

Selene-Ethylenelacticamides and N-Aryl-Propanamides as Broad-Spectrum Leishmanicidal Agents

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Curation
2.2. QSAR Modelling
2.3. VolSurf Descriptors
2.4. Homology Modelling
2.5. Molecular Docking Studies
2.6. Predicting In Silico ADMET Properties
2.7. In Vitro Assay
2.8. Molecular Dynamics
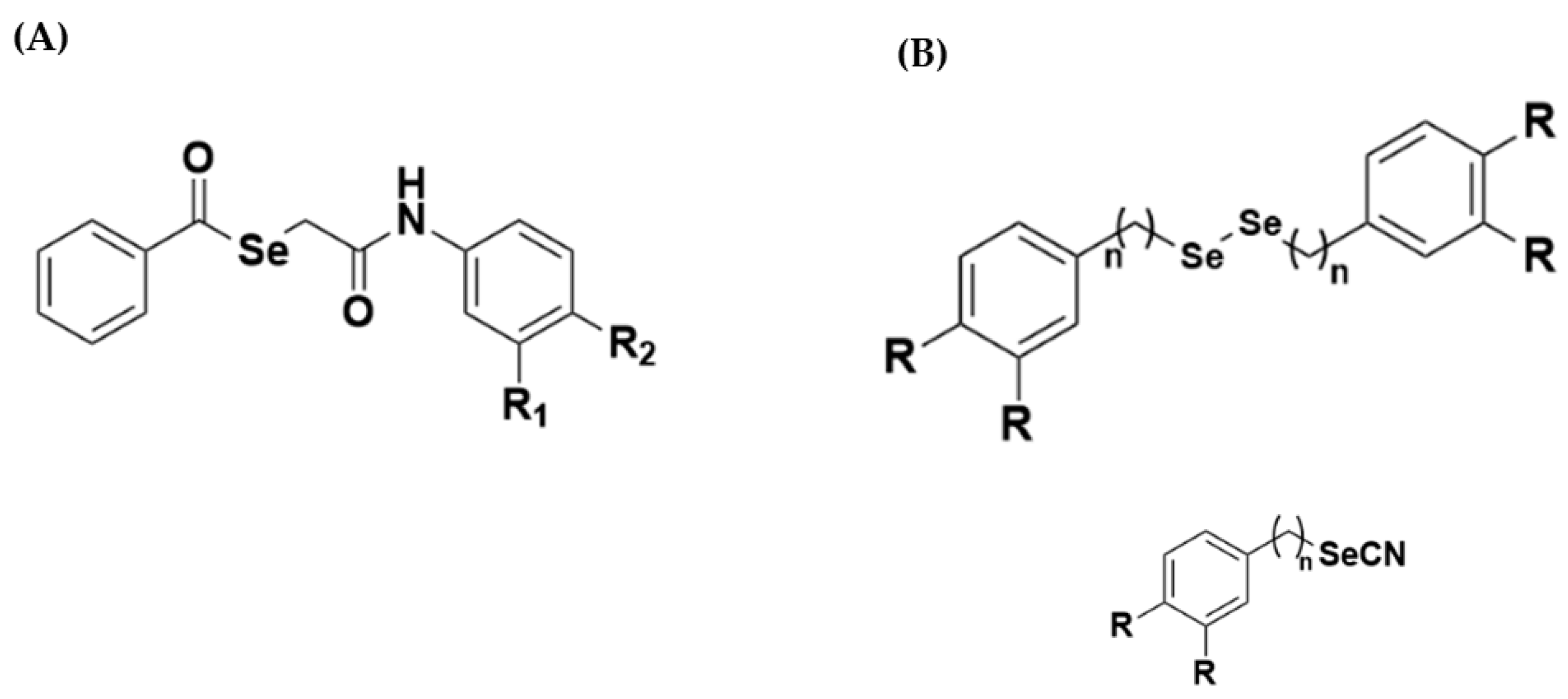
2.9. Major Signs Characterizing N-Arylpropanamide Compounds
2.10. Main Identification Signs of Selene-Ethylenelacticamides and Reaction Yield
3. Results and Discussion
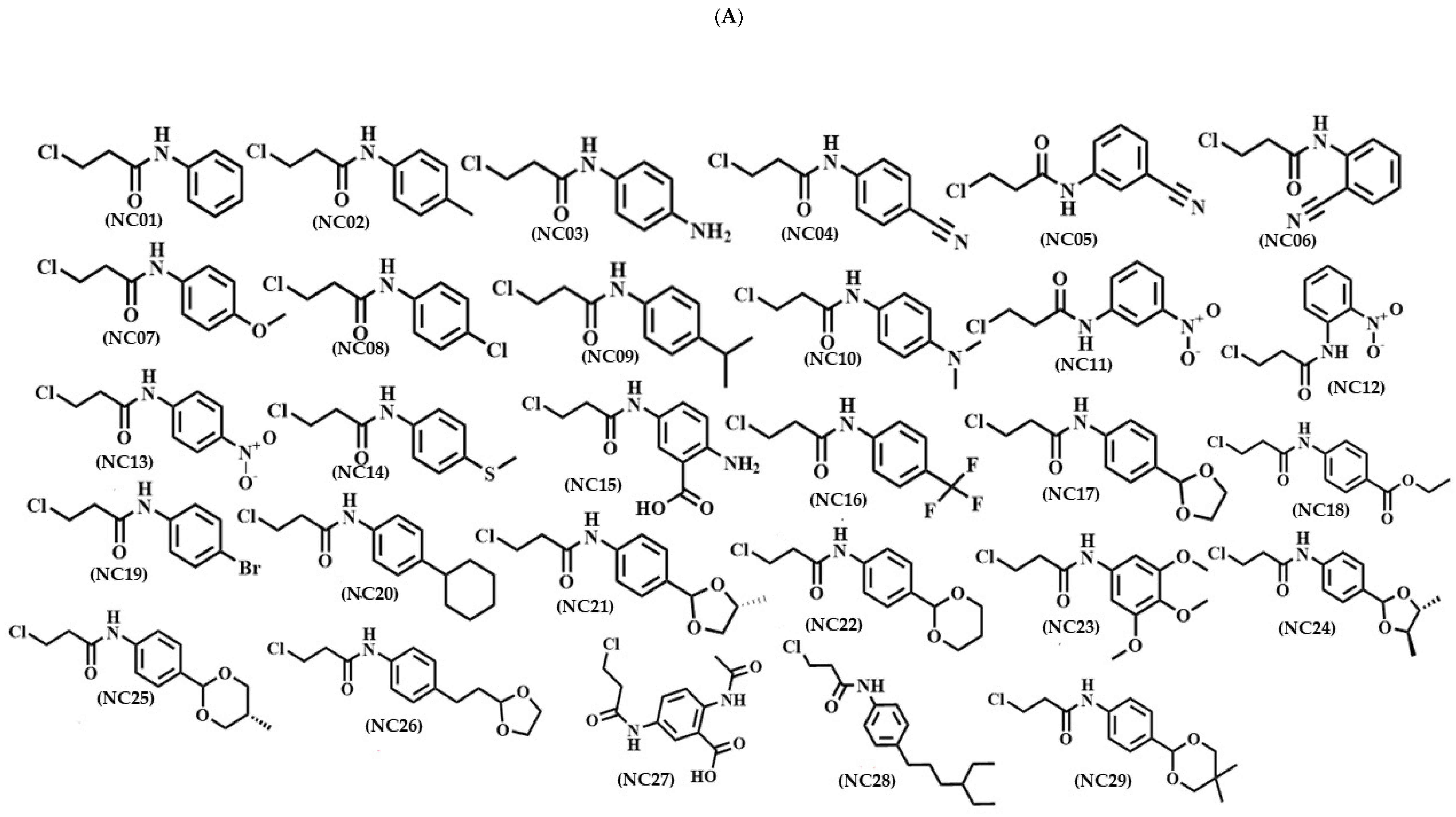
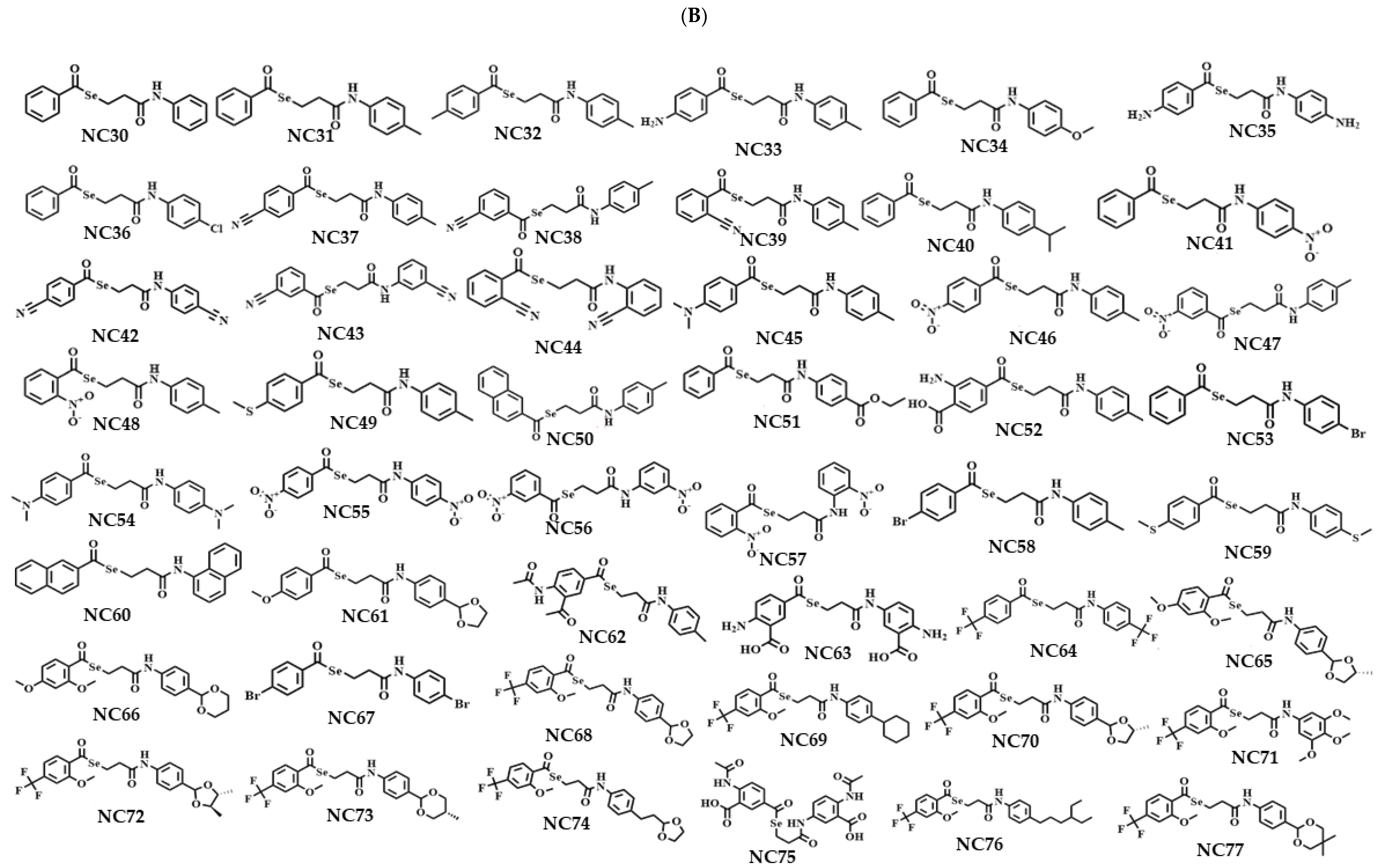
3.1. Compounds in Study
