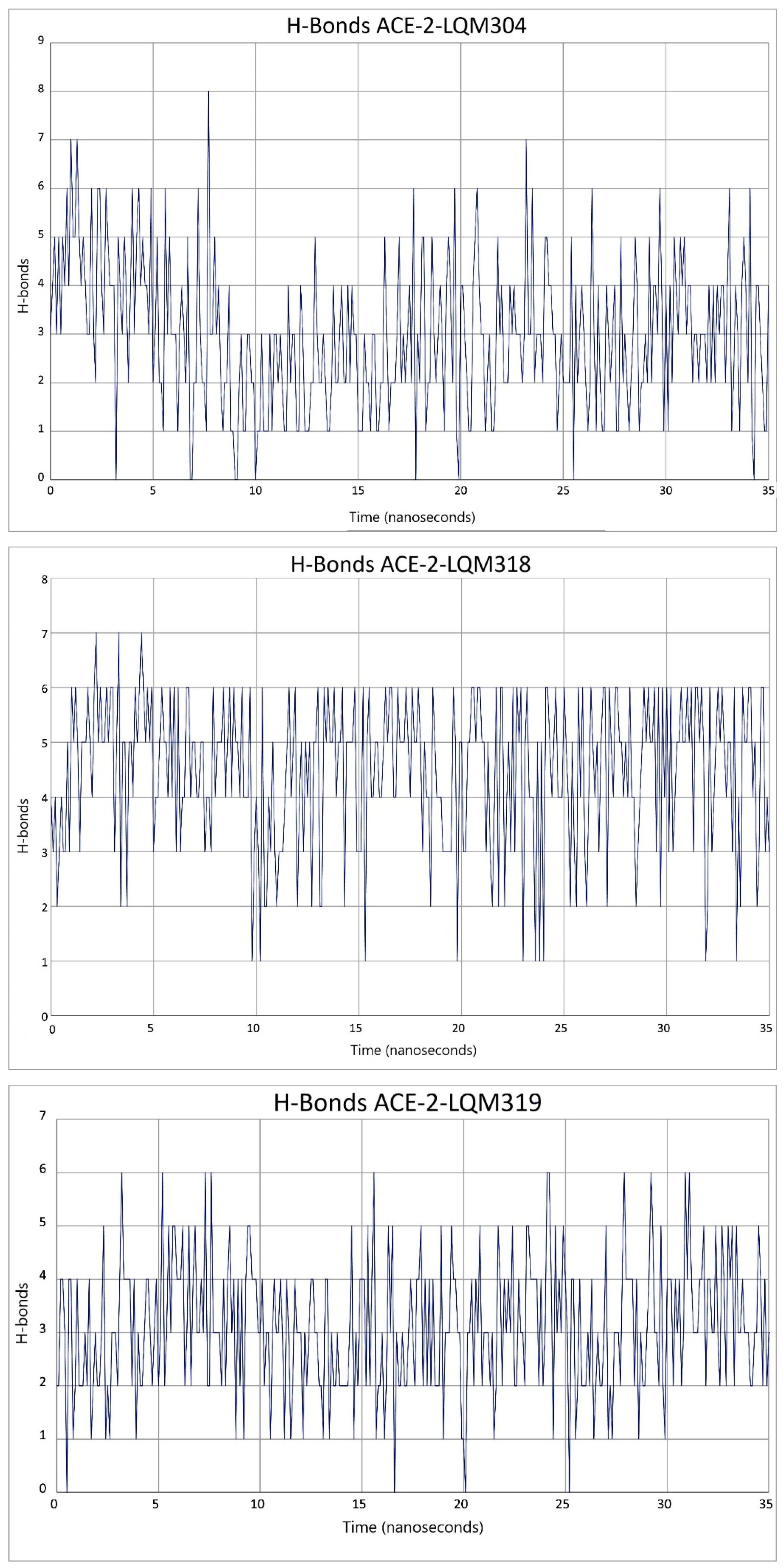
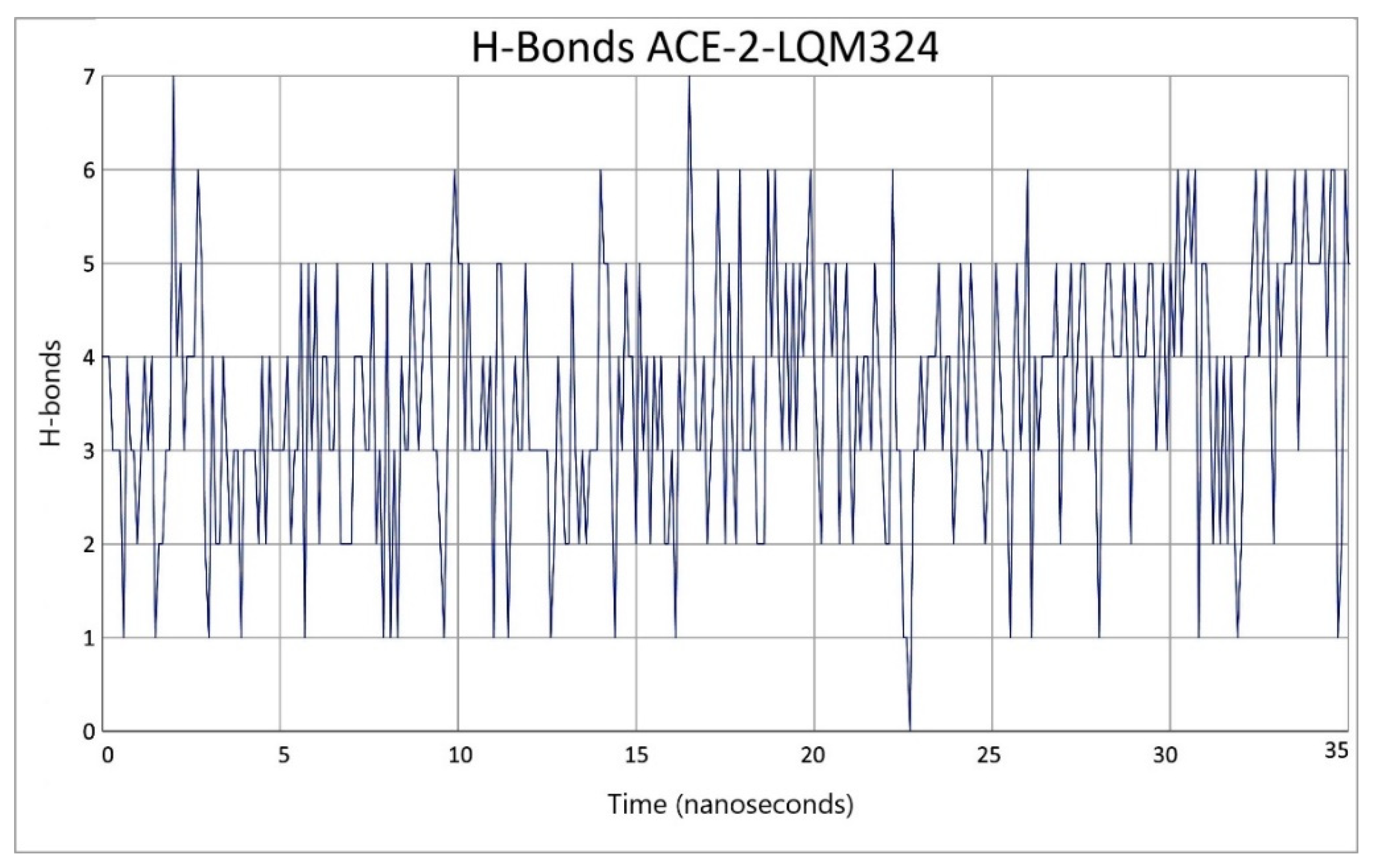
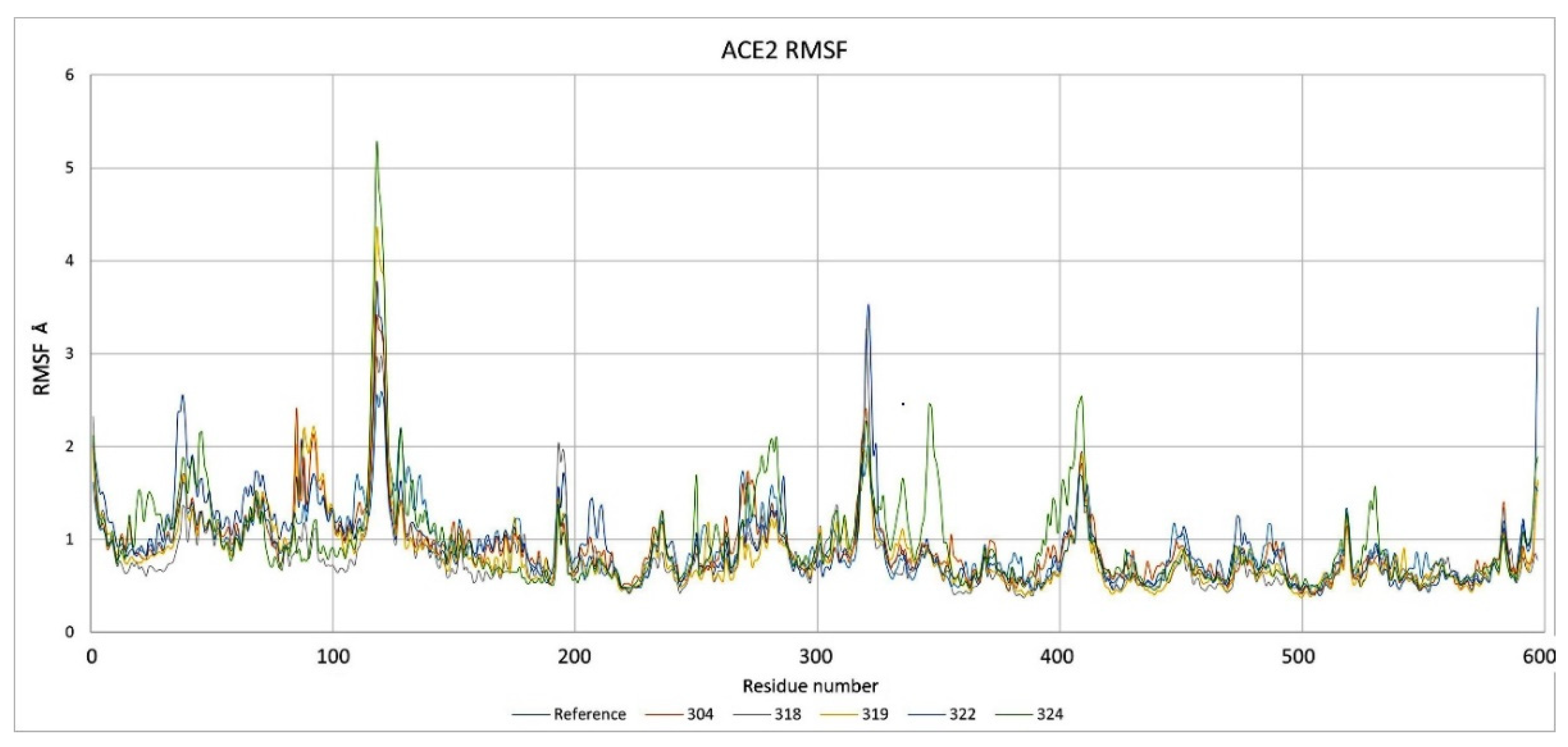
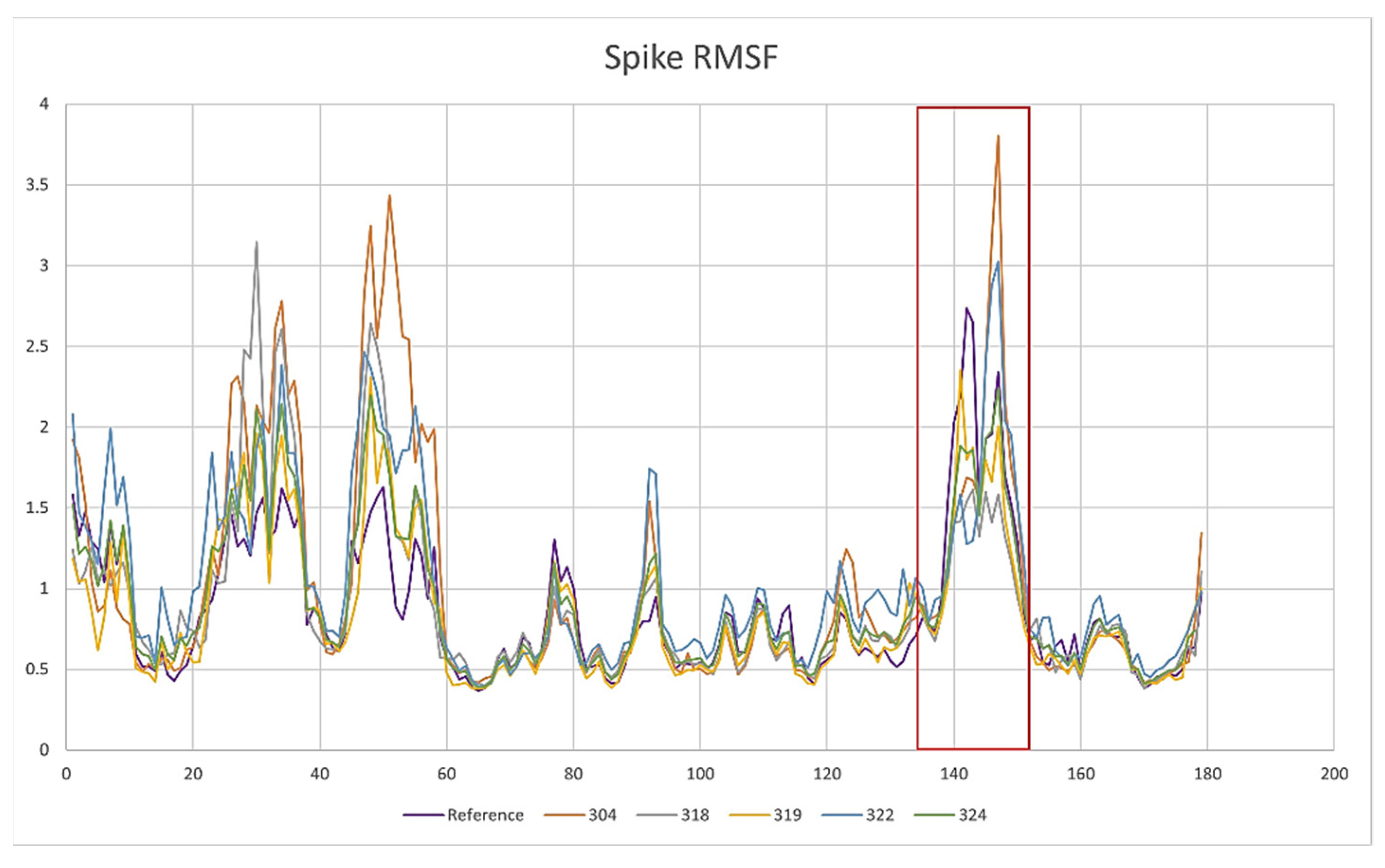
2.3. Description of the Dynamics of Protein–Protein Systems
