
Anthroponotic-Based Transfer of Staphylococcus to Dog: A Case Study
by
 ,
,
Massimiliano Orsini
1,*, 
Sara Petrin
1,
Michela Corrò
2,
Giulia Baggio
1,
Elena Spagnolo
2 and
Carmen Losasso
1,* 1
Laboratory of Microbial Ecology and Genomics, Istituto Zooprofilattico Sperimentale delle Venezie, Viale dell’Università 10, 35020 Legnaro, Italy
2
Department of Diagnostics in Animal Health, Istituto Zooprofilattico Sperimentale delle Venezie, Viale dell’Università 10, 35020 Legnaro, Italy
*
Authors to whom correspondence should be addressed.
Pathogens 2022, 11(7), 802; https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11070802
Submission received: 6 June 2022
/
Revised: 14 July 2022
/
Accepted: 14 July 2022
/
Published: 15 July 2022
(This article belongs to the Special Issue Exploring the Epidemiology, Pathogenicity, and Therapeutic Options of Staphylococcus spp.)
Abstract
:Although usually harmless, Staphylococcus spp. can cause nosocomial and community-onset skin and soft tissue infections in both humans and animals; thus, it is considered a significant burden for healthcare systems worldwide. Companion animals have been identified as potential reservoirs of pathogenic Staphylococcus with specific reference to Methicillin Resistant Staphylococcus aureus (MRSA). In this study, we investigated the circulation and the genetic relationships of a collection of Staphylococcus spp. isolates in a family composed of four adults (a mother, father, grandmother, and grandfather), one child, and a dog, which were sampled over three years. The routes of transmission among humans and between humans and the dog werelyzed. The results displayed the circulation of many Staphylococcus lineages, belonging to different species and sequence types (ST) and being related to both human and pet origins. However, among the observed host-switch events, one of them clearly underpinnthroponotic route from a human to a dog. This suggests that companion animals can potentially have a role as a carrier of Staphylococcus, thus posing a serious concern about MRSA spreading within human and animal microbial communities.
1. Introduction
Staphylococcus aureus (S. aureus) is a common bacterium present on the skin and mucous membranes in 20–30% of human healthy subjects [1]. Although usually harmless, it can be the cause of nosocomial and community-onset skin and soft tissue infections both in humans and animals; thus, it is considered a significant burden for healthcare systems worldwide [2].
Some strains of S. aureus can develop resistance to beta-lactam antibiotics such as penicillin, which are widely used to treat human infections [3]. These strains are known as methicillin-resistant Staphylococcus aureus (MRSA).
Humans become infected by S. aureus mainly through direct contact with infected persons or with medical instruments and equipment. In recent years, companion animals, such as dogs, cats, and horses, have been identified as potential reservoirs of S. aureus and specifically of MRSA [4]. This assumption derives from many case reports that have documented S. aureus infections in animal owners, associated with colonization through genetically related strains in their pets [5]. On the other hand, there are suggestions that S. aureus carriage is not sustained for long periods by companion animal hosts in a clean environment [4].
It is a matter of fact that S. aureus exhibits tropisms to many distinct animal hosts [6]. The infection is often maintained in both humans and lower vertebrate animals and transmitted in either direction (amphixenoses). Host switching is not infrequent; it is often accompanied by specific genetic signatures such as a loss of phages, which are known to play a role in human colonization [7]. The anthroponotic transmission is not infrequent either [8], and can be the cause of the human-to-human spread of potentially virulent strains via animal-supported routes [9]. In addition to those already mentioned routes of infection, a third hypothesis is plausible, and it consists of the possibility of an anthroponotic-based transfer of S. aureus and MRSA to companion animals. In this case, anthroponosis refers to a human infectious disease that can be naturally transmitted to other animals, and it is considered the reverse of the zoonosis, where a pathogen or a parasite infects primarily an animal but can also infect and cause disease in humans. Thus, based on the existing evidence, whether companion animals are true reservoirs of S. aureus and in particular MRSA or whether they should only be considered as contaminated living vectors is a topic that is still debated and is worthy of further study.
In this study, we tested this hypothesis by investigating the circulation and the genetic relationships of isolates of Staphylococcus spp. in a family composed of four adults (a mother, father, grandmother, and grandfather), one child, and a dog, which were sampled because of recurrent or relapsing folliculitis, which was resolved through general and local therapy. The routes of transmission among humans and between humans and the dog were also estimated.
2. Results
2.1. Clinical Context
A single family, composed of a father, mother, grandfather, grandmother, a child, and a dog, was enrolled and regularly sampled in the period 2017–2019. Three adults suffered from recurrent folliculitis: the father had recurrent folliculitis affecting several body districts since 2017, and he was treated with cephalosporin (400 mg die for ten days through oral administration) and chlorhexidine (1% topic daily administration) and then the infection was resolved in 2018; the grandfather suffered from scrotal folliculitis in 2018, which required surgery, and a recurrent skin folliculitis in the same year, which was successfully treated in the same way; and the grandmother had purulent folliculitis in various body areas in 2018, which resolved in 2019 as well. The mother and the child were asymptomatic.
2.2. Dataset Description
A total of 25 isolates were collected in the present study (Table 1). Seven samples were collected from the dog and were identified as S. pseudintermedius (n = 4), S. aureus (n = 2), and S. capitis (n = 1). The remaining isolates were identified as S. aureus (n = 13), S. epidermidis (n = 3), S. lugdunensis (n = 1), and S. warneri (n = 1), and were collected from different subjects from the same family over the three-year period considered.
2.3. Genomic Analysis
The draft assemblies ranged in size from 2.46 to 2.92 Mb with a range of 2312 to 3276 protein coding sequences (full assembly metrics are described in Supplementary Table S1). The 7-loci MLST analysis returned a reliable ST for all S. aureus isolates, which belonged to five different STs: ST8 was identified in 2017; ST8, ST10, ST22, ST59, ST7204, and ST707 were identified in 2018; and ST10, ST22, and ST59 were identified in 2019 (Table 1). The S. epidermidis strains showed a more heterogeneous profile, with a strain belonging to ST5, one belonging to ST153, and one belonging to ST1133 (Table 1, Supplementary Table S1). Interestingly, two novel S. pseudintermedius STs (ST2168 and ST2169) and one new S. aureus ST (ST7204) were identified and submitted to the pubMLST database [https://pubmlst.org accessed on 15 January 2022]. All these new STs were isolated from the dog in 2017, 2018, and 2019. A MLST scheme is not available for S. warnerii and S. capitis.
