1. Introduction
Diabetes is a chronic disease generally caused by a dysfunctional pancreas that does not produce enough insulin or by the body’s ineffective use of the insulin produced. Both issues result in an increase in plasma glucose levels. Insufficient insulin secretion causes glucose metabolism disorders, such as reduced glucose entering the cells, reduced glycogen synthesis, weakened tricarboxylic acid (TCA) circulation, glucose accumulation in the blood, and reduced glucose utilization in the liver, muscle, fat, and other tissues. Along with these changes, the muscular and hepatic glycogen breakdown diminishes and gluconeogenesis increases, resulting in an increase in glucose production, leading to hyperglycemia. In addition, diabetes is often accompanied by lipid metabolic disorders, leading to multiple cardio-cerebrovascular diseases. Diabetes mellitus is often accompanied by dyslipidemia, which is a result of insulin resistance. Therefore, patients with diabetes who also have dyslipidemia need effective lipid-lowering treatment in addition to hypoglycemic therapy.
Depending on the different types of cells and oxygen supply, glucose catabolism mainly includes anaerobic oxidation, aerobic oxidation, and the pentose phosphate pathway. After high carbohydrate intake, blood glucose rises rapidly. It thus stimulates insulin secretion to promote cell uptake and the use of glucose. Moreover, insulin is the main hormone regulating fatty acid synthesis. By stimulating the activity of protein phosphatase, insulin dephosphorylates acetyl CoA carboxylase and promotes fatty acid synthesis. In addition, insulin promotes fatty acid synthesis and increases fat synthesis. In patients with diabetes, the ability of tissue cells to absorb glucose is decreased. The intracellular energy supply is thus insufficient. This may lead to increased hunger, increased eating, and an increased accumulation of glucose in the serum, consequently causing hyperglycemia [
1]. Dyslipidemia caused by diabetes is closely related to insulin resistance, visceral obesity, and non-alcoholic fatty liver disease. In insulin-resistant individuals, excessive fatty acids flow into the liver, resulting in an excessive accumulation of very low-density lipoprotein [
2]. Dyslipidemia caused by diabetes is typically a mixed hyperlipidemia, which is characterized moderately by increased low-density lipoprotein levels, increased total cholesterol levels, and decreased high-density lipoprotein levels [
3].
Before taking insulin treatment into practice, bacterial infection was recognized as one of the most serious complications of diabetes mellitus, and it has been considered a critical cause of morbidity and mortality among diabetic patients. With the increased clinical use of insulin and antibiotics in treatment, the mortality rate of diabetes complicated with infection has reduced; nevertheless, the risk of diabetic patients dying from bacterial-related infections remains high. The reported standardized mortality rate is 1.83 [
4].
Staphylococcus aureus (
S. aureus) is an important human pathogen. Clinical infections related to
S. aureus are a major global health problem and have been exacerbated by the emergence and spread of drug-resistant strains [
5]. More importantly, recent evidence has revealed that the most predominant aerobic organism causing diabetic infections is
S. aureus, accounting for 18.7% of cases [
6]. In addition,
S. aureus and MRSA colonization rates are approximately two times higher in diabetic patients than in patients without diabetes [
7,
8]. Therefore,
S. aureus infections are most likely a crucial factor for the significant morbidity and mortality in patients with hyperglycemia [
7]. This is probably attributed to the frequent complication of foot ulcers in diabetic patients [
9]. It is thus critically important to obtain a deeper understanding of the molecular mechanism of
S. aureus infection in patients with diabetes. Furthermore, some recent studies conducted in type 2 diabetic mice have shown that
S. aureus infection induces insulin resistance, impairs glucose tolerance, and elevates blood glucose levels by secreting eLtaS protein, which binds to insulin [
10]. Nonetheless, the exact effects of
S. aureus infection on glucose and lipid metabolism and its molecular mechanism are not currently fully understood. A search for new molecular targets and innovative therapeutic strategies is thus urgently needed.