3.2. Quantitative Structure-Activity Relationship (QSAR) Modeling
Consensus Analysis
- Leishmania amazonensis
- Leishmania infantum
- Leishmania braziliensis
- Leishmania major
3.3. VolSurf Descriptor Principal Component Analysis
3.4. Toxicity Risk and Bioavailability In Silico Calculations
3.5. Selection of Molecules for Organic Synthesis
3.6. In Vitro Activity Assessment
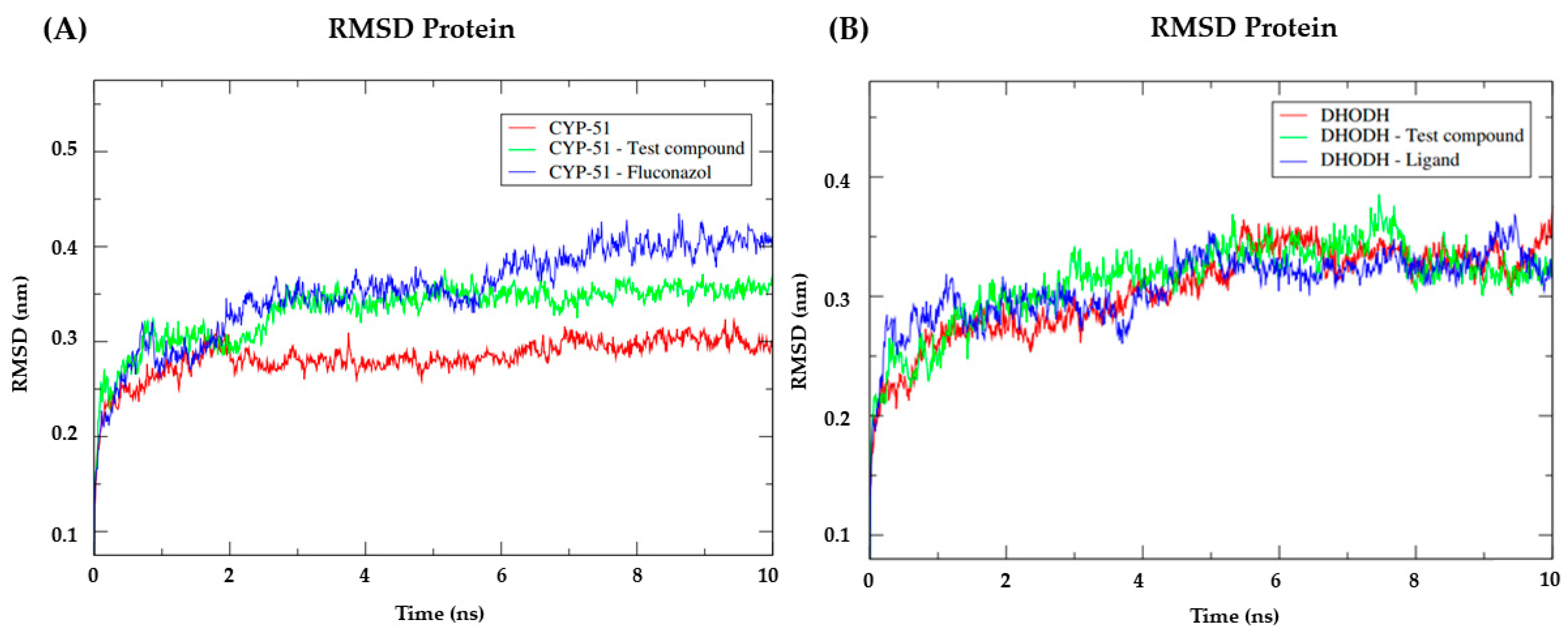
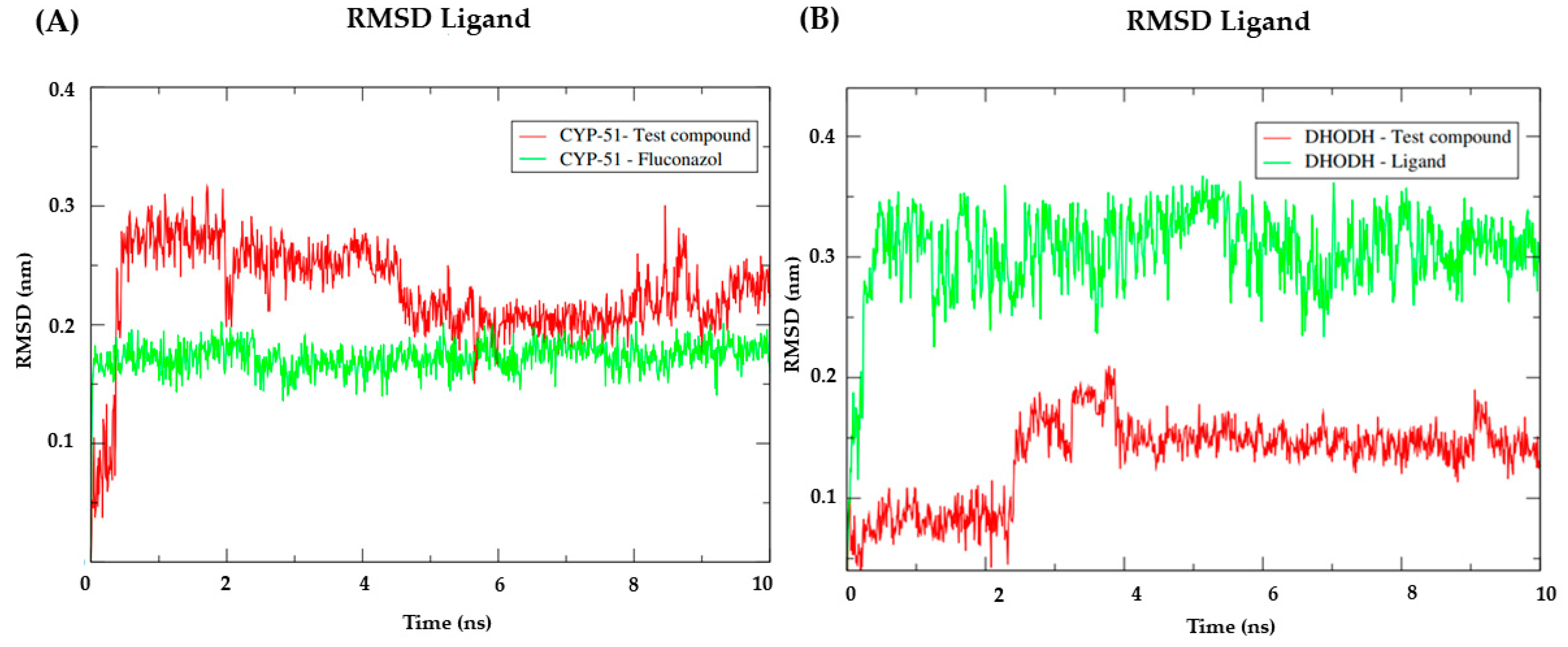
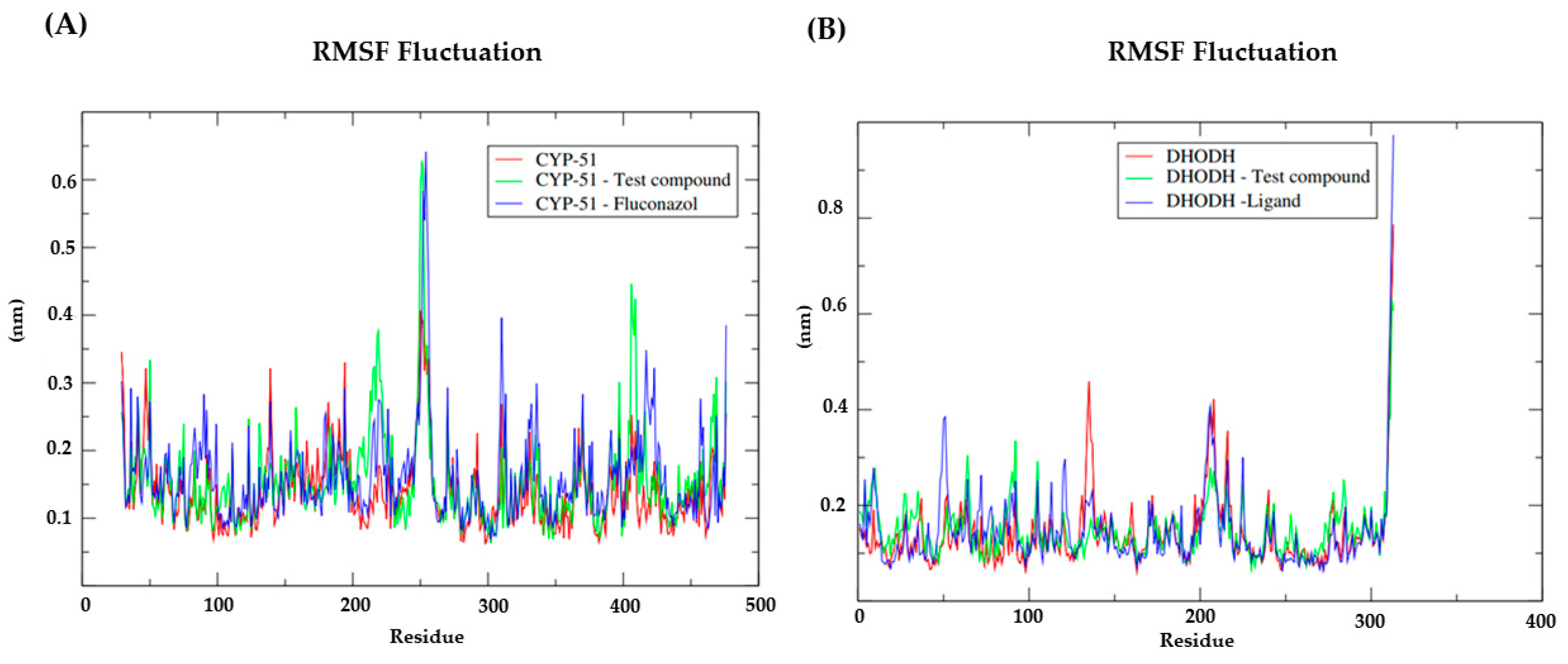
3.7. Molecular Dynamics
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). The NTD road map: Together towards 2030. Control Negl. Trop. Dis. 2020, 1, 2–12. [Google Scholar]