2.4. Antibiotic Resistance Profiling
The results of the presence of antibiotic-resistance genes showed a generalized resistance to the beta-lactam antibiotics, encoded by the blaZ gene, with the exclusion of the S. warnerii and the S. lugdunensis strains (Table 2). Two profiles of genes carrying the resistance to aminoglycoside antibiotics have been observed. In the first, shared by S. pseudointermedius and S. aureus ST8 strains, the resistance is encoded by the ant(6)-ia and the aph(3)-III genes; a second pattern consisting of aadD, aac(6)-aph(2), and cat(pC221) genes was observed in S. epidermidis strains. The presence of the cat(pC221) gene was also observed in the two S. pseudointermedius ST63 strains. The Fosfomycin resistance gene fosB was only detected in the three S. epidermidis strains. The mecA gene, conferring resistance to methicillin, was found in all the S. aureus ST8 strains, including the strain isolated from the dog; moreover, it was found in an S. epidermidis ST5 strain (Sep_19_GM_ST5_150797). The two genes mphC and msrA, conferring resistance to macrolides and to erythromycin and streptogramin B, respectively, were found in S. aureus ST8 and one ST10 strain isolated in 2019, in addition to the S. epidermidis Sep_17_CH_ST1133_123222 strain. One erythromycin inducible gene (ermB) conferring resistance to streptogramin and macrolides was found in all the S. pseudointermedius strains, while the ermC gene was found in one S. epidermidis strain (Sep_19_GM_ST5_150797). Finally, the S. epidermidis Sep_19_GM_ST5_150797 strain showed the presence of two genes (vgaA and vgaALC) conferring resistance to streptogramin A antibiotics and related compounds.
2.5. Prophages Analysis
A total of 10 different intact prophages were identified in the whole dataset (Table 3, extended results are described in Supplementary Table S2). The analysis revealed that prophages showed a strictly species-specific and ST-specific pattern, with the richest clusters being S. aureus ST10 and S. aureus ST8, with 4 and 3 different prophages detected, respectively (Table 3). The S. capitis Sca_19_DO_STX_150837 sample showed the presence of a single, intact prophage (Staphy_StB12_NC_020490) that was not observed in any other sample. A different pattern was observed for the two S. aureus samples isolated from the dog (Sau_18_DO_ST7204_153686-1 and Sau_17_DO_ST8_123220), belonging to ST7204 and ST8, respectively. Interestingly, the former, despite differing from the sample isolated from the child in the same period (Sau_18_CH_ST707_153686-4) by only one allele (arcC), shared the same prophage pattern with it.
2.6. Plasmids Content
A total of 13 plasmid replicons were observed (Table 4), and for the majority of them it was possible to identify a pattern of presence linked to the sequence type. For example, the rep19, rep7c, and the rep16 plasmid replicons were observed in the S. aureus ST8 samples only, while the rep16 plasmid replicon was further observed in the S. aureus ST707 and ST7204 samples, together with the repUS5 plasmid replicon as well, and shared with the Sau_19_MO_ST10_150824 strain. The S. epidermidis strain Sep_19_GM_ST5_150797 displayed a unique pattern consisting of rep7a, repUS19, repUS12, and rep5b. Interestingly, the rep7a plasmid replicon was observed in samples of different species; namely, it was present in S. epidermidis (3/3), S. lugdunensis (1/1), S. pseudintermedius (2/4), and S. aureus ST8 (1/5) isolates. On average, when present, one to three plasmid replicons were identified in the samples, and only one sample (Sep_19_GM_ST5_150797) carried four plasmid replicons. Conversely, samples belonging to S. aureus ST22 and ST59, in addition to two S. aureus ST10 isolates, did not show any contig-carrying plasmid replicons. Some other plasmid replicons, such as rep20, rep39, repUS35, and rep22, were sporadically observed in Sca_19_DO_STX_150837, Sep_17_CH_ST1133_123222, Swa_19_FA_STX_150815, and Sep_17_CH_ST1133_123222, respectively.
2.7. IEC Genes
The results of the IEC gene sequence search at the protein level are shown in Table 5. The scn gene was found in all samples, highlighting the presence of an IEC-converting gene (Beta-C-Phi-s); on the contrary, the sea and sep genes were not detected in any samples. The sak gene was found in all samples except for the two S. aureus ST59 samples, while the chp gene was absent only in the S. aureus ST10 samples. The predominant IEC variant [10] was type B (sak, chp, and scn), followed by type C (chp, scn), and type E (sak, and scn).
2.8. Phylogenetic Analysis
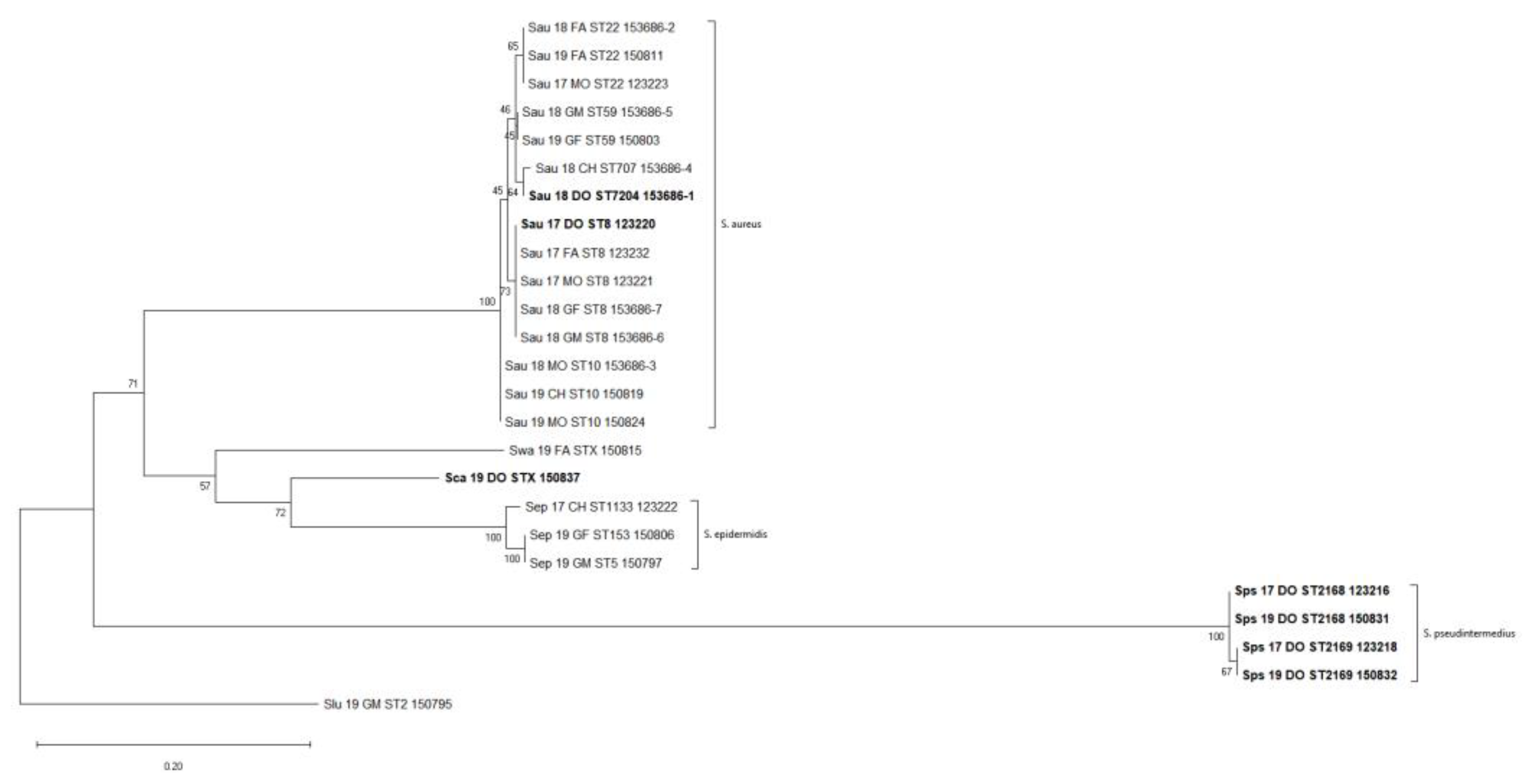