It is well-known that catabolite control protein A (CcpA) is a master regulator of carbon catabolite repression in
S. aureus [
11]. In the presence of glucose or other preferred carbon sources, CcpA forms a complex with S46-phosphorylated Hpr protein. The CcpA–Hpr-S46(P) complex binds with the catabolite-responsive element (
cre) sequences of diverse genes to activate or repress their expression. CcpA represses the TCA cycle and is also instrumental in
S. aureus pathogenesis [
12,
13,
14]. In addition, the pyruvate dehydrogenase complex (PDC) is a multi-enzyme complex of the mitochondria and is implicated in glycolysis with the Krebs cycle. PDC is thus essential in producing acetyl CoA from glucose and regulates fuel consumption. The core PDC comprises three catalytic enzymes: pyruvate dehydrogenase (PDH), dihydrolipoamide acetyltransferase (DLAT), and dihydrolipoamide dehydrogenase (DLD). [
15] The activity of PDC is regulated by pyruvate dehydrogenase kinases (PDKs). The main PDK subtypes expressed in the liver are PDK1, PDK2, and PDK4. PDK4 is a key regulator of PDC activity as well as pyruvate oxidation and glucose balance in vivo. It also inhibits PDC activity by phosphorylating PDH [
16,
17].
Considering the background information discussed above, we hypothesized that when S. aureus invades the body, CcpA protein may profoundly affect the glucose and lipid metabolism of the host in a high-glucose environment. In the present study, type 1 diabetic mice induced by streptozotocin injection and WRL68 human hepatocytes and subjected to high-glucose treatment were used as a model to investigate the potential effects of CcpA on abnormal metabolic phenotypes of diabetes. In addition, we studied the underlying mechanism for the first time to understand whether the phenotypes are associated with the activation of the liver’s signal transducer and activator of transcription 5 (STAT5)/PDK4 pathway. The study may provide significant insights into the development of antimicrobial agents targeting CcpA and their potential use in patients with diabetes.
2. Methods
2.1. Strains and Plasmids
The bacterial strains and plasmids used in this study are listed in
Table 1. The
S. aureus strain XN108 was isolated from a burn patient on 4 March 2004. RN4220 and Mu50 were kindly provided by Dr. Xiancai Rao (Army Medical University, China). The
S. aureus Δ
CcpA strain was constructed using an allelic replacement strategy as follows. Briefly, the
CcpA gene was amplified by PCR from the genomic DNA of XN108 using the primer pairs pBT2–
CcpA–5′ and pBT2–
CcpA–3′. Then, it was cloned into the temperature-sensitive shuttle vector pBT2, which is widely used for markerless double homologous recombination. The pBT2–Δ
CcpA plasmid was transformed into the
S. aureus strain RN4220, extracted, purified, and then electro-transformed into XN108 to generate XN108 ΔCcpA after recombination. The Δ
CcpA mutation after allelic exchange was confirmed by Sanger sequencing. The mutant Mu50 Δ
CcpA was constructed using similar allelic replacement techniques.
2.2. Animals and the Establishment of Streptozotocin-Induced Diabetic Mice
The ethics committee of Guangzhou Medical University (Guangzhou, China) approved the protocols for the use and care of animals (approval number: GY2017–040). The handling and treatment of mice were conducted in strict accordance with Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines for reporting experiments involving animals.
Adult male C57BL/6 mice (7–8 weeks, 20.0 g ± 2.0 g) were purchased from the Guangdong Medical Laboratory Animal Center (Guangzhou, China). Mice were housed in separate cages in a controlled environment (12-h/12-h day/night cycle, 50–70% humidity, and 26 °C temperature) and had ad libitum access to food and water. One-week acclimatization was performed to minimize stress.
