
Intranasal Immunization with Liposome-Displayed Receptor-Binding Domain Induces Mucosal Immunity and Protection against SARS-CoV-2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
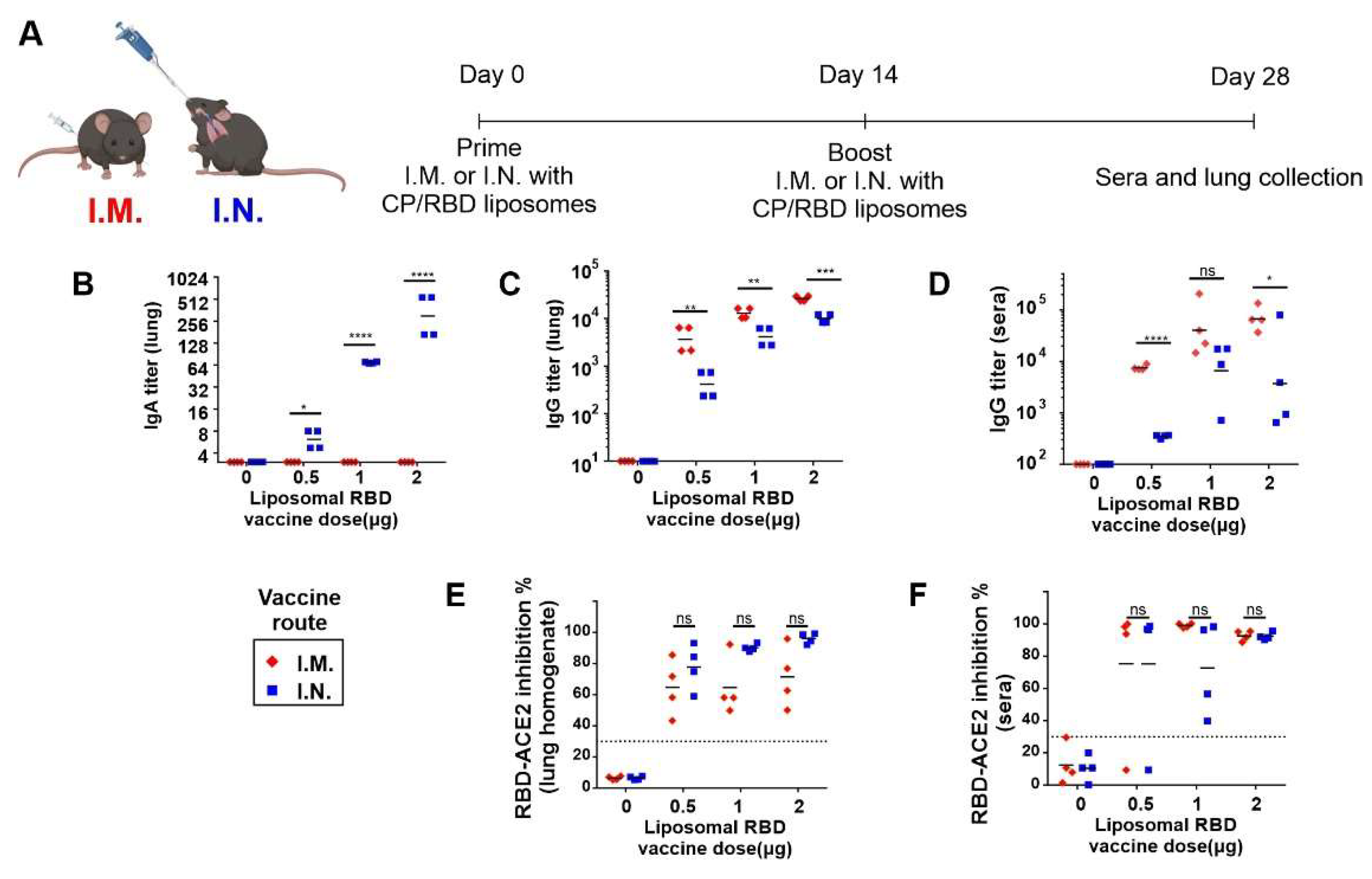
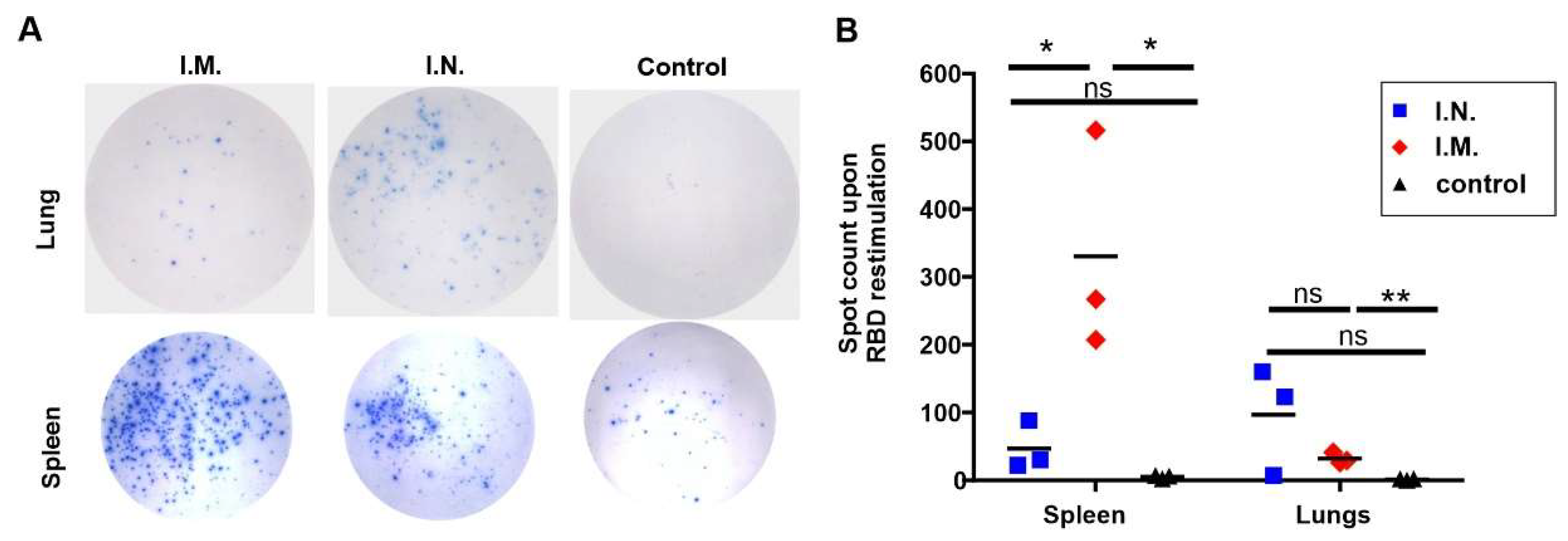
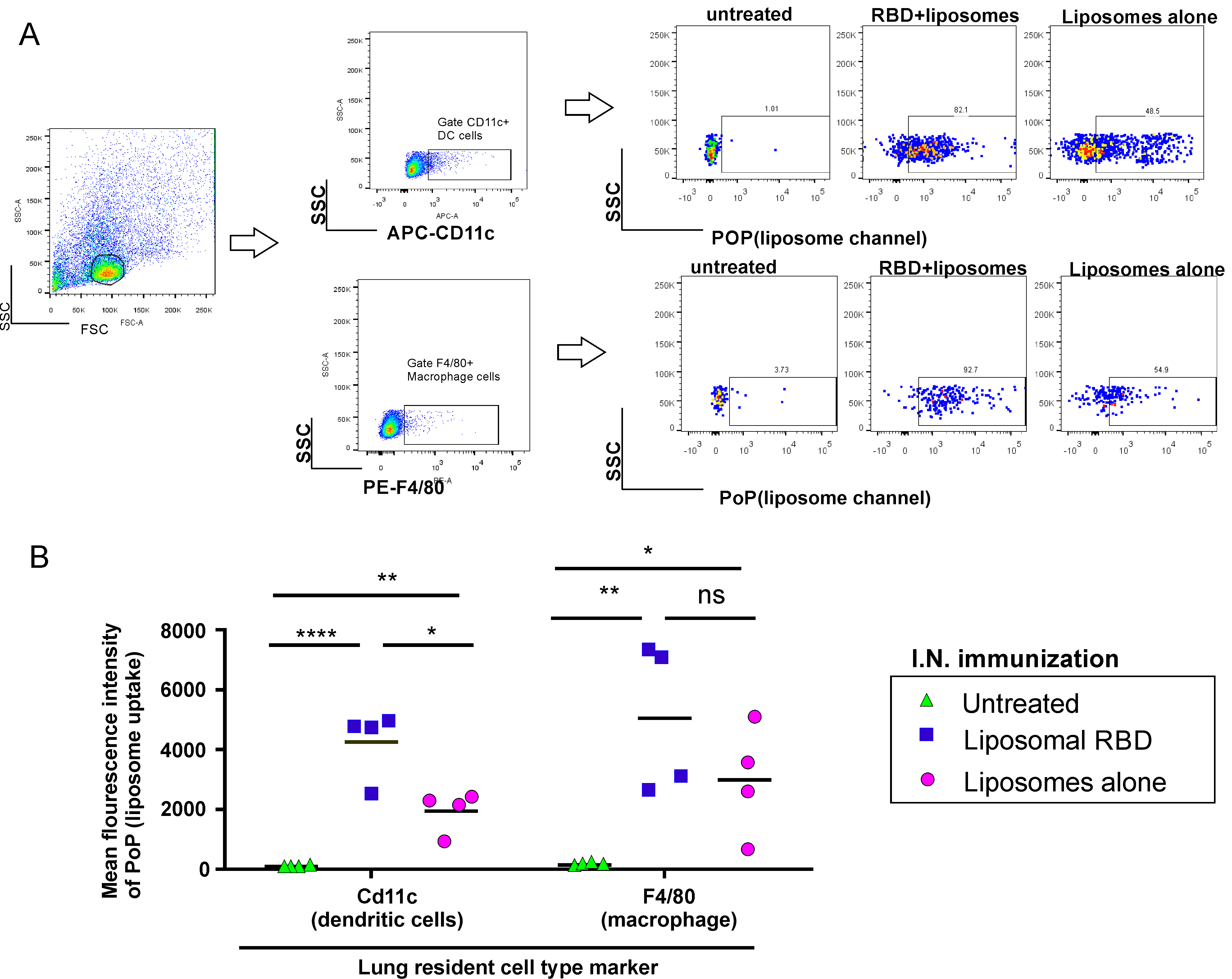
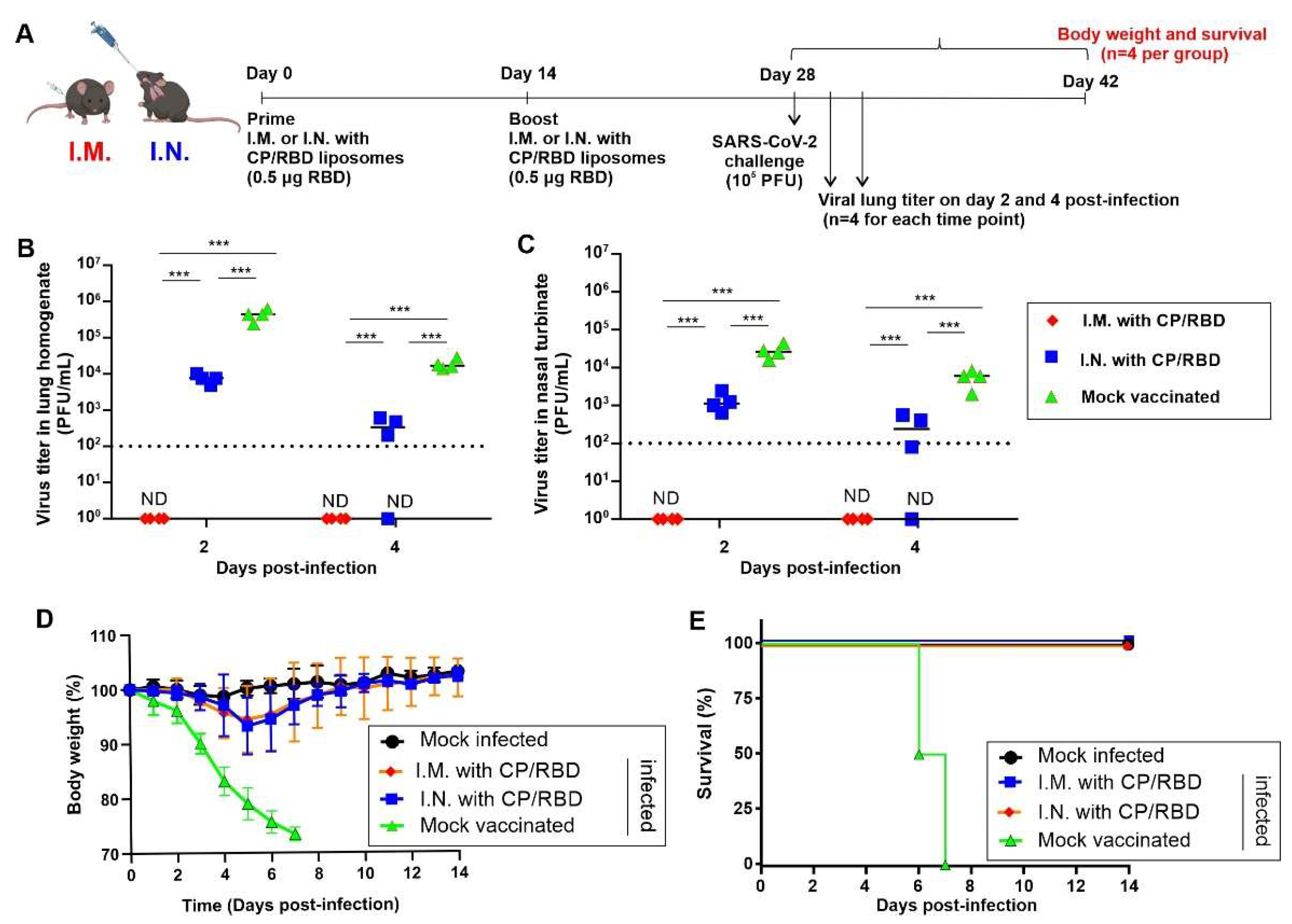
2. Results
3. Discussion
4. Conclusions
5. Experimental
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chan, J.F.; Yuan, S.; Kok, K.H.; To, K.K.