Single nucleotide polymorphisms (SNPs) were taken from 171 informative sites in the core genome within the entire dataset. A maximum-likelihood phylogenetic tree from this matrix was built, which resolved three major clades and three single leaves that agree with species-wide phylogeny (Figure 1). The largest clade encompassed all the S. aureus strains, irrespective of the isolation source (dog or human), which were further grouped according to the ST. A second clade grouped all the S. pseudointermedius strains; finally, a small clade included all the S. epidermidis strains.
To better investigate the intraspecies relationships, the SNP matrixes were re-calculated for those clusters that exhibited multi-host or possible host-switching events only, i.e., the S. aureus and S. epidermidis clusters, while the S. pseudointermedius cluster was considered as the baseline for a species recurrently isolated from a single-host (Supplementary Table S3). The distance matrix for the S. aureus isolates only, reporting the total number of different SNPs between any pair of isolates, showed a distance of about five SNPs per year with some deviations depending on the specific ST. Indeed, the SNP distance reached the lower value in the case of the S. aureus ST59 and ST10 strains that displayed a minimum value of three and two SNPs/year, respectively. On the contrary, the isolates belonging to ST8 and ST22 showed a minimum value of 4 and 11 SNPs/year, respectively.
A possible host-switching event was observed within the S. aureus clade where the closest related strains (Sau_17_DO_ST8_123220) and (SAU_17_FA_ST8_123232), only differing by four core genome SNPs, belonged to the dog and the father, respectively (Supplementary Table S3). Assuming a mutation rate of 8 SNPs per year [11] for S. aureus ST8, we suggest about 6 months of divergence for the ST8 strains.
Interestingly, the SNP variation analysis revealed a different genomic stability between S. aureus and S. pseudintermedius over time. In particular, within the S. aureus clusters, the ST8, ST10, and ST59 displayed lower genomic differences in terms of SNPs variation. Conversely, the ST22 strains were less genomically similar, as they showed the highest SNP differences per year (i.e., 11 SNPs). Surprisingly, the S. aureus ST707 and the ST7204 strains SNP distance was one of the lowest (i.e., 5 SNPs) found within the investigated dataset, despite belonging to different STs.
In regard to the S. pseudintermedius strains, the ST2169 can be compared to S.aureus ST22 strains in terms of genomic stability as a difference of 10 SNPs was found among them. The highest differences in terms of SNPs can be seen among ST2168 strains, with a difference of about 21 SNPs per year.
3. Discussion
Of the 25 Staphylococcus isolates collected in the present study, all but the four S. pseudintermedius strains were of a typically human origin [12,13,14], even though the S. aureus strains were isolated from both humans and the family dog. On the contrary, S. pseudintermedius, typically colonizing dogs’ skin [13], was isolated during the entire sampling time span from the dog only. This suggests the possibility for the unidirectional transfer of S. aureus circulating in the human reservoir from humans to dogs and not the other way around.
S. aureus ST8 that colonized the mother, the father, and the dog in 2017 and the grandparents in 2018, is known to be a pandemic clonal lineage of hypervirulent, community-acquired, methicillin-resistant S. aureus that includes the well-known PFGE strain USA300 [14]. This ST is the predominant MRSA isolated in North America and some parts of Europe [15] and it is known to be multiresistant and mecA-positive. Notably, this ST was spread both in symptomatic (father, grandmother, and grandfather) and asymptomatic (mother) humans. This poses serious concerns regarding (i) the possibility for such a virulent ST to provoke folliculitis, (ii) the possibility for asymptomatic people to spread this ST to immunocompromised people, and (iii) the possibility for this ST to be transferred from humans to pets and vice-versa. Since conserved prophage and plasmid replicon patterns were shared by Sau_17_MO_ST8_123221 Sau_17_DO_ST8_123220 Sau_18_GM_ST8_153686-6, it is possible to speculate a transfer from the mother to the dog.
The S. aureus ST59, isolated from the grandparents in two different sampling sessions (2018 and 2019), is the most prevalent sequence type isolated in Eastern Asia, Europe, and North America, representing the major cause of skin and soft tissue infections, as well as being described as a commensal colonizer [16,17]. Thus, we cannot exclude that the severe symptoms displayed by both grandparents could be ascribable to ST8 and/or ST59. In addition, in our dataset, this ST, as well as ST8, resulted in being multiresistant, with particular reference to beta-lactamases, tetracycline, methicillin, aminoglycosides macrolides, erythromycin, and streptogramin B.