Type 1 diabetes mellitus was induced by intraperitoneal injection of streptozotocin (Sigma-Aldrich, St. Louis, MO, USA) at a dose of 45 mg/kg/day for 5 days [
18]. Age-matched male C57 mice were used as controls and given the same volume of citrate buffer. The level of plasma glucose was determined using a glucometer (OneTouch
TM Ultra Mini
@ Blood Glucose Monitoring System, LLC, USA) three days after the final injection. Mice with a plasma glucose level of >16.7 mmol/L were defined as having DM.
2.3. Experimental Conditions for the Mice with Diabetes
Four weeks after DM induction, the mice were randomly divided into three groups: (1) the diabetic group (diabetic + vehicle, n = 8), which received intraperitoneal injections of PBS; (2) a diabetic group wherein the jugular vein was injected with 1 × 106 CFU S. aureus XN108 ΔCcpA (diabetic + XN108 ΔCcpA, n = 9); and (3) a diabetic group wherein the jugular vein was injected with 1 × 106 CFU wild-type S. aureus XN108 (diabetic + WT, n = 9). The control mice were randomly divided into three groups similar to those of the diabetic mice (non-diabetic + vehicle, non-diabetic + ΔCcpA, and non-diabetic + WT; n = 9 for each group). All mice were fed a normal diet and had ad libitum access to drinking water during the experiment.
2.4. Histological Measurements
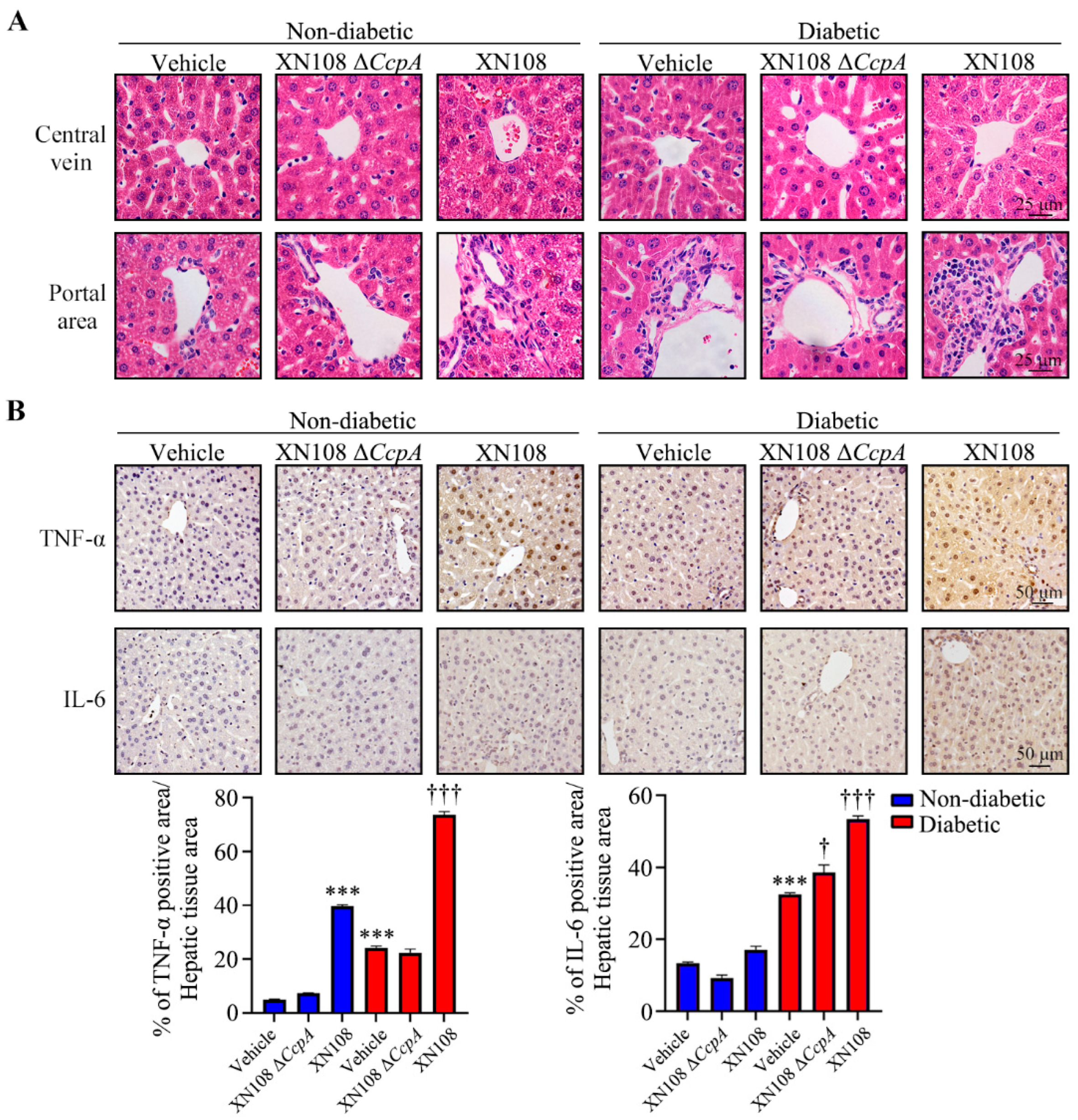
Murine livers were fixed with 4% paraformaldehyde and immersed in paraffin. For hematoxylin and eosin and immunohistochemical staining [
19], paraffin-embedded hepatic tissues from the mice in each group were cut into 4 μm sections using a microtome. After three cycles of deparaffinization in xylene and tissue rehydration, antigen retrieval was performed in Tris buffer (pH 9.0; Dako, Santa Clara, CA, USA) by heating to 99 °C for 20 min. The endogenous peroxidase activity was quenched with 3% H