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.; Poon, R.W.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef]
- Rothe, C.; Schunk, M.; Sothmann, P.; Bretzel, G.; Froeschl, G.; Wallrauch, C.; Zimmer, T.; Thiel, V.; Janke, C.; Guggemos, W.; et al. Transmission of 2019-nCoV Infection from an Asymptomatic Contact in Germany. N. Engl. J. Med. 2020, 382, 970–971. [Google Scholar] [CrossRef] [PubMed]
- Mabrouk, M.T.; Huang, W.C.; Martinez-Sobrido, L.; Lovell, J.F. Advanced Materials for SARS-CoV-2 Vaccines. Adv. Mater. 2022, 34, e2107781. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Allergy, N.I.O.; Diseases, I.; ModernaTX, I. Safety and Immunogenicity Study of 2019-nCoV Vaccine (mRNA-1273) for Prophylaxis of SARS-CoV-2 Infection (COVID-19). 2020. Available online: https://ClinicalTrials.gov/show/NCT04283461 (accessed on 2 January 2020).
- Allergy, N.I.O.; Diseases, I.; ModernaTX, I. Safety and Immunogenicity Study of a SARS-CoV-2 (COVID-19) Variant Vaccine (mRNA-1273.351) in Naïve and Previously Vaccinated Adults. 2021. Available online: https://ClinicalTrials.gov/show/NCT04785144 (accessed on 5 March 2021).
- ANRS, Emerging Infectious Diseases; Institut National de la Santé Et de la Recherche Médicale, France. BNT162b2 Vaccination With Two Doses in COVID-19 Negative Adult Volunteers and With a Single Dose in COVID-19 Positive Adult Volunteers. 2021. Available online: https://ClinicalTrials.gov/show/NCT04824638 (accessed on 1 April 2021).