- Valverde, R. Doenças Negligenciadas. FIOCRUZ-Fundação Nacional Osvaldo Cruz 2018. Available online: https://agencia.fiocruz.br/doen%C3%A7as-negligenciadas (accessed on 10 November 2021).
- Catorze, M.G.B. Leishmaniose e SIDA. Med. Cutan. Ibero. Lat. Am. 2005, 33, 237–250. [Google Scholar]
- Paternina-Gómez, M.; Díaz-Olmos, Y.; Paternina, L.E.; Bejarano, E.E. High prevalence of infection with in (Kinetoplastea: Trypanosomatidae) in dogs in northern Colombia. Biomédica 2013, 33, 375–382. [Google Scholar] [PubMed] [Green Version]
- Cochero, S.; Anaya, Y.; Díaz, Y.; Paternina, M.; Luna, A.; Paternina, L.; Bejarano, E.E. Infección natural de Lutzomyia cayennensis cayennensis con parásitos tripanosomatídeos (Kinetoplastida: Trypanosomatidae) en Los Montes de María, Colombia. Rev. Cubana Med. Trop. 2007, 59, 35–39. [Google Scholar]
- Marcondes, M.; Rossi, C.N. Leishmaniose visceral no Brasil. Braz. J. Vet. Res. Anim. Sci. 2013, 50, 341–352. [Google Scholar] [CrossRef] [Green Version]
- BRASIL. Secretaria de Vigilância em Saúde Vigilância em saúde no Brasil 2013–2019. Boletim Epidemiológico 2019, 7, 639–704. [Google Scholar]
- FIOCRUZ as Leishmanioses 1997. Available online: http://www.dbbm.fiocruz.br/tropical/leishman/leishext/index.htm (accessed on 10 November 2021).