It is important to highlight that the SAU_18_CH_ST707_153686-4 and SAU_18_DO_ST7204_153686-1 strains, isolated from the child and the dog, respectively, despite belonging to different STs (707 and 7204), were very close in terms of SNPs and shared the same prophage profiles, antibiotic resistance genes, and plasmid profile, and were the only two strains displaying a common mobilome. This, together with the remark that ST7204 was isolated for the first time in this study from a dog, led us to hypothesize that this strain could have been transferred to the dog via the child.
In addition, this study revealed the same IEC type in both dog and human strains, thus suggesting the possibility that all the S. aureus found in the dog had a human source. IEC genes are involved in host immune evasion and provide S. aureus with a unique mechanism to adapt to and counteract the human host [18].
Although ubiquitous on human skin, S. epidermidis was only isolated from the child’s mouth in 2017 and from the grandparents’ noses in 2019. The three isolates belonged to different STs, and this suggests their independent origin. Even though S. epidermidis is one of the major representative taxa of human skin microbiota, it is considered an opportunistic pathogen and has been found to be a carrier and reservoir for antibiotic-resistance genes, particularly those that do not impose a major fitness cost on the bacteria, such as methicillin resistance coding elements [19,20]. Accordingly, there is evidence suggesting the possibility of the transfer of methicillin resistance cassettes from S. epidermidis to S. aureus [21]. The three isolates characterized herein shared the presence of aminoglycosides, Fosfomycin, methicillin, macrolides, erythromycin, and streptogramin A and B resistance genes. While S. epidermidis infections rarely develop into severe diseases, their frequency, and the fact that they are multiresitent and thus extremely difficult to treat, represent a serious burden for public health.
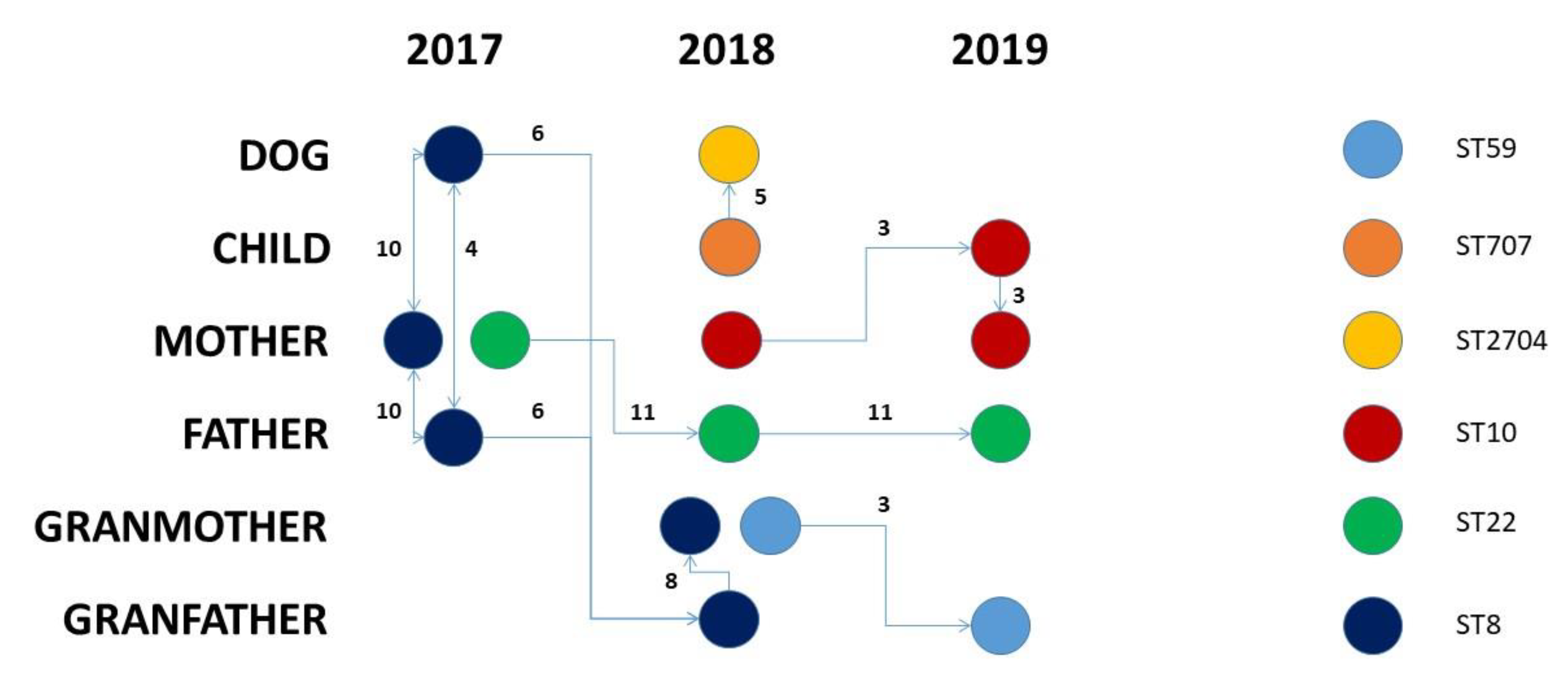
Taking into consideration the SNP distances, we have drawn a hypothetical transmission path for the S. aureus strains that has been summarized in Figure 2. Within the investigated family from 2017 and 2019, five different S. aureus STs had circulated. The S. aureus ST8 strains circulated in 2017 and 2018 between the family members, excluding the child. The three ST8 strains isolated in 2017 differed for 4–10 SNPs; the two ST8 strains isolated from the grandparents in 2018 differed by 6 and 14 SNPs from the closest 2017 strains and just 8 SNPs were among them. From a conservative point of view, this suggests a unidirectional transfer from grandfather to grandmother (Figure 2).
Conversely, in the case of the S. aureus ST10, the circulation might have originated from the mother in 2018, where the isolates persisted with a number of SNPs equal to three and then it was transferred to the child. The S. aureus ST22 originated from the mother in 2017 and then moved to the father in 2018 where it persisted until 2019 with a slightly higher SNP distance (11 SNPs). The S. aureus ST59 was probably transferred to the grandfather from the grandmother after a divergence of three SNPs. Finally, the S. aureus ST707-ST2704 circulated between the child and the dog, and the two strains remained closely related as they differed by only five SNPs.
This evidence, together with the presence of the same antibiotic resistance gene pattern, EIC type variant, and plasmid and prophage patterns provide the proof for the anthroponotic transmission of S. aureus (ST707-ST2704) from the child to the family dog and suggest that companion animals can potentially have a role in the spread of Staphylococcus between humans and animals, thus posing a serious concern about MRSA spreading within human and animal microbial communities.