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cardenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B.; et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. N. Engl. J. Med. 2021, 384, 2187–2201. [Google Scholar] [CrossRef]
- Falsey, A.R.; Sobieszczyk, M.E.; Hirsch, I.; Sproule, S.; Robb, M.L.; Corey, L.; Neuzil, K.M.; Hahn, W.; Hunt, J.; Mulligan, M.J.; et al. Phase 3 Safety and Efficacy of AZD1222 (ChAdOx1 nCoV-19) Covid-19 Vaccine. N. Engl. J. Med. 2021, 385, 2348–2360. [Google Scholar] [CrossRef]
- Li, J.; Hou, L.; Guo, X.; Jin, P.; Wu, S.; Zhu, J.; Pan, H.; Wang, X.; Song, Z.; Wan, J.; et al. Heterologous AD5-nCOV plus CoronaVac versus homologous CoronaVac vaccination: A randomized phase 4 trial. Nat. Med. 2022, 28, 401–409. [Google Scholar] [CrossRef]
- Parida, S.P.; Sahu, D.P.; Singh, A.K.; Alekhya, G.; Subba, S.H.; Mishra, A.; Padhy, B.M.; Patro, B.K. Adverse events following immunization of COVID-19 (Covaxin) vaccine at a tertiary care center of India. J. Med. Virol. 2022, 94, 2453–2459. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, Y.; Wang, Y.; Wang, H.; Yang, Y.; Gao, G.F.; Tan, W.; Wu, G.; Xu, M.; Lou, Z.; et al. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBIBP-CorV: A randomised, double-blind, placebo-controlled, phase 1/2 trial. Lancet Infect. Dis. 2021, 21, 39–51. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, Y.; Wang, Y.; Wang, H.; Yang, Y.; Gao, G.F.; Tan, W.; Wu, G.; Xu, M.; Lou, Z.; et al. Safety and immunogenicity of an inactivated COVID-19 vaccine, BBIBP-CorV, in people younger than 18 years: A randomised, double-blind, controlled, phase 1/2 trial. Lancet Infect. Dis. 2022, 22, 196–208. [Google Scholar] [CrossRef]
- Yang, S.; Li, Y.; Dai, L.; Wang, J.; He, P.; Li, C.; Fang, X.; Wang, C.; Zhao, X.; Huang, E.; et al. Safety and immunogenicity of a recombinant tandem-repeat dimeric RBD-based protein subunit vaccine (ZF2001) against COVID-19 in adults: Two randomised, double-blind, placebo-controlled, phase 1 and 2 trials. Lancet Infect. Dis. 2021, 21, 1107–1119. [Google Scholar] [CrossRef]
- Dunkle, L.M.; Kotloff, K.L.; Gay, C.L.; Anez, G.; Adelglass, J.M.; Hernandez, A.Q.B.; Harper, W.L.; Duncanson, D.M.; McArthur, M.A.; Florescu, D.F.; et al. Efficacy and Safety of NVX-CoV2373 in Adults in the United States and Mexico. N. Engl. J. Med. 2022, 386, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Granwehr, B.P. In adults who had not had COVID-19, Novavax vaccine had 90% efficacy at >/=7 d after the second dose. Ann. Intern. Med. 2022, 175, JC52. [Google Scholar] [CrossRef]
- Su, F.; Patel, G.B.; Hu, S.; Chen, W. Induction of mucosal immunity through systemic immunization: Phantom or reality? Hum. Vaccin. Immunother. 2016, 12, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zeng, C.; Cox, T.M.; Li, C.; Son, Y.M.; Cheon, I.S.; Wu, Y.; Behl, S.; Taylor, J.J.; Chakraborty, R.; et al. Respiratory mucosal immunity against SARS-CoV-2 following mRNA vaccination. Sci. Immunol. 2022, eadd4853. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.P.; Vora, L.K.; Pandya, A.K.; Patravale, V.B. Intranasal vaccines for SARS-CoV-2: From challenges to potential in COVID-19 management. Drug Discov. Today 2021, 26, 2619–2636. [Google Scholar] [CrossRef]