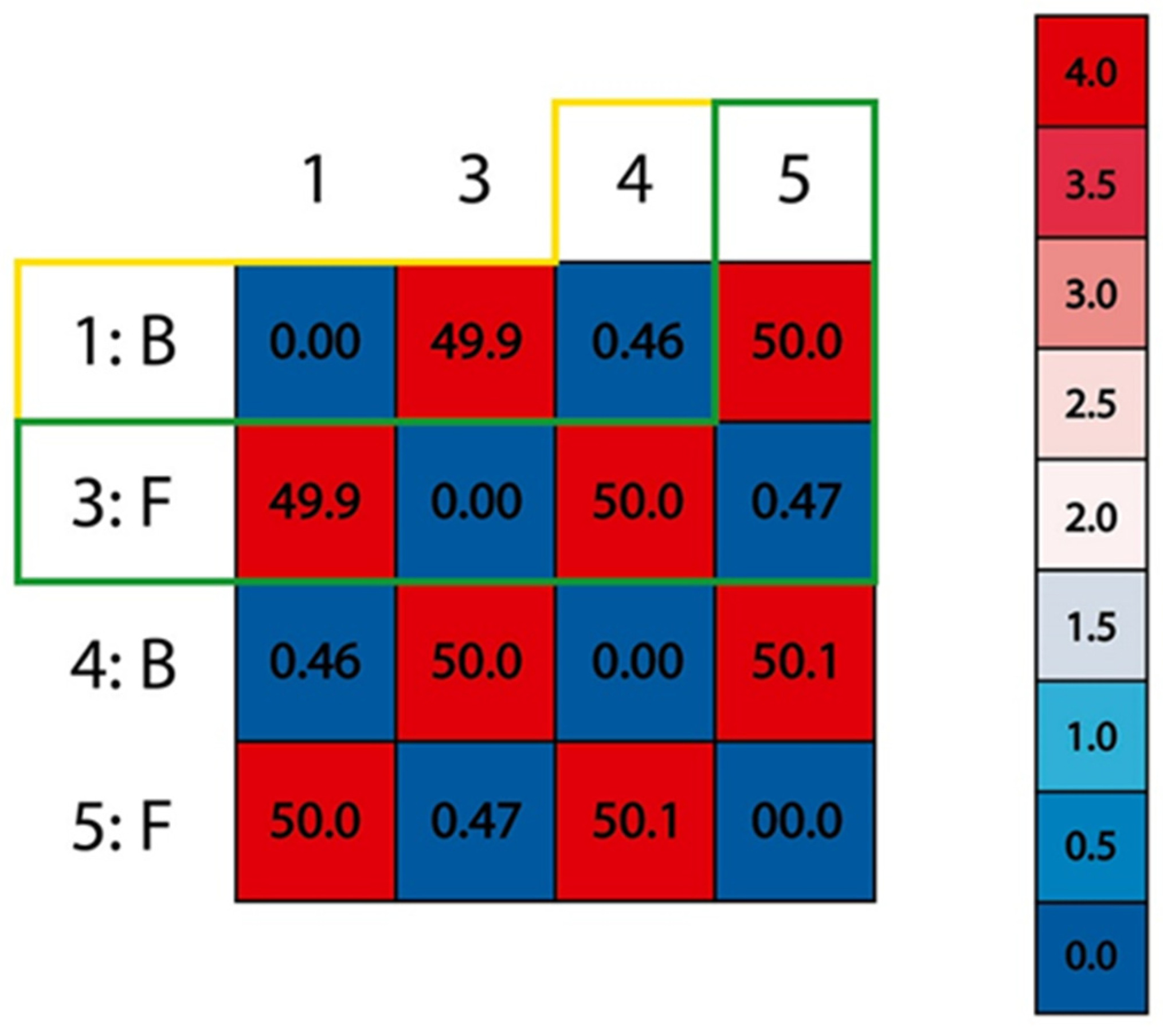
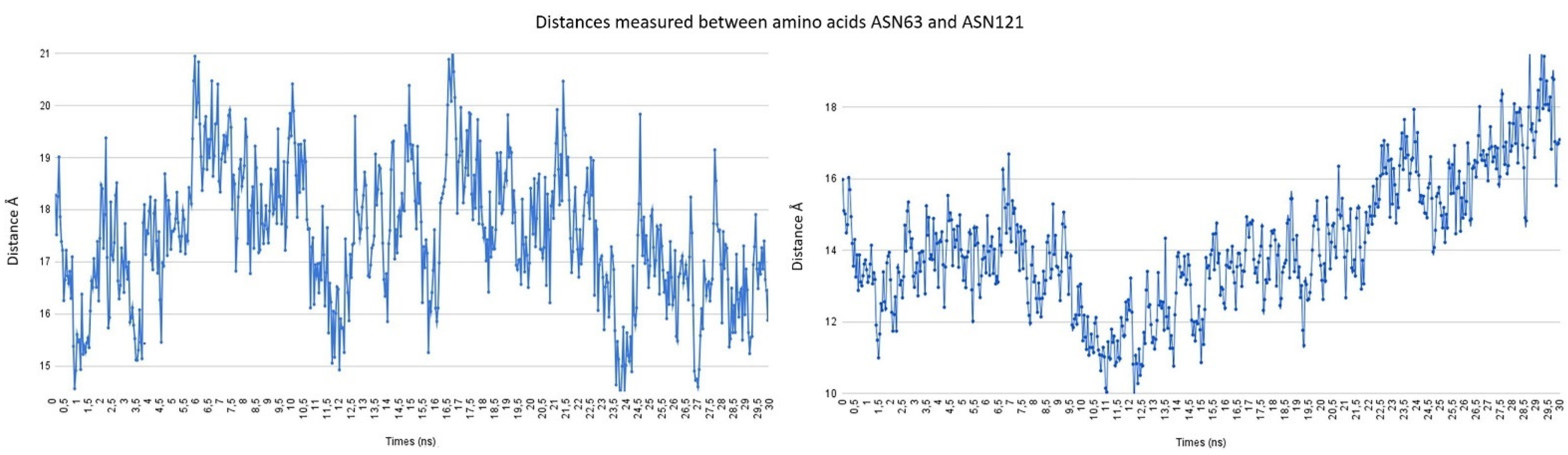
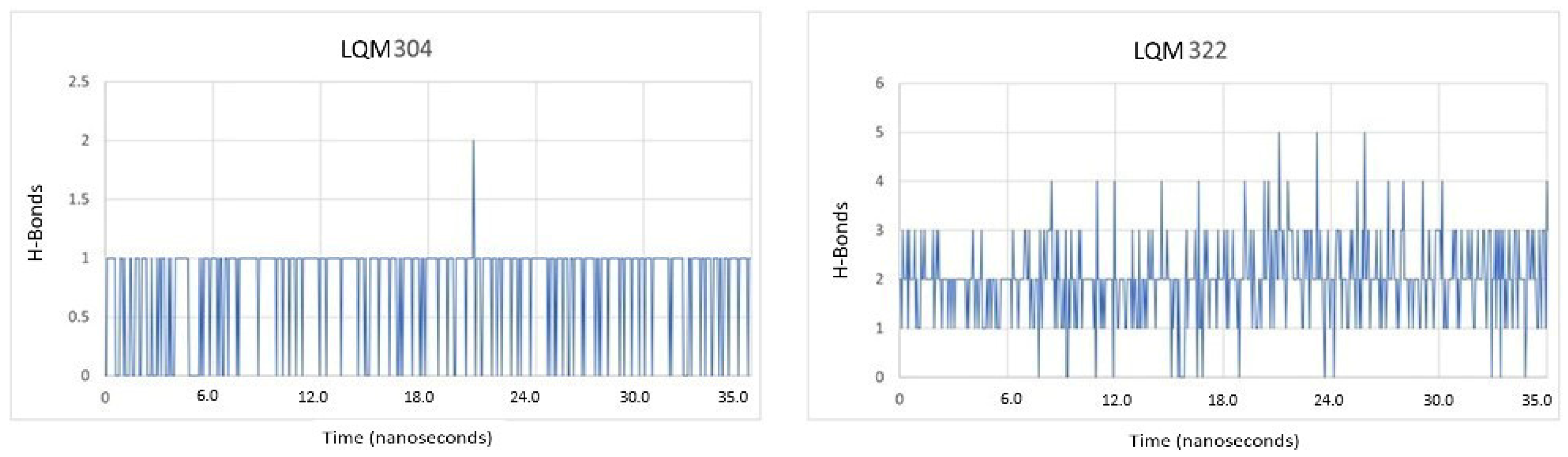
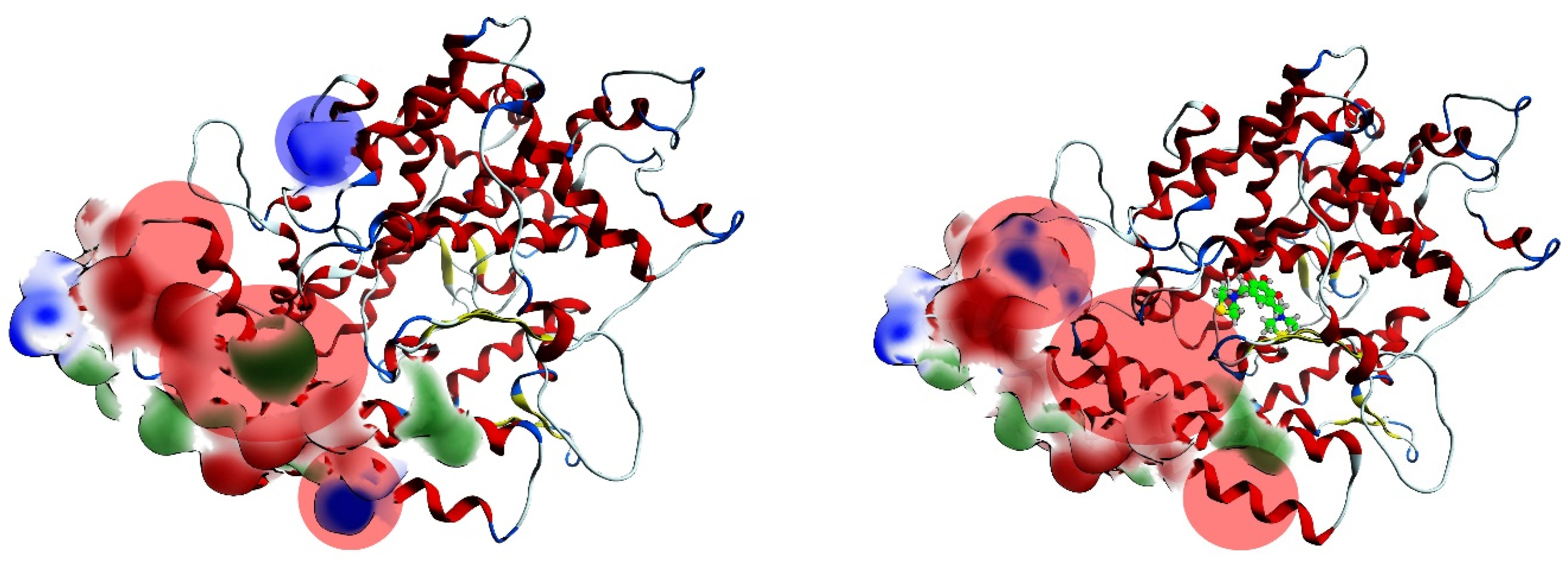
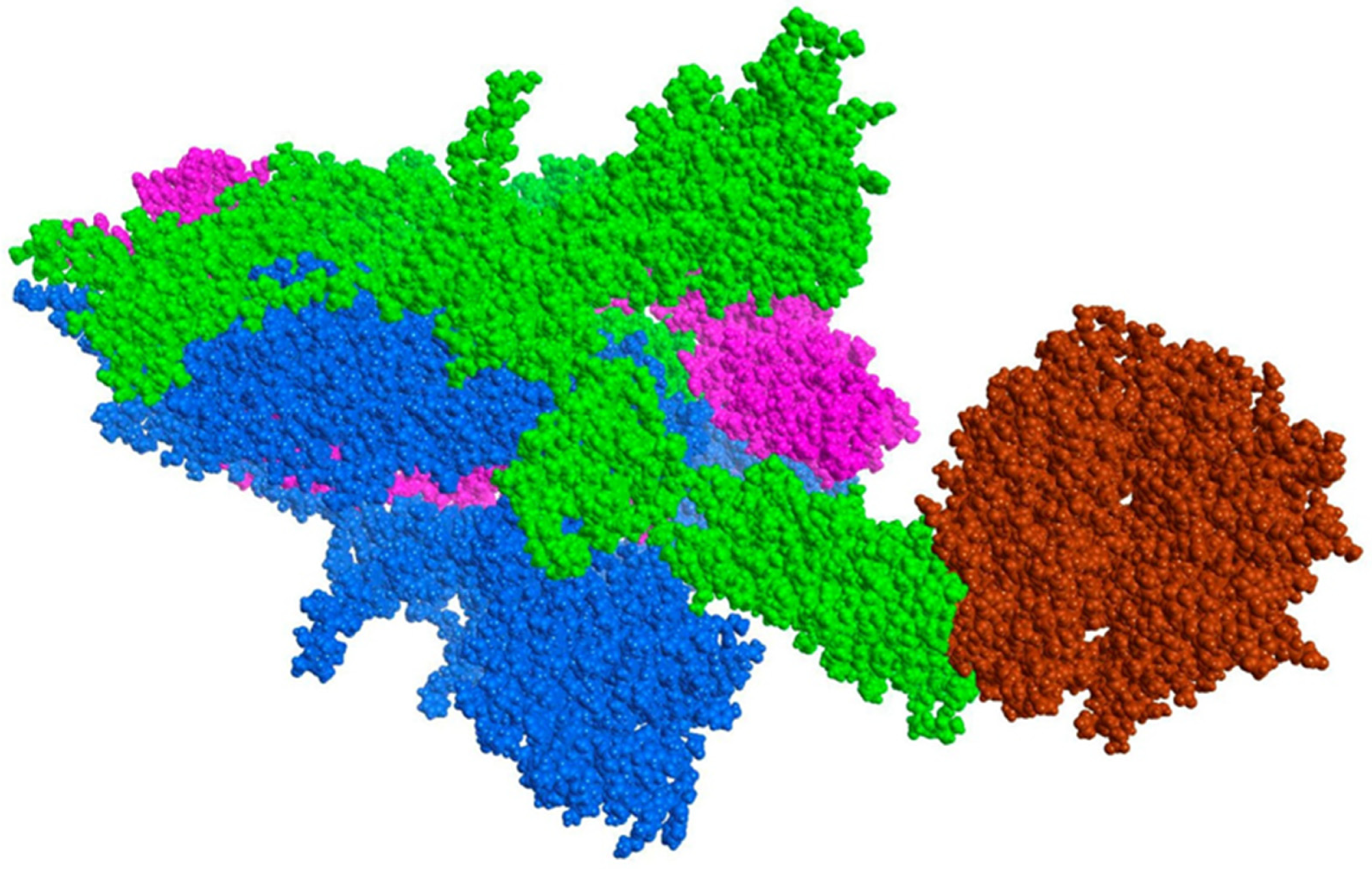
2.4. Procedure for the Analysis of “Patches” of the Interaction Surfaces
3. Materials and Methods
3.1. Preparation of Protein Structures
3.2. Validation Process of Molecular Protein–Ligand Coupling
3.3. Validation Process of Molecular Protein–Protein Coupling
3.4. Molecular Docking Protocol
3.5. Molecular Dynamics Performed for ACE2–In Systems
3.6. Description of the Dynamics of Protein–Protein Systems
- Heating for 1 ns, temperature gradient of 0 K to 300 K.
- Equilibrium of 4 ns at 300 K and 1 atm.
- Production 40 ns at 300 K and 1 atm.
3.7. Free Binding Energy Calculations
3.8. Procedure for the Analysis of “Patches” of the Interaction Surfaces
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19. 11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 18 July 2021).
- World Health Organization. Weekly Epidemiological Update on COVID-19—13 July 2021. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---13-july-2021 (accessed on 18 July 2021).