4. Materials and Methods
4.1. Study Design
A single family, composed of father, mother, grandfather, grandmother, a child, and a dog, was yearly sampled in the period 2017–2019. Sterile test tubes together with swabs (Modified Amies Medium, Meus s.r.l. Piove di Sacco, Italy) were used to collect samples from human skin lesions, intact skin, and mucous membranes (hands, mouth, and nostrils) in symptomatic subjects and from mucous membranes (hands, mouth, and nostrils) in asymptomatic ones. In dogs, mouth, ear canals, axillary region, and groin were sampled in the same way. Swabs were stored in refrigerated boxes at 6–8 °C and were cultured within 24/36 h of collection.
The dog was monitored as the suspected cause of the infection by the attending physician, but it never showed skin infections. The study was ethically approved by IZSVe ethical committee (opinion n. CE_IZSVE 02/2022).
4.2. Microbiological Investigations
Bacterial isolation was performed according to standard laboratory culture techniques. Briefly, each swab was inoculated onto the following solid media: nutrient blood agar (Blood Agar Base, Biolife, Milano, Italy; supplemented with 5% defibrinated sheep blood, Allevamento Blood, Teramo, Italy), Enterobacteriaceae selective medium (McConkey agar, Oxoid, Basingstoke, UK), methicillin-resistant Staphylococci selective medium, (CHROMagar® MRSA II, BD BBL™, Heidelberg, Germany), and nutrient broth (Mueller–Hinton broth, Biokar Diagnostics, Alonne, FR, enriched with 6.5% sodium chloride, Sigma-Aldrich s.r.l., Milano, Italy), and then incubated at 37 °C in aerobic conditions for 24–48 h.
In case of the absence of microbial growth on the plates after 24 h of incubation, in the presence of turbidity of the broth, seeding onto methicillin-resistant Staphylococci selective medium was carried out as described above.
Staphylococcal colonies were recognized on nutritive medium, according to colony morphology, Gram stain appearance and cellular morphology, and catalase and coagulase tube tests. Suspected pink to mauve colonies grown on the methicillin-resistant Staphylococci selective medium were suspected and confirmed as being MRSA with a multiplex-PCR targeting nuc and mecA genes [22]. S. aureus DSMZ 11729 was used as positive control.
Species identification was performed by MALDI-TOF MS: Microflex LT instrument (MALDI Biotyper, Bruker Daltonics) equipped with FlexControl software (version 3.3, Bruker Daltonics).
4.3. DNA Extraction and Whole-Genome Sequencing
Genomic DNA was extracted from single colonies cultured on Blood Agar plates, using the QIAamp DNA Mini kit (QIAGEN), according to the manufacturer’s protocol, and quantified with a Qubit 3.0 Fluorometer (Life Technologies). Libraries for whole genome sequencing were prepared starting from 1 ng of genomic DNA with the Nextera XT DNA sample preparation kit (Illumina). Paired-end high-throughput sequencing (2 × 251 bp) was performed on a MiSeq sequencing platform, using the v3 chemistry.
4.4. Genome Assembly and Annotation
De novo contigs were generated for each genome using SPAdes (version 3.13) via parameters for 2 × 251 Illumina reads [23]. Contigs longer than 200 bp were retained. Chromosome assemblies were annotated by Prokka (version 1.11) [24]. The genus and species were confirmed by using the online version of kmerFinder tool (version 3.2) [25]. The multi-locus sequence type (MLST) was derived from the assemblies using the MLST web application (version 2.0) [26]. Antibiotic resistance genes were identified using the ResFinder web application (version 4.1) [27] accessed in March 2020. Finally, integrated prophages were searched using the PHASTER web server (accessed in March 2020) [28], while potential plasmidic contigs were checked by PlasmidFinder web application (version 2.1) [29]. Immune evasion cluster (IEC) genes sequences were obtained from Genebank using accession numbers reported in the work of Ahmadrajabiet and colleagues [10]. IEC genes sequences, at protein level, were searched by similarity using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi accessed on 20 November 2021) against annotated protein coding genes returned by Prokka.
4.5. Accession Number(s)
The genome sequence data from this study have been uploaded to Genebank under the bioproject accession number PRJNA762216.
4.6. Phylogenetic Analysis
The phylogenetic analysis was conducted on the alignments of the core genome SNP matrix as returned by KSNP3 software (version 3.91) [30], using a kmer value equal to 21. The alignments were imported into MEGA X phylogenetic package [31] to derive SNP distance among isolates; further, a Maximum Likelihood phylogenetic tree was calculated using GTR gamma-4 substitution model.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/pathogens11070802/s1, Table S1: Assembly metrics and extended MLST; Table S2: Extended Phaster Results; Table S3: S.aureus, S. epidermidis, and S. pseudointermedius intra species core-SNP distances.
Author Contributions
Conceptualization, M.O. and C.L.; methodology, M.O. and C.L.; formal analysis, M.O. and G.B.; investigation, S.P., MC., M.O. and E.S.; data curation, M.O. and G.B.; writing—original draft preparation, M.O. and C.L.; writing—review and editing, M.O., S.P. and C.L.; visualization, M.O.; supervision, C.L.; funding acquisition, M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received funding from Italian Ministry of Health Grant n. RC IZSVE 16/18.
Institutional Review Board Statement
The study was ethically approved by IZSVe ethical committee (opinion n. CE_IZSVE 02/2022).