- Russell, M.W.; Moldoveanu, Z.; Ogra, P.L.; Mestecky, J. Mucosal Immunity in COVID-19: A Neglected but Critical Aspect of SARS-CoV-2 Infection. Front. Immunol. 2020, 11, 611337. [Google Scholar] [CrossRef]
- Zhou, R.; Wang, P.; Wong, Y.-C.; Xu, H.; Lau, S.-Y.; Liu, L.; Mok, B.W.-Y.; Peng, Q.; Liu, N.; Woo, K.-F.; et al. Nasal prevention of SARS-CoV-2 infection by intranasal influenza-based boost vaccination in mouse models. eBioMedicine 2022, 75, 103762. [Google Scholar] [CrossRef]
- Du, Y.; Xu, Y.; Feng, J.; Hu, L.; Zhang, Y.; Zhang, B.; Guo, W.; Mai, R.; Chen, L.; Fang, J.; et al. Intranasal administration of a recombinant RBD vaccine induced protective immunity against SARS-CoV-2 in mouse. Vaccine 2021, 39, 2280–2287. [Google Scholar] [CrossRef]
- Hassan, A.O.; Shrihari, S.; Gorman, M.J.; Ying, B.; Yaun, D.; Raju, S.; Chen, R.E.; Dmitriev, I.P.; Kashentseva, E.; Adams, L.J.; et al. An intranasal vaccine durably protects against SARS-CoV-2 variants in mice. Cell Rep. 2021, 36, 109452. [Google Scholar] [CrossRef] [PubMed]
- Limited, B.B.I. Safety and Immunogenicity of an Intranasal SARS-CoV-2 Vaccine (BBV154) for COVID-19. 2021. Available online: https://ClinicalTrials.gov/show/NCT04751682 (accessed on 25 February 2021).
- Langel, S.N.; Johnson, S.; Martinez, C.I.; Tedjakusuma, S.N.; Peinovich, N.; Dora, E.G.; Kuehl, P.J.; Irshad, H.; Barrett, E.G.; Werts, A.; et al. Adenovirus type 5 SARS-CoV-2 vaccines delivered orally or intranasally reduced disease severity and transmission in a hamster model. Sci. Transl. Med. 2022, 14, eabn6868. [Google Scholar] [CrossRef] [PubMed]
- Vaxart. Safety and Immunogenicity Trial of an Oral SARS-CoV-2 Vaccine (VXA-CoV2-1) for Prevention of COVID-19 in Healthy Adults. 2020. Available online: https://ClinicalTrials.gov/show/NCT04563702 (accessed on 24 September 2020).
- Lung, P.; Yang, J.; Li, Q. Nanoparticle formulated vaccines: Opportunities and challenges. Nanoscale 2020, 12, 5746–5763. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.K.; Huszthy, P.C.; Gopalakrishnan, R.P.; Jacobsen, J.T.; Fauskanger, M.; Tveita, A.A.; Grodeland, G.; Bogen, B. Enhanced germinal center reaction by targeting vaccine antigen to major histocompatibility complex class II molecules. NPJ Vaccines 2019, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Fredriksen, A.B.; Sandlie, I.; Bogen, B. Targeted DNA vaccines for enhanced induction of idiotype-specific B and T cells. Front. Oncol. 2012, 2, 154. [Google Scholar] [CrossRef]
- Shao, S.; Geng, J.; Yi, H.A.; Gogia, S.; Neelamegham, S.; Jacobs, A.; Lovell, J.F. Functionalization of cobalt porphyrin–phospholipid bilayers with his-tagged ligands and antigens. Nat. Chem. 2015, 7, 438. [Google Scholar] [CrossRef]
- Huang, W.-C.; Deng, B.; Lin, C.; Carter, K.A.; Geng, J.; Razi, A.; He, X.; Chitgupi, U.; Federizon, J.; Sun, B. A malaria vaccine adjuvant based on recombinant antigen binding to liposomes. Nat. Nanotechnol. 2018, 13, 1174–1181. [Google Scholar] [CrossRef]
- McLeod, B.; Mabrouk, M.T.; Miura, K.; Ravichandran, R.; Kephart, S.; Hailemariam, S.; Pham, T.P.; Semesi, A.; Kucharska, I.; Kundu, P.; et al. Vaccination with a structure-based stabilized version of malarial antigen Pfs48/45 elicits ultra-potent transmission-blocking antibody responses. Immunity 2022. [Google Scholar] [CrossRef]