2O
2, and non-specific binding was blocked with 10% non-immune goat serum (Life Technologies, Waltham, MA, USA); this was followed by the incubation of samples with primary antibodies at 4 °C overnight. TNF-α antibody and IL-6 antibody (Abcam, Cambridge, UK) were used at a 1:200 dilution. The signal was amplified using the Histostain SP Kit (Life Technologies, Waltham, MA, USA) and detected with diaminobenzidine (DAB) substrate (Dako, Santa Clara, CA, USA). The color development of DAB for diseased and control livers was microscopically monitored in parallel with the same reaction time. Hematoxylin was used as a counterstain. The intensity of TNF-α and IL-6 was quantified with the 3 sets of IHC straining images by Image J software (ImageJ bundled with 64-bit Java version 1.8.0_172). Negative controls were treated similarly, except for the omission of primary antibodies.
2.5. Determination of the Level of Random Plasma Glucose, Fasting Plasma Glucose, and Glucose Tolerance
The random plasma glucose level and fasting plasma glucose level were measured using a glucose analyzer (OneTouch Ultra Mini Blood Glucose Monitoring System, Johnson Co., New Brunswick, NJ, USA). Glucose tolerance was determined using intraperitoneal glucose tolerance tests (IPGTTs) in overnight-fasted mice [
20]. After the intraperitoneal injection of glucose (2 g/kg), the plasma glucose level in blood samples obtained from the caudal vein was measured at 0, 30, 60, 90, and 120 min using the glucometer. The area under the blood glucose curve (AUC) was measured using the trapezoidal rule [
21].