- De Araujo-Pereira, T.; de Pita-Pereira, D.; Moreira, R.B.; Silva-Galdino, T.; Duarte, M.P.; Brazil, R.P.; Britto, C. Molecular diagnosis of cutaneous leishmaniasis in an endemic area of Acre State in the Amazonian Region of Brazil. Rev. Soc. Bras. Med. Trop. 2018, 51, 376–381. [Google Scholar] [CrossRef]
- Dorval, M.E.M.C.; Oshiro, E.T.; Cupollilo, E.; Castro, A.C.C.D.; Alves, T.P. Ocorrência de leishmaniose tegumentar americana no Estado do Mato Grosso do Sul associada à infecção por Leishmania (Leishmania) amazonensis. Rev. Soc. Bras. Med. Trop. 2006, 39, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Benchimol, J.L. Leishmanioses Do Novo Numa Perspectiva Histórica Global e Global Dos Anos 1930 Aos 1960. História, Ciências, Saúde Manguinhos. 2020, 27, 1–29. Available online: https://www.scielo.br/j/hcsm/a/wMYtWHsn5ycBdMBtNJ6cNRP/?format=pdf&lang=en (accessed on 3 December 2021).
- BRASIL. Mitelfosina para o Tratamento da Leishmaniose Tegumentar – Relatório de recomendação. CONITEC–Comissão Nacional de Incorporação de Tecnologias no Sistema Único de Saúde. Ministério Saúde Brasília Distrito Federal 2018, 34, 2–28. [Google Scholar]
- BRASIL. Portaria No 56, 30 de Outubro de 2018; Ministério da Saúde-MS: Brasília, Distrito Federal. 2018. Available online: http://138.68.60.75/images/portarias/outubro2018/dia31/portaria56.pdf (accessed on 3 December 2021).
- Uliana, S.R.B.; Trinconi, C.T.; Coelho, A.C. Chemotherapy of leishmaniasis: Present challenges. Parasitology 2018, 145, 464–480. [Google Scholar] [CrossRef]
- Silva, D.G.; Gillespie, J.R.; Ranade, R.M.; Herbst, Z.M.; Nguyen, U.T.T.; Buckner, F.S.; Montanari, C.A.; Gelb, M.H. New class of antitrypanosomal agents based on imidazopyridines. ACS Med. Chem. Lett. 2017, 8, 766–770. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Luis, J.A.; da Silva Souza, H.D.; Lira, B.F.; da Silva Alves, F.; de Athayde-Filho, P.F.; de Souza Lima, T.K.; Rocha, J.C.; Mendonça Junior, F.J.B.; Scotti, L.; Scotti, M.T. Combined structure- and ligand-based virtual screening aiding discovery of selenoglycolicamides as potential multitarget agents against Leishmania species. J. Mol. Struct. 2019, 1198, 1–12. [Google Scholar] [CrossRef]
- Plano, D.; Baquedano, Y.; Moreno-Mateos, D.; Font, M.; Jiménez-Ruiz, A.; Palop, J.A.; Sanmartín, C. Selenocyanates and diselenides: A new class of potent antileishmanial agents. Eur. J. Med. Chem. 2011, 46, 3315–3323. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, 1100–1107. [Google Scholar] [CrossRef] [Green Version]
- ChEMBL Data Base. EMBL-EBI: European Molecular Biology Laboratory. Cambridgeshire, UK. 2019. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 3 December 2021).
- Sherbiny, F. Synthesis, Biological Evaluation and Binding Studies of New Flavone Derivatives as Adenosine A2b Receptor Antagonists. Al-Azhar J. Pharm. Sci. 2016, 53, 73–89. [Google Scholar] [CrossRef]
- Huang, M.-F.N.; Luis, J.A.S.; Silva, A.P.D.; Rocha, J.C.; Lima, T.K.S.; Scotti, M.T.; Scotti, L.; Oliveira, R.F.D.; Souza, H.D.S.; Athayde-Filho, P.F.D. Synthesis, in silico Study and Antileishmanial Evaluation of New Selenides Derived from 7-Chloro-quinoline and N-Phenylacetamides. J. Braz. Chem. Soc. 2021, 32, 712–721. [Google Scholar]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but Verify II: A Practical Guide to Chemogenomics Data Curation. J. Chem. Inf. Model. 2016, 56, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Fourches, D.; Muratov, E.; Tropsha, A. Curation of chemogenomics data. Nat. Chem. Biol. 2015, 11, 1. Available online: https://0-www-nature-com.brum.beds.ac.uk/articles/nchembio.1881 (accessed on 15 December 2021). [CrossRef]
- Dong, J.; Yao, Z.J.; Zhu, M.-F.; Wang, N.-N.; Lu, B.; Chen, A.F.; Lu, A.-P.; Miao, H.; Zeng, W.-B.; Cao, D.-S. ChemSAR: An online pipelining platform for molecular SAR modeling. J. Cheminform. 2017, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Standardizer. ChemAxon. Budapest, Hungary. 2018. Available online: https://chemaxon.com/ (accessed on 3 December 2021).
- Dos Santos Maia, M.; Rodrigues, G.C.S.; De Sousa, N.F.; Scotti, M.T.; Scotti, L.; Mendonça-Junior, F.J.B. Identification of New Targets and the Virtual Screening of Lignans against Alzheimer’s Disease. Oxid. Med. Cell Longev. 2020, 2020, 3098673. [Google Scholar] [CrossRef]
- Dos Santos Maia, M.; de Sousa, N.F.; Rodrigues, G.C.S.; Monteiro, A.F.M.; Scotti, M.T.; Scotti, L. Lignans and Neolignans anti-tuberculosis identified by QSAR and Molecular Modeling. Comb. Chem. High Throughput Screen. 2020, 23, 504–516. [Google Scholar] [CrossRef]
- Dragon 7.0. Kode Informatics srl. Pisa, Italy. 2019. Available online: https://chm.kode-solutions.net/pf/dragon-7-0/ (accessed on 3 December 2021).
- Volsurf Program V 1.7.0. Molecular Discovery. Borehamwood, United Kingdom. 2003. Available online: https://www.moldiscovery.com/about/ (accessed on 3 December 2021).