- Huang, W.C.; Deng, B.; Mabrouk, M.T.; Seffouh, A.; Ortega, J.; Long, C.; Miura, K.; Wu, Y.; Lovell, J.F. Particle-based, Pfs230 and Pfs25 immunization is effective, but not improved by duplexing at fixed total antigen dose. Malar. J. 2020, 19, 309. [Google Scholar] [CrossRef]
- Federizon, J.; Feugmo, C.G.T.; Huang, W.C.; He, X.; Miura, K.; Razi, A.; Ortega, J.; Karttunen, M.; Lovell, J.F. Experimental and Computational Observations of Immunogenic Cobalt Porphyrin Lipid Bilayers: Nanodomain-Enhanced Antigen Association. Pharmaceutics 2021, 13, 98. [Google Scholar] [CrossRef]
- Huang, W.C.; Deng, B.; Seffouh, A.; Ortega, J.; Long, C.A.; Suresh, R.V.; He, X.; Miura, K.; Lee, S.M.; Wu, Y.; et al. Antibody response of a particle-inducing, liposome vaccine adjuvant admixed with a Pfs230 fragment. NPJ Vaccines 2020, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Federizon, J.; Frye, A.; Huang, W.C.; Hart, T.M.; He, X.; Beltran, C.; Marcinkiewicz, A.L.; Mainprize, I.L.; Wills, M.K.B.; Lin, Y.P.; et al. Immunogenicity of the Lyme disease antigen OspA, particleized by cobalt porphyrin-phospholipid liposomes. Vaccine 2020, 38, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Mabrouk, M.T.; Huang, W.C.; Deng, B.; Li-Purcell, N.; Seffouh, A.; Ortega, J.; Atilla-Gokcumen, G.E.; Long, C.A.; Miura, K.; Lovell, J.F. Lyophilized, antigen-bound liposomes with reduced MPLA and enhanced thermostability. Int. J. Pharm. 2020, 589, 119843. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Mabrouk, M.T.; Zhou, L.; Baba, M.; Tachibana, M.; Torii, M.; Takashima, E.; Locke, E.; Plieskatt, J.; King, C.R.; et al. Vaccine co-display of CSP and Pfs230 on liposomes targeting two Plasmodium falciparum differentiation stages. Commun. Biol. 2022, 5, 773. [Google Scholar] [CrossRef]
- Huang, W.C.; Zhou, S.; He, X.; Chiem, K.; Mabrouk, M.T.; Nissly, R.H.; Bird, I.M.; Strauss, M.; Sambhara, S.; Ortega, J.; et al. SARS-CoV-2 RBD Neutralizing Antibody Induction is Enhanced by Particulate Vaccination. Adv. Mater. 2020, 32, e2005637. [Google Scholar] [CrossRef]
- Mabrouk, M.T.; Chiem, K.; Rujas, E.; Huang, W.C.; Jahagirdar, D.; Quinn, B.; Surendran Nair, M.; Nissly, R.H.; Cavener, V.S.; Boyle, N.R.; et al. Lyophilized, thermostable Spike or RBD immunogenic liposomes induce protective immunity against SARS-CoV-2 in mice. Sci. Adv. 2021, 7, eabj1476. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-nCoV Spike in the Prefusion Conformation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.M.A.; van Haperen, R.; Osterhaus, A.; van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020, 11, 2251. [Google Scholar] [CrossRef]
- Ter Meulen, J.; van den Brink, E.N.; Poon, L.L.; Marissen, W.E.; Leung, C.S.; Cox, F.; Cheung, C.Y.; Bakker, A.Q.; Bogaards, J.A.; van Deventer, E.; et al. Human monoclonal antibody combination against SARS coronavirus: Synergy and coverage of escape mutants. PLoS Med. 2006, 3, e237. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Lam, E.C.; St Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. medRxiv 2021. [Google Scholar] [CrossRef]
- Oladunni, F.S.; Park, J.-G.; Pino, P.A.; Gonzalez, O.; Akhter, A.; Allué-Guardia, A.; Olmo-Fontánez, A.; Gautam, S.; Garcia-Vilanova, A.; Ye, C.; et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat. Commun. 2020, 11, 6122. [Google Scholar] [CrossRef]