Informed Consent Statement
Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
The genome sequence data from this study have been uploaded to Genebank under the bioproject accession number PRJNA762216.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Park, J.Y.; Seo, K.S. Staphylococcus aureus. In Food Microbiology: Fundamentals and Frontiers; American Society for Microbiology: Washington, DC, USA, 2022; pp. 555–584. [Google Scholar] [CrossRef]
- Hidron, A.I.; Low, C.E.; Honig, E.G.; Blumberg, H.M. Emergence of community-acquired meticillin-resistant Staphylococcus aureus strain USA300 as a cause of necrotising community-onset pneumonia. Lancet Infect. Dis. 2009, 9, 384–392. [Google Scholar] [CrossRef]
- Lee, A.S.; De Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Prim. 2018, 4, 18033. [Google Scholar] [CrossRef] [PubMed]
- Petinaki, E.; Spiliopoulou, I. Methicillin-resistant Staphylococcus aureus colonization and infection risks from companion animals: Current perspectives. Vet. Med. Res. Rep. 2015, 6, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Bierowiec, K.; Płoneczka-Janeczko, K.; Rypuła, K. Is the Colonisation of Staphylococcus aureus in Pets Associated with Their Close Contact with Owners? PLoS ONE 2016, 11, e0156052. [Google Scholar] [CrossRef]
- Balasubramanian, D.; Harper, L.; Shopsin, B.; Torres, V.J. Staphylococcus aureus pathogenesis in diverse host environments. Pathog. Dis. 2017, 75, ftx005. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Cerquetella, M.; Attili, A.R. Amphixenosic Aspects of Staphylococcus aureus Infection in Man and Animals. Curr. Top. Microbiol. Immunol. 2017, 409, 297–323. [Google Scholar] [CrossRef]
- Matuszewska, M.; Murray, G.G.R.; Harrison, E.M.; Holmes, M.A.; Weinert, L.A. The Evolutionary Genomics of Host Specificity in Staphylococcus aureus. Trends Microbiol. 2020, 28, 465–477. [Google Scholar] [CrossRef]
- Yu, F.; Cienfuegos-Gallet, A.V.; Cunningham, M.H.; Jin, Y.; Wang, B.; Kreiswirth, B.N.; Chen, L. Molecular Evolution and Adaptation of Livestock-Associated Methicillin-Resistant Staphylococcus aureus (LA-MRSA) Sequence Type 9. mSystems 2021, 6, e0049221. [Google Scholar] [CrossRef]
- Ahmadrajabi, R.; Layegh-Khavidaki, S.; Kalantar-Neyestanaki, D.; Fasihi, Y. Molecular analysis of immune evasion cluster (IEC) genes and intercellular adhesion gene cluster (ICA) among methicillin-resistant and methicillin-sensitive isolates of Staphylococcus aureus. J. Prev. Med. Hyg. 2017, 58, E308–E314. [Google Scholar] [CrossRef]
- Uhlemann, A.-C.; Dordel, J.; Knox, J.R.; Raven, K.E.; Parkhill, J.; Holden, M.T.G.; Peacock, S.J.; Lowy, F.D. Molecular tracing of the emergence, diversification, and transmission of S. aureus sequence type 8 in a New York Community. Proc. Natl. Acad. Sci. USA 2014, 111, 6738–6743. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, J.R. Evolution of Staphylococcus aureus during human colonization and infection. Infect. Genet. Evol. 2014, 21, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Maali, Y.; Badiou, C.; Martins-Simões, P.; Hodille, E.; Bes, M.; Vandenesch, F.; Lina, G.; Diot, A.; Laurent, F.; Trouillet-Assant, S. Understanding the virulence of Staphylococcus pseudintermedius: A major role of pore-forming toxins. Front. Cell. Infect. Microbiol. 2018, 8, 221. [Google Scholar] [CrossRef] [PubMed]
- Strauß, L.; Stegger, M.; Akpaka, P.E.; Alabi, A.; Breurec, S.; Coombs, G.; Egyir, B.; Larsen, A.R.; Laurent, F.; Monecke, S.; et al. Origin, evolution, and global transmission of community-acquired Staphylococcus aureus ST8. Proc. Natl. Acad. Sci. USA 2017, 114, E10596–E10604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monecke, S.; Coombs, G.; Shore, A.C.; Coleman, D.C.; Akpaka, P.; Borg, M.; Chow, H.; Ip, M.; Jatzwauk, L.; Jonas, D.; et al. A Field Guide to Pandemic, Epidemic and Sporadic Clones of Methicillin-Resistant Staphylococcus aureus. PLoS ONE 2011, 6, e17936. [Google Scholar] [CrossRef]
- McClure, J.A.; Lakhundi, S.; Niazy, A.; Dong, G.; Obasuyi, O.; Gordon, P.; Chen, S.; Conly, J.M.; Zhang, K. Staphylococcus aureus ST59: Concurrent but Separate Evolution of North American and East Asian Lineages. Front. Microbiol. 2021, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Higuchi, W.; Otsuka, T.; Baranovich, T.; Enany, S.; Saito, K.; Isobe, H.; Dohmae, S.; Ozaki, K.; Takano, M.; et al. Novel Characteristics of Community-Acquired Methicillin-Resistant Staphylococcus aureus Strains Belonging to Multilocus Sequence Type 59 in Taiwan. Antimicrob. Agents Chemother. 2008, 52, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Rooijakkers, S.H.M.; van Kessel, K.P.M.; van Strijp, J.A.G. Staphylococcal innate immune evasion. Trends Microbiol. 2005, 13, 596–601. [Google Scholar] [CrossRef]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Flowers, L.; Grice, E.A. The Skin Microbiota: Balancing Risk and Reward. Cell Host Microbe 2020, 28, 190–200. [Google Scholar] [CrossRef]
- Bloemendaal, A.L.A.; Brouwer, E.C.; Fluit, A.C. Methicillin Resistance Transfer from Staphylocccus epidermidis to Methicillin-Susceptible Staphylococcus aureus in a Patient during Antibiotic Therapy. PLoS ONE 2010, 5, e11841. [Google Scholar] [CrossRef]
- Louie, L.; Goodfellow, J.; Mathieu, P.; Glatt, A.; Louie, M.; Simor, A.E. Rapid Detection of Methicillin-Resistant Staphylococci from Blood Culture Bottles by Using a Multiplex PCR Assay. J. Clin. Microbiol. 2002, 40, 2786–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling Genomes and Mini-metagenomes from Highly Chimeric Reads. Lect. Notes Comput. Sci. 2013, 7821, 158–170. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Hasman, H.; Saputra, D.; Sicheritz-Ponten, T.; Lund, O.; Svendsen, C.A.; Frimodt-Møller, N.; Aarestrup, F.M. Rapid Whole-Genome Sequencing for Detection and Characterization of Microorganisms Directly from Clinical Samples. J. Clin. Microbiol. 2014, 52, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids. Antimicrob using PlasmidFinder and plasmid multilocus sequence typing. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Gardner, S.N.; Slezak, T.; Hall, B.G. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 2015, 31, 2877–2878. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
Figure 1.
Whole Dataset SNP-based Phylogenetic Tree. The evolutionary history was inferred by using the Maximum Likelihood method and General Time Reversible model. The tree with the highest log likelihood (−1000.76) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. There was a total of 171 positions in the final dataset. Samples isolated from the dog are reported in bold.
Figure 1.
Whole Dataset SNP-based Phylogenetic Tree. The evolutionary history was inferred by using the Maximum Likelihood method and General Time Reversible model. The tree with the highest log likelihood (−1000.76) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. There was a total of 171 positions in the final dataset. Samples isolated from the dog are reported in bold.