- Cruciani, G.; Crivori, P.; Carrupt, P.A.; Testa, B. Molecular fields in quantitative structure-permeation relationships: The VolSurf approach. J. Mol. Struct. THEOCHEM 2000, 503, 17–30. [Google Scholar] [CrossRef]
- Scotti, L.; Fernandes, M.B.; Muramatsu, E.; Pasqualoto, K.F.M.; Emereciano, V.D.P.; Tavares, L.C.; da Silva, M.S.; Scotti, M.T. Self-organizing maps and VolSurf approach to predict aldose reductase inhibition by flavonoid compounds. Braz. J. Pharmacogn. 2011, 21, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer Jr, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. Eur. J. Biochem. 1977, 80, 319–324. [Google Scholar] [CrossRef]
- Protein Data Bank (RCSB-PDB). Brookhaven National Laboratory. Nova York, EUA. 1977. Available online: https://www.rcsb.org/ (accessed on 14 April 2021).
- Modeller 9.20-Program for Comparative Protein Structure Modelling by Satisfaction of Spatial Restraints. University of California San Francisco–USFC. San Francisco, EUA. 2020. Available online: https://salilab.org/modeller/ (accessed on 14 April 2021).
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Akula, N.V.; Kumar, S.; Singh, V.; Tiwari, M. Homology modeling and QSAR analysis of 1, 3, 4-thiadiazole and 1, 3, 4-triazole derivatives as carbonic anhydrase inhibitors. Indian J. Biochem. Biophys. 2010, 47, 234–242. [Google Scholar] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef]
- What If. Available online: http://swift.cmbi.ru.nl/servers/html/index.html (accessed on 14 April 2021).
- Mollegro Virtual Docker 6.0. CLC Bio Company. Odder, Denmark. 2022. Available online: http://molexus.io/about/ (accessed on 22 February 2021).
- Discovery Studio 3.5. Biovia. San Diego, EUA. 2022. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 22 February 2021).
- Rorije, E.; Aldenberg, T.; Buist, H.; Kroese, D.; Schüürmann, G. The OSIRIS Weight of Evidence approach: ITS for skin sensitisation. Regul. Toxicol. Pharmacol. 2013, 67, 146–156. [Google Scholar] [CrossRef] [PubMed]
- OSIRIS 5.0 DATA WARRIOR Program. Open Molecules. Available online: https://openmolecules.org/datawarrior/ (accessed on 22 February 2021).
- Rodrigues, K.A.D.F.; Dias, C.N.D.S.; Neris, P.L.D.N.; Rocha, J.D.C.; Scotti, M.T.; Scotti, L.; Mascarenhas, S.R.; Veras, R.C.; De Medeiros, I.A.; Keesen, T.D.S.L.; et al. 2-Amino-thiophene derivatives present antileishmanial activity mediated by apoptosis and immunomodulation in vitro. Eur. J. Med. Chem. 2015, 106, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. 1964, 68, 441–451. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Alves, V.; Braga, R.; Muratov, E.; Andrade, C. Quimioinformática: Uma Introdução. Quim. Nova 2017, 41, 202–212. [Google Scholar] [CrossRef]
- Lunghini, F.; Marcou, G.; Azam, P.; Horvath, D.; Patoux, R.; Van Miert, E.; Varnek, A. Consensus models to predict oral rat acute toxicity and validation on a dataset coming from the industrial context. SAR QSAR Environ. Res. 2019, 30, 879–897. [Google Scholar] [CrossRef]
- Benfenati, E.; Chaudhry, Q.; Gini, G.; Dorne, J. Lou Integrating in silico models and read-across methods for predicting toxicity of chemicals: A step-wise strategy. Environ. Int. 2019, 131, 1–15. [Google Scholar] [CrossRef]
- BRASIL. Risco Químico. Fundação Nacional Osvaldo Cruz–FIOCRUZ. Rio de Janeiro, RJ. 2018. Available online: http://www.fiocruz.br/biosseguranca/Bis/lab_virtual/riscos_quimicos.html (accessed on 22 February 2021).
- Mugesh, G.; du Mont, W.-W.; Sies, H. Chemistry of biologically important synthetic organoselenium compounds. Chem. Rev. 2001, 101, 2125–2180. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Zeni, G.; Rocha, J.B.T. Organoselenium and organotellurium compounds: Toxicology and pharmacology. Chem. Rev. 2004, 104, 6255–6286. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Lauri, G. Prediction of intestinal permeability. Adv. Drug Deliv. Rev. 2002, 54, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Navia, M.A.; Chaturvedi, P.R. Design principles for orally bioavailable drugs. Drug Discov. Today 1996, 1, 179–189. [Google Scholar] [CrossRef]
- Li, A.P. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov. Today 2001, 6, 357–366. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.F.; Maltarollo, V.G.; Oliveira, P.R.; Silva, A.B.F. Advances and Perspectives in Applying Deep Learning for Drug Design and Discovery. Front. Robot. AI 2019, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Dello Iacono, L.; Di Pisa, F.; Mangani, S. Crystal Structure of the Ternary Complex of Leishmania Major Pteridine Reductase 1 with the Cofactor NADP+/NADPH and the Substrate Folic Acid. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2022, 78, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Villafraz, O.; Baudouin, H.; Mazet, M.; Kulyk, H.; Dupuy, J.-W.; Pineda, E.; Botté, C.; Inaoka, D.K.; Portais, J.-C.; Bringaud, F. The Trypanosome UDP-Glucose Pyrophosphorylase Is Imported by Piggybacking into Glycosomes, Where Unconventional Sugar Nucleotide Synthesis Takes Place. MBio 2021, 12, 1–22. [Google Scholar] [CrossRef]