- Lovell, J.F.; Jin, C.S.; Huynh, E.; Jin, H.; Kim, C.; Rubinstein, J.L.; Chan, W.C.; Cao, W.; Wang, L.V.; Zheng, G. Porphysome nanovesicles generated by porphyrin bilayers for use as multimodal biophotonic contrast agents. Nat. Mater. 2011, 10, 324–332. [Google Scholar] [CrossRef]
- Afkhami, S.; D'Agostino, M.R.; Zhang, A.; Stacey, H.D.; Marzok, A.; Kang, A.; Singh, R.; Bavananthasivam, J.; Ye, G.; Luo, X.; et al. Respiratory mucosal delivery of next-generation COVID-19 vaccine provides robust protection against both ancestral and variant strains of SARS-CoV-2. Cell 2022, 185, 896–915.e819. [Google Scholar] [CrossRef]
- Wong, T.Y.; Lee, K.S.; Russ, B.P.; Horspool, A.M.; Kang, J.; Winters, M.T.; Allison Wolf, M.; Rader, N.A.; Miller, O.A.; Shiflett, M.; et al. Intranasal administration of BReC-CoV-2 COVID-19 vaccine protects K18-hACE2 mice against lethal SARS-CoV-2 challenge. NPJ Vaccines 2022, 7, 36. [Google Scholar] [CrossRef]
- Van Doremalen, N.; Purushotham, J.N.; Schulz, J.E.; Holbrook, M.G.; Bushmaker, T.; Carmody, A.; Port, J.R.; Yinda, C.K.; Okumura, A.; Saturday, G.; et al. Intranasal ChAdOx1 nCoV-19/AZD1222 vaccination reduces viral shedding after SARS-CoV-2 D614G challenge in preclinical models. Sci. Transl. Med. 2021, 13, abh0755. [Google Scholar] [CrossRef]
- An, X.; Martinez-Paniagua, M.; Rezvan, A.; Fathi, M.; Singh, S.; Biswas, S.; Pourpak, M.; Yee, C.; Liu, X.; Varadarajan, N. Single-dose intranasal vaccination elicits systemic and mucosal immunity against SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Park, J.G.; Oladunni, F.S.; Rohaim, M.A.; Whittingham-Dowd, J.; Tollitt, J.; Hodges, M.D.J.; Fathallah, N.; Assas, M.B.; Alhazmi, W.; Almilaibary, A.; et al. Immunogenicity and protective efficacy of an intranasal live-attenuated vaccine against SARS-CoV-2. iScience 2021, 24, 102941. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e1415. [Google Scholar] [CrossRef]
- Szabo, P.A.; Dogra, P.; Gray, J.I.; Wells, S.B.; Connors, T.J.; Weisberg, S.P.; Krupska, I.; Matsumoto, R.; Poon, M.M.L.; Idzikowski, E.; et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity 2021, 54, 797–814.e796. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhou, S.; Huang, W.C.; Seffouh, A.; Mabrouk, M.T.; Morgan, M.T.; Ortega, J.; Abrams, S.I.; Lovell, J.F. A Potent Cancer Vaccine Adjuvant System for Particleization of Short, Synthetic CD8(+) T Cell Epitopes. ACS Nano 2021, 15, 4357–4371. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, W.-C.; Chiem, K.; Martinez-Sobrido, L.; Lovell, J.F. Intranasal Immunization with Liposome-Displayed Receptor-Binding Domain Induces Mucosal Immunity and Protection against SARS-CoV-2. Pathogens 2022, 11, 1035. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11091035
Huang W-C, Chiem K, Martinez-Sobrido L, Lovell JF. Intranasal Immunization with Liposome-Displayed Receptor-Binding Domain Induces Mucosal Immunity and Protection against SARS-CoV-2. Pathogens. 2022; 11(9):1035. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11091035
Chicago/Turabian StyleHuang, Wei-Chiao, Kevin Chiem, Luis Martinez-Sobrido, and Jonathan F. Lovell. 2022. "Intranasal Immunization with Liposome-Displayed Receptor-Binding Domain Induces Mucosal Immunity and Protection against SARS-CoV-2" Pathogens 11, no. 9: 1035. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11091035