Figure 2.
Evolutionary hypothesis. The figure shows the evolutionary hypothesis regarding the transmission of S. aureus strains among the investigated subjects over time. Links among samples are placed based on allelic distances, while arrows are placed according to the sampling date when possible.
Figure 2.
Evolutionary hypothesis. The figure shows the evolutionary hypothesis regarding the transmission of S. aureus strains among the investigated subjects over time. Links among samples are placed based on allelic distances, while arrows are placed according to the sampling date when possible.


{kind=link}
{kind=link}
Table 1.
Dataset. The table reports the strains involved in this study, the species, the date of collection, and the source of isolation in terms of family member and body district. Seven Loci Sequence Type was derived from the assemblies (see Supplementary Table S1 for extended results). ND: Not Determined (Schema not available).
Table 1.
Dataset. The table reports the strains involved in this study, the species, the date of collection, and the source of isolation in terms of family member and body district. Seven Loci Sequence Type was derived from the assemblies (see Supplementary Table S1 for extended results). ND: Not Determined (Schema not available).
| Sample | Organism | Collection Date | Family Member, District | ST |
|---|---|---|---|---|
| Sau_17_FA_ST8_123232 | S. aureus | 2017 | father, skin | ST8 |
| Sau_17_MO_ST8_123221 | S. aureus | 2017 | mother, mouth | ST8 |
| Sau_17_DO_ST8_123220 | S. aureus | 2017 | dog, armpit | ST8 |
| Sau_18_GF_ST8_153686-7 | S. aureus | 2018 | grandfather, nose | ST8 |
| Sau_18_GM_ST8_153686-6 | S. aureus | 2018 | grandmother, armpit | ST8 |
| Sau_18_MO_ST10_153686-3 | S. aureus | 2018 | mother, nose | ST10 |
| Sau_19_MO_ST10_150824 | S. aureus | 2019 | mother, nose | ST10 |
| Sau_19_CH_ST10_150819 | S. aureus | 2019 | child, nose | ST10 |
| Sau_17_MO_ST22_123223 | S. aureus | 2017 | mother, nose | ST22 |
| Sau_18_FA_ST22_153686-2 | S. aureus | 2018 | father, nose | ST22 |
| Sau_19_FA_ST22_150811 | S. aureus | 2019 | father, nose | ST22 |
| Sau_18_GM_ST59_153686-5 | S. aureus | 2018 | grandmother, nose | ST59 |
| Sau_19_GF_ST59_150803 | S. aureus | 2019 | grandfather, nose | ST59 |
| Sau_18_CH_ST707_153686-4 | S. aureus | 2018 | child, nose | ST707 |
| Sau_18_DO_ST7204_153686-1 | S. aureus | 2018 | dog, mouth | ST7204 |
| Sca_19_DO_STX_150837 | S. capitis | 2019 | dog, armpit | ND |
| Sep_19_GF_ST153_150806 | S. epidermidis | 2019 | grandfather, nose | ST153 |
| Sep_17_CH_ST1133_123222 | S. epidermidis | 2017 | child, mouth | ST1133 |
| Sep_19_GM_ST5_150797 | S. epidermidis | 2019 | grandmother, nose | ST5 |
| Slu_19_GM_ST2_150795 | S. lugdunensis | 2019 | grandmother, nose | ST2 |
| Sps_17_DO_ST2169_123218 | S. pseudintermedius | 2017 | dog, mouth | ST2169 |
| Sps_19_DO_ST2169_150832 | S. pseudintermedius | 2019 | dog, foreskin | ST2169 |
| Sps_17_DO_ST2168_123216 | S. pseudintermedius | 2017 | dog, mouth | ST2168 |
| Sps_19_DO_ST2168_150831 | S. pseudintermedius | 2019 | dog, mouth | ST2168 |
| Swa_19_FA_STX_150815 | S. warneri | 2019 | father, nose | ND |
Table 2.
Antibiotic-Resistance Genes. The table reports antibiotic-resistance genes as detected by the ResFinder webserver.
Table 2.
Antibiotic-Resistance Genes. The table reports antibiotic-resistance genes as detected by the ResFinder webserver.
| Sample | blaZ | ant(6)-Ia | aph(3)-III | aadD | aac(6)-aph(2) | cat(pC221) | fosB | fusB | mecA | mph(C) | msr(A) | erm(C) | erm(B) | vga(A) | vga(A)LC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sau_17_FA_ST8_123232 | x | x | x | x | x | x | |||||||||
| Sau_17_MO_ST8_123221 | x | x | x | x | x | x | |||||||||
| Sau_17_DO_ST8_123220 | x | x | x | x | x | x | |||||||||
| Sau_18_GF_ST8_153686-7 | x | x | x | x | x | x | |||||||||
| Sau_18_GM_ST8_153686-6 | x | x | x | x | x | x | |||||||||
| Sau_18_MO_ST10_153686-3 | x | ||||||||||||||
| Sau_19_MO_ST10_150824 | x | x | x | ||||||||||||
| Sau_19_CH_ST10_150819 | x | ||||||||||||||
| Sau_17_MO_ST22_123223 | x | ||||||||||||||
| Sau_18_FA_ST22_153686-2 | x | ||||||||||||||
| Sau_19_FA_ST22_150811 | x | ||||||||||||||
| Sau_18_GM_ST59_153686-5 | x | ||||||||||||||
| Sau_19_GF_ST59_150803 | x | ||||||||||||||
| Sau_18_CH_ST707_153686-4 | x | ||||||||||||||
| Sau_18_DO_ST7204_153686-1 | x | ||||||||||||||
| Sca_19_DO_STX_150837 | x | ||||||||||||||
| Sep_19_GF_ST153_150806 | x | x | x | x | |||||||||||
| Sep_17_CH_ST1133_123222 | x | x | x | x | x | x | |||||||||
| Sep_19_GM_ST5_150797 | x | x | x | x | x | x | x | x | x | x | |||||
| Slu_19_GM_ST2_150795 | x | ||||||||||||||
| Sps_17_DO_ST2169_123218 | x | x | x | x | |||||||||||
| Sps_19_DO_ST2169_150832 | x | x | x | x | |||||||||||
| Sps_17_DO_ST2168_123216 | x | x | x | x | x | ||||||||||
| Sps_19_DO_ST2168_150831 | x | x | x | x | x | ||||||||||
| Swa_19_FA_STX_150815 |
Table 3.