- Boschi, D.; Pippione, A.C.; Sainas, S.; Lolli, M.L. Dihydroorotate Dehydrogenase Inhibitors in Anti-Infective Drug Research. Eur. J. Med. Chem. 2019, 183, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Maamri, S.; Benarous, K.; Yousfi, M. Identification of 3-Methoxycarpachromene and Masticadienonic Acid as New Target Inhibitors against Trypanothione Reductase from Leishmania Infantum Using Molecular Docking and ADMET Prediction. Molecules 2021, 26, 3335. [Google Scholar] [CrossRef]
- Vincendeau, P.; Gobert, A.P.; Daulouède, S.; Moynet, D.; Mossalayi, M.D. Arginases in Parasitic Diseases. Trends Parasitol. 2003, 19, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Boitz, J.M.; Gilroy, C.A.; Olenyik, T.D.; Paradis, D.; Perdeh, J.; Dearman, K.; Davis, M.J.; Yates, P.A.; Li, Y.; Riscoe, M.K. Arginase Is Essential for Survival of Leishmania Donovani Promastigotes but Not Intracellular Amastigotes. Infect. Immun. 2017, 85, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairlamb, A.H.; Cerami, A. Metabolism and Functions of Trypanothione in the Kinetoplastida. Annu. Rev. Microbiol. 1992, 46, 695–729. [Google Scholar] [CrossRef] [PubMed]
- Badirzadeh, A.; Taheri, T.; Abedi-Astaneh, F.; Taslimi, Y.; Abdossamadi, Z.; Montakhab-Yeganeh, H.; Aghashahi, M.; Niyyati, M.; Rafati, S. Arginase Activity of Leishmania Isolated from Patients with Cutaneous Leishmaniasis. Parasite Immunol. 2017, 39, 1–10. [Google Scholar] [CrossRef]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.B. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, E.R.; Come, J.A.A.D.S.S.; Brogi, S.; Calderone, V.; Chemi, G.; Campiani, G.; Oliveira, T.M.F.D.S.; Pham, T.-N.; Pudlo, M.; Girard, C. Cinnamides Target Leishmania Amazonensis Arginase Selectively. Molecules 2020, 25, 1–19. [Google Scholar]
- Lepesheva, G.I.; Friggeri, L.; Waterman, M.R. CYP51 as Drug Targets for Fungi and Protozoan Parasites: Past, Present and Future. Parasitology 2018, 145, 1820–1836. [Google Scholar] [CrossRef]
- Costa, C.H.S.D.; Bichara, T.W.; Gomes, G.C.; Dos Santos, A.M.; da Costa, K.S.; Lima, A.H.L.E.; Alves, C.N.; Lameira, J. Unraveling the Conformational Dynamics of Glycerol 3-Phosphate Dehydrogenase, a Nicotinamide Adenine Dinucleotide-Dependent Enzyme of Leishmania Mexicana. J. Biomol. Struct. Dyn. 2021, 39, 2044–2055. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Nes, W.D.; Waterman, M.R.; Lepesheva, G.I. Substrate Preferences and Catalytic Parameters Determined by Structural Characteristics of Sterol 14α-Demethylase (CYP51) from Leishmania Infantum. J. Biol. Chem. 2011, 286, 26838–26848. [Google Scholar] [CrossRef] [Green Version]
- Saccoliti, F.; Angiulli, G.; Pupo, G.; Pescatori, L.; Madia, V.N.; Messore, A.; Colotti, G.; Fiorillo, A.; Scipione, L.; Gramiccia, M. Inhibition of Leishmania Infantum Trypanothione Reductase by Diaryl Sulfide Derivatives. J. Enzyme Inhib. Med. Chem. 2017, 32, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Biradar, P.; Patil, V.; Joshi, H.; Khanal, P.; Mallapur, S. Experimental Validation and Network Pharmacology Evaluation to Decipher the Mechanism of Action of Erythrina variegata L. Bark against Scopolamine-Induced Memory Impairment in Rats. Adv. Tradit. Med. 2020, 22, 193–206. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Fragments, Network Biology and Designing Multiple Ligands. Drug Discov. Today 2007, 12, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Viana, J.D.O.; Félix, M.B.; Maia, M.D.S.; Serafim, V.D.L.; Scotti, L.; Scotti, M.T. Drug Discovery and Computational Strategies in the Multitarget Drugs Era. Brazilian J. Pharm. Sci. 2018, 54, 1–25. [Google Scholar]
- Steinbrenner, H. Interference of Selenium and Selenoproteins with the Insulin-Regulated Carbohydrate and Lipid Metabolism. Free Radic. Biol. Med. 2013, 65, 1538–1547. [Google Scholar] [CrossRef]
- Chibli, L.A.; Schmidt, T.J.; Nonato, M.C.; Calil, F.A.; Da Costa, F.B. Natural Products as Inhibitors of Leishmania Major Dihydroorotate Dehydrogenase. Eur. J. Med. Chem. 2018, 157, 852–866. [Google Scholar] [CrossRef]
- Brannigan, J.A.; Roberts, S.M.; Bell, A.S.; Hutton, J.A.; Hodgkinson, M.R.; Tate, E.W.; Leatherbarrow, R.J.; Smith, D.F.; Wilkinson, A.J. Diverse Modes of Binding in Structures of Leishmania Major N-Myristoyltransferase with Selective Inhibitors. IUCrJ 2014, 1, 250–260. [Google Scholar] [CrossRef] [Green Version]