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Zhao, J.; Qiu, J.; Aryal, S.; Hackett, J.L.; Wang, J. The RNA architecture of the SARS-CoV-2 3′-untranslated region. Viruses 2020, 12, 1473. [Google Scholar] [CrossRef]
- Miao, Z.; Tidu, A.; Eriani, G.; Martin, F. Secondary structure of the SARS-CoV-2 5′-UTR. RNA Biol. 2021, 18, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A structural view of SARS-CoV-2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.J.R.; Alves da Silva, C.T.; Mendes, R.P.G.; Pena, L. Role of nonstructural proteins in the pathogenesis of SARS-CoV-2. J. Med. Virol. 2020, 92, 1427–1429. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Zhang, Y.; Ge, J.; Zheng, L.; Gao, Y.; Wang, T.; Jia, Z.; Wang, H.; Huang, Y.; Li, M.; et al. Architecture of a SARS-CoV-2 mini replication and transcription complex. Nat. Commun. 2020, 11, 5874. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef]
- Dinesh, D.C.; Chalupska, D.; Silhan, J.; Koutna, E.; Nencka, R.; Veverka, V.; Boura, E. Structural basis of RNA recognition by the SARS-CoV-2 nucleocapsid phosphoprotein. PLoS Pathog. 2020, 16, e1009100. [Google Scholar] [CrossRef]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Martí, D.; Torras, J.; Bertran, O.; Turon, P.; Alemán, C. Temperature effect on the SARS-CoV-2: A molecular dynamics study of the spike homotrimeric glycoprotein. Comput. Struct. Biotechnol. J. 2021, 19, 1848–1862. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e819. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Zhang, M.; Xiao, J.; Deng, A.; Zhang, Y.; Zhuang, Y.; Hu, T.; Li, J.; Tu, H.; Li, B.; Zhou, Y. Transmission dynamics of an outbreak of the COVID-19 delta variant B. 1.617. 2—Guangdong province, China, May–June 2021. China CDC Wkly. 2021, 3, 584–586. [Google Scholar] [CrossRef]
- Awadasseid, A.; Wu, Y.; Tanaka, Y.; Zhang, W. SARS-CoV-2 variants evolved during the early stage of the pandemic and effects of mutations on adaptation in Wuhan populations. Int. J. Biol. Sci. 2021, 17, 97–106. [Google Scholar] [CrossRef]
- Haas, E.J.; Angulo, F.J.; McLaughlin, J.M.; Anis, E.; Singer, S.R.; Khan, F.; Brooks, N.; Smaja, M.; Mircus, G.; Pan, K.; et al. Impact and effectiveness of mRNA BNT162b2 vaccine against SARS-CoV-2 infections and COVID-19 cases, hospitalisations, and deaths following a nationwide vaccination campaign in Israel: An observational study using national surveillance data. Lancet 2021, 397, 1819–1829. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc. Natl. Acad. Sci. USA 2021, 118, e2024302118. [Google Scholar] [CrossRef]
- Han, N.; Hwang, W.; Tzelepis, K.; Schmerer, P.; Yankova, E.; MacMahon, M.; Lei, W.; Katritsis, N.M.; Liu, A.; Felgenhauer, U.; et al. Identification of SARS-CoV-2–induced pathways reveals drug repurposing strategies. Sci. Adv. 2021, 7, eabh3032. [Google Scholar] [CrossRef]
- Singh, N.; Decroly, E.; Khatib, A.-M.; Villoutreix, B.O. Structure-based drug repositioning over the human TMPRSS2 protease domain: Search for chemical probes able to repress SARS-CoV-2 Spike protein cleavages. Eur. J. Pharm. Sci. 2020, 153, 105495. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.K.; Chaurasia, H.; Kumari, P.; Som, A.; Mishra, R.; Srivastava, R.; Naaz, F.; Singh, A.; Singh, R.K. Design, synthesis, and molecular dynamics simulation studies of quinoline derivatives as protease inhibitors against SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 1–24. [Google Scholar] [CrossRef]
- Sakkiah, S.; Guo, W.; Pan, B.; Ji, Z.; Yavas, G.; Azevedo, M.; Hawes, J.; Patterson, T.A.; Hong, H. Elucidating interactions between SARS-CoV-2 trimeric spike protein and ACE2 using homology modeling and molecular dynamics simulations. Front. Chem. 2021, 8, 1247. [Google Scholar] [CrossRef]
- Delgado, J.M.; Duro, N.; Rogers, D.M.; Tkatchenko, A.; Pandit, S.A.; Varma, S. Molecular basis for higher affinity of SARS-CoV-2 spike RBD for human ACE2 receptor. Proteins 2021, 89, 1134–1144. [Google Scholar] [CrossRef]
- Hakmi, M.; Bouricha, E.M.; Jihane, A.; Lmimouni, B.; Harti, J.; Belyamani, L.; Ibrahimi, A.; Ibrahimi, P. In silico exploration of small-molecule α-helix mimetics as inhibitors of SARS-COV-2 attachment to ACE2. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Zhuo, L.-S.; Wang, M.-S.; Yang, J.-F.; Xu, H.-C.; Huang, W.; Shang, L.-Q.; Yang, G.-F. Insights into SARS-CoV-2: Medicinal Chemistry Approaches to Combat Its Structural and Functional Biology. Top. Curr. Chem. 2021, 379, 23. [Google Scholar] [CrossRef]
- Vázquez-Valadez, V.H.; Carrillo, C.I.; Ramírez, C.N.; Abrego, V.H.; López Castañares, R.; Angeles, E. Evaluation of the inhibition of angiotensin-converting enzyme by new thiomorpholine compounds using capillary zone electrophoresis. J. Mex. Chem. Soc. 2018, 62, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Lazim, R. Application of conventional molecular dynamics simulation in evaluating the stability of apomyoglobin in urea solution. Sci. Rep. 2017, 7, 44651. [Google Scholar] [CrossRef] [Green Version]
- Pace, C.N.; Fu, H.; Lee Fryar, K.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K.; et al. Contribution of hydrogen bonds to protein stability. Protein. Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Chen, H.; Cai, W.; Shao, X.; Chipot, C. BFEE2: Automated, streamlined, and accurate absolute binding free-energy calculations. J. Chem. Inf. Modeling 2021, 61, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- DeChancie, J.; Houk, K.N. The origins of femtomolar protein–ligand binding: Hydrogen-bond cooperativity and desolvation energetics in the biotin–(strept)avidin binding site. J. Am. Chem. Soc. 2007, 129, 5419–5429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preißner, R.; Goede, A.; Frömmel, C. Dictionary of interfaces in proteins (DIP). Data bank of complementary molecular surface patches11Edited by R. Huber. J. Mol. Biol. 1998, 280, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE) 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2021.