Intact Prophage Sequences. The table reports the intact prophage sequences only as predicted by PHASTER web server. For brevity, the “Staphy” prefix was removed from the prophage names.
Table 3.
Intact Prophage Sequences. The table reports the intact prophage sequences only as predicted by PHASTER web server. For brevity, the “Staphy” prefix was removed from the prophage names.
| S. aureus | 69 | 11 | phi2958PVL | StauST398_2 | phiJB | phi2958PVL | P282 | YMC/09/04/R1988 |
|---|---|---|---|---|---|---|---|---|
| Sau_18_MO_ST10_153686-3 | x | x | ||||||
| Sau_19_MO_ST10_150824 | x | x | ||||||
| Sau_19_CH_ST10_150819 | x | x | ||||||
| Sau_18_CH_ST707_153686-4 | x | |||||||
| Sau_18_DO_ST7204_153686-1 | x | |||||||
| Sau_17_FA_ST8_123232 | ||||||||
| Sau_17_MO_ST8_123221 | x | x | ||||||
| Sau_17_DO_ST8_123220 | x | x | ||||||
| Sau_18_GM_ST8_153686-6 | x | x | ||||||
| Sau_18_GF_ST8_153686-7 | x | x | ||||||
| S. epidermidis | StB20_like | Ipla5 | ||||||
| Sep_17_CH_ST1133_123222 | ||||||||
| Sep_19_GF_ST153_150806 | x | x | ||||||
| Sep_19_GM_ST5_150797 |
Table 4.
Plasmid content. The table reports the plasmid sequences as detected by the plasmid finder web server.
Table 4.
Plasmid content. The table reports the plasmid sequences as detected by the plasmid finder web server.
| Sample | rep19 | rep39 | rep7a | repUS43 | rep7c | rep16 | repUS5 | rep20 | repUS35 | rep22 | repUS19 | repUS12 | rep5b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sau_17_DO_ST8_123220 | x | x | x | ||||||||||
| Sau_17_FA_ST8_123232 | x | x | x | x | |||||||||
| Sau_18_GF_ST8_153686-7 | x | x | x | ||||||||||
| Sau_18_GM_ST8_153686-6 | x | x | x | ||||||||||
| Sau_17_MO_ST8_123221 | x | x | x | ||||||||||
| Sau_18_CH_ST707_153686-4 | x | x | |||||||||||
| Sau_18_DO_ST7204_153686-1 | x | x | |||||||||||
| Sau_18_FA_ST22_153686-2 | |||||||||||||
| Sau_17_MO_ST22_123223 | |||||||||||||
| Sau_19_FA_ST22_150811 | |||||||||||||
| Sau_19_MO_ST10_150824 | x | x | |||||||||||
| Sau_18_MO_ST10_153686-3 | |||||||||||||
| Sau_19_CH_ST10_150819 | |||||||||||||
| Sau_19_GF_ST59_150803 | |||||||||||||
| Sau_18_GM_ST59_153686-5 | |||||||||||||
| Sca_19_DO_STX_150837 | x | ||||||||||||
| Sep_17_CH_ST1133_123222 | x | x | x | ||||||||||
| Sep_19_GF_ST153_150806 | x | ||||||||||||
| Sep_19_GM_ST5_150797 | x | x | x | x | |||||||||
| Slu_19_GM_ST2_150795 | x | ||||||||||||
| Sps_17_DO_ST2169_123218 | x | ||||||||||||
| Sps_17_DO_ST2168_123216 | x | ||||||||||||
| Sps_19_DO_ST2169_150832 | x | ||||||||||||
| Sps_19_DO_ST2168_150831 | x | ||||||||||||
| Swa_19_FA_STX_150815 | x |
Table 5.
IEC Genes. The table reports the presence of IEC Genes, including the IEC variant.
| Sample | scn | sep | sea | sak | chp | IEC-Variant | Sample |
|---|---|---|---|---|---|---|---|
| Sau_17_DO_ST8_123220 | x | x | x | B | Sau_17_DO_ST8_123220 | ||
| Sau_17_FA_ST8_123232 | x | x | x | B | Sau_17_FA_ST8_123232 | ||
| Sau_18_GF_ST8_153686-7 | x | x | x | B | Sau_18_GF_ST8_153686-7 | ||
| Sau_18_GM_ST8_153686-6 | x | x | x | B | Sau_18_GM_ST8_153686-6 | ||
| Sau_17_MO_ST8_123221 | x | x | x | B | Sau_17_MO_ST8_123221 | ||
| Sau_18_CH_ST707_153686-4 | x | x | x | B | Sau_18_CH_ST707_153686-4 | ||
| Sau_18_DO_ST7204_153686-1 | x | x | x | B | Sau_18_DO_ST7204_153686-1 | ||
| Sau_18_FA_ST22_153686-2 | x | x | x | B | Sau_18_FA_ST22_153686-2 | ||
| Sau_17_MO_ST22_123223 | x | x | x | B | Sau_17_MO_ST22_123223 | ||
| Sau_19_FA_ST22_150811 | x | x | x | B | Sau_19_FA_ST22_150811 | ||
| Sau_19_MO_ST10_150824 | x | x | E | Sau_19_MO_ST10_150824 | |||
| Sau_18_MO_ST10_153686-3 | x | x | E | Sau_18_MO_ST10_153686-3 | |||
| Sau_19_CH_ST10_150819 | x | x | E | Sau_19_CH_ST10_150819 | |||
| Sau_19_GF_ST59_150803 | x | x | C | Sau_19_GF_ST59_150803 | |||
| Sau_18_GM_ST59_153686-5 | x | x | C | Sau_18_GM_ST59_153686-5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Orsini, M.; Petrin, S.; Corrò, M.; Baggio, G.; Spagnolo, E.; Losasso, C. Anthroponotic-Based Transfer of Staphylococcus to Dog: A Case Study. Pathogens 2022, 11, 802. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11070802
AMA Style
Orsini M, Petrin S, Corrò M, Baggio G, Spagnolo E, Losasso C. Anthroponotic-Based Transfer of Staphylococcus to Dog: A Case Study. Pathogens. 2022; 11(7):802. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11070802
Chicago/Turabian StyleOrsini, Massimiliano, Sara Petrin, Michela Corrò, Giulia Baggio, Elena Spagnolo, and Carmen Losasso. 2022. "Anthroponotic-Based Transfer of Staphylococcus to Dog: A Case Study" Pathogens 11, no. 7: 802. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11070802
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.