- Bell, A.S.; Yu, Z.; Hutton, J.A.; Wright, M.H.; Brannigan, J.A.; Paape, D.; Roberts, S.M.; Sutherell, C.L.; Ritzefeld, M.; Wilkinson, A.J. Novel Thienopyrimidine Inhibitors of Leishmania N-Myristoyltransferase with on-Target Activity in Intracellular Amastigotes. J. Med. Chem. 2020, 63, 7740–7765. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative Molecular Field Analysis (CoMFA). 1. Effect of Shape on Binding of Steroids to Carrier Proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef]
- Yusuf, D.; Davis, A.M.; Kleywegt, G.J.; Schmitt, S. An Alternative Method for the Evaluation of Docking Performance: RSR vs RMSD. J. Chem. Inf. Model. 2008, 48, 1411–1422. [Google Scholar] [CrossRef]
- Schneider, N.; Hindle, S.; Lange, G.; Klein, R.; Albrecht, J.; Briem, H.; Beyer, K.; Claußen, H.; Gastreich, M.; Lemmen, C. Substantial Improvements in Large-Scale Redocking and Screening Using the Novel HYDE Scoring Function. J. Comput. Aided. Mol. Des. 2012, 26, 701–723. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specie | Validation | Specificity | Sensitivity | Accuracy | PPV | NPV | MCC | ROC |
|---|---|---|---|---|---|---|---|---|
| L. amazonensis | Test | 0.82 | 0.82 | 0.82 | 0.82 | 0.82 | 0.64 | 0.920 |
| Cross | 0.761 | 0.707 | 0.742 | 0.707 | 0.778 | 0.524 | 0.846 | |
| L. infantum | Test | 0.885 | 0.815 | 0.849 | 0.815 | 0.885 | 0.70 | 0.904 |
| Cross | 0.733 | 0.785 | 0.759 | 0.785 | 0.733 | 0.519 | 0.849 | |
| L. braziliensis | Test | 0.79 | 0.85 | 0.821 | 0.85 | 0.79 | 0.642 | 0.878 |
| Cross | 0.885 | 0.704 | 0.792 | 0.704 | 0.885 | 0.597 | 0.877 | |
| L. major | Test | 0.885 | 0.873 | 0.879 | 0.873 | 0.885 | 0.758 | 0.932 |
| Cross | 0.846 | 0.815 | 0.83 | 0.815 | 0.849 | 0.661 | 0.882 |
| Specie | Validation | Specificity | Sensitivity | Accuracy | PPV | NPV | MCC | ROC |
|---|---|---|---|---|---|---|---|---|
| L. amazonensis | Test | 0.713 | 0.786 | 0.75 | 0.762 | 0.737 | 0.50 | 0.854 |
| Cross | 0.745 | 0.755 | 0.75 | 0.755 | 0.745 | 0.50 | 0.841 | |
| L. infantum | Test | 0.762 | 0.841 | 0.802 | 0.841 | 0.762 | 0.605 | 0.887 |
| Cross | 0.846 | 0.667 | 0.755 | 0.667 | 0.846 | 0.52 | 0.853 | |
| L. braziliensis | Test | 0.786 | 0.844 | 0.816 | 0.825 | 0.806 | 0.632 | 0.897 |
| Cross | 0.731 | 0.778 | 0.755 | 0.764 | 0.745 | 0.509 | 0.860 | |
| L. major | Test | 0.816 | 0.811 | 0.814 | 0.815 | 0.813 | 0.628 | 0.897 |
| Cross | 0.82 | 0.794 | 0.806 | 0.806 | 0.613 | 0.613 | 0.876 |
| ID | L.b Pro (p%) | IC50 | L.i Pro (p%) | IC50 | L.m Pro (p%) | IC50 | L.a Pro (p%) | IC50 |
|---|---|---|---|---|---|---|---|---|
| NC01 | 8% | 17.6 | 59% | 26.89 | 26% | >50 | 18% | >50 |
| NC02 | 5% | >50 | 59% | >50 | 32% | >50 | 19% | >50 |
| NC07 | 5% | >50 | 58% | >50 | 37% | >50 | 10% | >50 |
| NC08 | 4% | >50 | 61% | >50 | 32% | >50 | 26% | >50 |
| NC09 | 16% | >50 | 58% | >50 | 32% | >50 | 19% | >50 |
| NC13 | 9% | >50 | 52% | >50 | 42% | >50 | 8% | >50 |
| NC18 | 25% | >50 | 54% | >50 | 46% | >50 | 14% | >50 |
| NC19 | 4% | >50 | 60% | >50 | 16% | >50 | 27% | >50 |
| NC30 | 44% | 7.92 | 69% | 10.4 | 58% | 13.97 | 64% | 14.5 |
| NC31 | 44% | 5.23 | 68% | 7.36 | 56% | 5.7 | 61% | 15.3 |
| NC34 | 45% | 2.1 | 67% | 3.28 | 49% | 4.84 | 72% | 7.28 |
| NC36 | 48% | 4.24 | 68% | 8.52 | 59% | 10.36 | 70% | 15.9 |
| NC40 | 58% | 5.85 | 74% | 6.6 | 60% | >50 | 68% | 8.2 |
| NC41 | 53% | >50 | 59% | >50 | 46% | >50 | 61% | >50 |
| NC51 | 61% | 3.71 | 68% | 6.43 | 47% | 9.85 | 63% | 16.4 |
| NC53 | 54% | 3.9 | 71% | 8.05 | 64% | 8.63 | 70% | 25.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Sousa, N.F.; da Silva Souza, H.D.; de Menezes, R.P.B.; da Silva Alves, F.; Acevedo, C.A.H.; de Lima Nunes, T.A.; Sessions, Z.L.; Scotti, L.; Muratov, E.N.; Mendonça-Junior, F.J.B.; et al. Selene-Ethylenelacticamides and N-Aryl-Propanamides as Broad-Spectrum Leishmanicidal Agents. Pathogens 2023, 12, 136. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens12010136
de Sousa NF, da Silva Souza HD, de Menezes RPB, da Silva Alves F, Acevedo CAH, de Lima Nunes TA, Sessions ZL, Scotti L, Muratov EN, Mendonça-Junior FJB, et al. Selene-Ethylenelacticamides and N-Aryl-Propanamides as Broad-Spectrum Leishmanicidal Agents. Pathogens. 2023; 12(1):136. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens12010136
Chicago/Turabian Stylede Sousa, Natália Ferreira, Helivaldo Diógenes da Silva Souza, Renata Priscila Barros de Menezes, Francinara da Silva Alves, Chonny Alexander Herrera Acevedo, Thaís Amanda de Lima Nunes, Zoe L. Sessions, Luciana Scotti, Eugene N. Muratov, Francisco Jaime Bezerra Mendonça-Junior, and et al. 2023. "Selene-Ethylenelacticamides and N-Aryl-Propanamides as Broad-Spectrum Leishmanicidal Agents" Pathogens 12, no. 1: 136. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens12010136