2.6. Transcriptome Sequencing
Liver tissues from twelve male C57BL/6 mice per group were dissected and immediately frozen in liquid nitrogen. Total RNA was extracted with TRIzol (Life Technologies, Waltham, MA, USA) reagent. RNA integrity (RNA integrity ≥ 7.9) and quantity were determined with an Agilent 2100 Bioanalyzer. Equal RNA amounts from individual mice were combined at 3 samples per pool, and 3 pools per group were analyzed in the Illumina HiSeqTM2000 at BGI Shenzhen (Beijing Genome Institute, Shenzhen 518083, China). All reads that passed quality metrics were mapped on the mouse transcriptome and genome using HISAT40/Bowtie241 tools. Then, normalization was performed with the mapped data, and the FPKM (fragments per kilobase per million mapped reads) was calculated using RESM software. An FDR (false discovery rate) < 0.01 and an absolute value of log2 Ratio ≥ 2 were used to identify differentially expressed genes (DEGs) in the diabetic + vehicle versus diabetic + XN108 ΔCcpA groups and the diabetic + vehicle versus diabetic + WT groups [
22].
2.7. Cell Culture
WRL-68 cells were purchased from Guang Zhou Jennio Biotech Co., Ltd., Guangzhou, China. The cells were cultured in Dulbecco’s modified Eagle medium (DMEM) with L-glutamine (Gibco, Gland island, NE, USA) supplemented with 10% v/v fetal bovine serum (FBS; Gibco, Waltham, MA, USA) and the bacterial strain (1.5 × 107 CFU/mL) at 37 °C and 5% CO2. The control group samples were cultured in DMEM with 10% v/v FBS.
2.8. Western Blotting
Hepatic protein samples and WRL68 cellular protein samples for Western blotting were extracted using 10× RIPA buffer (Cell Signaling, Danvers, MA, USA) supplemented with protease inhibitors. The following antibodies were purchased: rabbit anti-PDK1 (BS1291, Bioworld Technology, St Louis Park, MN, USA); rabbit anti-PDK2 (ab68164, Abcam, Cambridge, UK); rabbit anti-PDK4 (ab214938, Abcam, Cambridge, UK); rabbit anti-PDH (ab168379, Abcam, Cambridge, UK); rabbit anti-p-PDH (NB110-93479SS, NOVUS, Minneapolis, MN, USA); rabbit anti-PPARα (ab24509, Abcam, Cambridge, UK); rabbit anti-PGC-1α (ab191838, Abcam, Cambridge, UK); rabbit anti-FOXO1 (ab52857, Abcam, Cambridge, UK); rabbit anti-p-FOXO1 (9461, Cell Signaling, Danvers, MA, USA); rabbit anti-STAT5 (94205, Cell Signaling, Danvers, MA, USA); rabbit anti-p-STAT5 (ab32364, Abcam, Cambridge, UK); rabbit anti-β-actin (AP0060, Bioworld Technology, Minnesota, USA); and anti-rabbit IgG HRP-linked antibody (7074P2, Cell Signaling, Danvers, MA, USA). Herein, 1 μg/mL primary antibodies and 50 ng/mL secondary antibodies were used for blotting.
For Western blotting, the membrane was blocked with 5% skim milk in PBST for 1 h at 25 °C with gentle shaking. Next, the membrane was incubated with primary antibodies diluted in a solution containing 3% BSA in PBST overnight at 4°C with gentle shaking. Furthermore, the membrane was washed three times for 10 min in PBST with vigorous shaking. Then, at 25 °C, the membrane was incubated with secondary antibodies diluted in 5% skim milk in PBST for 1 h with gentle shaking. The membrane was again washed three times for 10 min in PBST with vigorous shaking. Detection was performed using SuperSignal
TM West Pico PLUS (Thermo, Waltham, MA, USA) [
23].