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–38. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.-Y.; Katsamba, P.S.; Sampson, J.M.; Schön, A.; Bimela, J.; et al. Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a pH-dependent switch to mediate endosomal positioning of receptor-binding domains. Cell Host Microbe 2020, 28, 867–879.e865. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Valadez, V.H.; Abrego, V.H.; Martínez, P.A.; Torres, G.; Zúñiga, O.; Escutia, D.; Vilchis, R.; Velázquez, A.M.; Martínez, L.; Ruiz, M.; et al. Docking studies of methylthiomorpholin phenols (LQM300 Series) with angiotensin-converting enzyme (ACE). Open Med. Chem. J. 2013, 7, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Peng, G.; Wilken, M.; Geraghty, R.J.; Li, F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2012, 287, 8904–8911. [Google Scholar] [CrossRef] [PubMed] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LQM304 | LQM318 | LQM319 | |
 |  |  | |
| LQM322 | LQM324 | ||
 |  | ||
| System (ACE2-LQM) | ΔG (kcal/mol) | Deviation ± (kcal/mol) |
|---|---|---|
| 304 | −4.91 | 0.21 |
| 318 | −4.98 | 0.20 |
| 319 | −5.71 | 0.41 |
| 322 | −5.59 | 0.26 |
| 324 | −4.89 | 0.29 |
| Area Å2 | % ASA | |
|---|---|---|
| Hydrophobic | 419.20 | 4.87 |
| Negative | 224.70 | 2.62 |
| Positive | 204.30 | 2.38 |
| ACE2 | 304 | 318 | 319 | 322 | 324 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AREA | % ASA | AREA | % ASA | AREA | % ASA | AREA | % ASA | AREA | % ASA | AREA | % ASA | |
| Hyd | 127.45 | 0.57 | 112.25 | 0.50 | 66.36 | 0.30 | 73.40 | 0.33 | 72.85 | 0.33 | 63.90 | 0.29 |
| Negative | 150.20 | 0.67 | 157.65 | 0.70 | 156.35 | 0.72 | 148.50 | 0.66 | 165.10 | 0.73 | 152.15 | 0.68 |
| Positive | 58.60 | 0.26 | 0.00 | 0.00 | 214.40 | 0.95 | 50.50 | 0.20 | 163.40 | 0.74 | 164.10 | 0.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Valadez, V.H.; Hernández-Serda, A.; Jiménez-Cabiedes, M.F.; Aguirre-Vidal, P.; González-Tapia, I.; Carreño-Vargas, L.; Alarcón-López, Y.A.; Espejel-Fuentes, A.; Martínez-Soriano, P.; Lugo Álvarez, M.; et al. Evaluation of Inhibitory Activity In Silico of In-House Thiomorpholine Compounds between the ACE2 Receptor and S1 Subunit of SARS-CoV-2 Spike. Pathogens 2021, 10, 1208. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10091208
Vázquez-Valadez VH, Hernández-Serda A, Jiménez-Cabiedes MF, Aguirre-Vidal P, González-Tapia I, Carreño-Vargas L, Alarcón-López YA, Espejel-Fuentes A, Martínez-Soriano P, Lugo Álvarez M, et al. Evaluation of Inhibitory Activity In Silico of In-House Thiomorpholine Compounds between the ACE2 Receptor and S1 Subunit of SARS-CoV-2 Spike. Pathogens. 2021; 10(9):1208. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10091208
Chicago/Turabian StyleVázquez-Valadez, Victor H., Alejandro Hernández-Serda, Ma. Fernanda Jiménez-Cabiedes, Pablo Aguirre-Vidal, Ingrid González-Tapia, Laura Carreño-Vargas, Yoshio A. Alarcón-López, Andrea Espejel-Fuentes, Pablo Martínez-Soriano, Miguel Lugo Álvarez, and et al. 2021. "Evaluation of Inhibitory Activity In Silico of In-House Thiomorpholine Compounds between the ACE2 Receptor and S1 Subunit of SARS-CoV-2 Spike" Pathogens 10, no. 9: 1208. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10091208