2.9. RNA Isolation and qRT-PCR
Total RNA from liver tissues was extracted using TRIzol (Life Technologies, USA). Complementary DNA (cDNA) was synthesized using the First Strand cDNA Synthesis Kit (Takara, Japan) according to the manufacturer’s instructions. RT-qPCR was performed in a 20 μL system using the QuantiTect SYBR Green PCR Kit (Takara, Osaka, Japan) on an ABI StepOneTM Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). The primer sequences used were as follows: PDK1, 5′-TTCCTGGACTTCGGGTCAGT-3′ (forward) and 5′-GTACGGATGGGGTCCTGAGA-3′ (reverse); PDK2, 5′-TTCAGCAAGTTCTCCCCGTC-3′ (forward) and 5′-GACATACCAGCTCTGCACCA-3′ (reverse); PDK4, 5′-AGCAGTAGTCGAAGATGCCTT-3′ (forward) and 5′-CACGATGTGGATTGGTTGGC-3′ (reverse); Pdhα1, 5′-GGGACGTCTGTTGAGAGAGC-3′ (forward) and 5′-TGTGTCCATGGTAGCGGTAA-3′ (reverse); and Glc6p, 5′-CTCTGGGTGGCAGTGGTCGG-3′ (forward) and 5′-AGGACCCACCAATACGGGCGT-3′ (reverse).
2.10. Measurement of Biochemical Indicators
Biochemical indicators—the levels of total cholesterol, triglycerides, and glycated serum protein—were determined using diagnostic kits (Dade Behring Holdings, Shanghai, China) with an auto-analyzer (Siemens Healthcare Diagnostics, Nuremberg, Germany).
2.11. Measurement of the Content of Pyruvic Acid
Pyruvic acid content was determined using a commercial quantitative kit (Sigma-Aldrich, USA) according to the manufacturer’s instructions.
2.12. Statistical Analyses
Data are represented as the mean ± standard error of the mean. Differences between groups were analyzed using SPSS v18.0 (IBM, New York, NY, USA). One-way analysis of variance with Bonferroni post hoc testing was used for comparing multiple groups. A p-value of <0.05 was considered significant.
4. Discussion
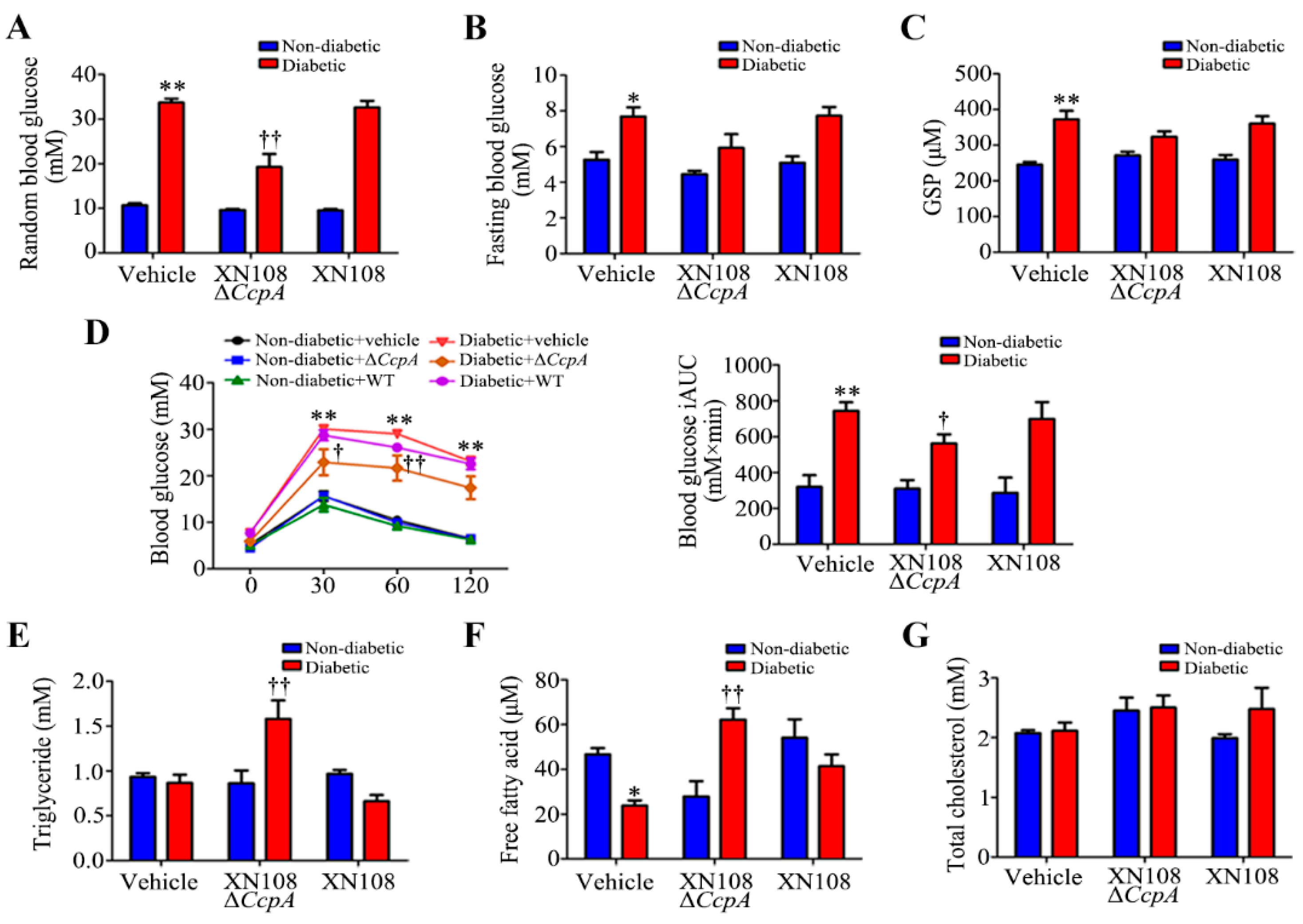
After the intravenous injection of CcpA-knockout S. aureus in type 1 diabetic mice, the random blood glucose level was found to decrease significantly. The blood lipids and serum free fatty acids also increased significantly. Meanwhile, we also found a decrease in the random blood glucose level in type 2 diabetic mice infected with CcpA-knockout S. aureus. However, the metabolic phenotype changes in type 1 diabetic mice were more obvious, so we focused on type 1 diabetes in this study. The specific mechanisms by which CcpA-knockout S. aureus affects type 2 diabetic mice will also be explored in future studies. The results in type 1 diabetic mice suggest that the CcpA protein in the S. aureus-infected mice profoundly affected the glucose and lipid metabolism of the host in a high-glucose environment. Furthermore, type 1 diabetic mice and WRL68 human hepatocytes subjected to high-glucose treatments were used as research models to verify that CcpA-knockout S. aureus infection activated the signal transducer and activator of the transcription 5 (STAT5)/PDK4 pathway of the liver by secreting proteins, resulting in abnormal metabolic phenotypes of diabetes.
CcpA is a very important global regulatory protein and typically exists in Gram-positive bacteria, such as
S. aureus,
Streptococcus spp., and
Bacillus subtilis. Previous studies have shown that during the exponential growth period of
S. aureus, more than 150 genes are regulated by CcpA [
28]. It is also known that CcpA not only regulates the expression of metabolic genes in
S. aureus but also controls staphylotoxin secretion, biofilm formation, and antibiotic resistance [
29,
30]. Considering the important role of CcpA in the virulence of
S. aureus, it could be a feasible antimicrobial target. The virulence of
S. aureus may be reduced via chemical inhibition in the interaction of
CcpA with the Cre DNA region [
31]. In the present study, type 1 diabetic mice were treated with
CcpA-knockout
S. aureus strains with different genetic backgrounds. After two weeks, evident damage and edema of the hepatocytes in the diabetic mice infected with the wild-type strain XN108 was observed, along with extensive inflammatory cell infiltration in diabetic mice infected with XN108 Δ
CcpA. The immunohistochemistry results also showed that the expression of inflammatory factors, such as TNF-α and IL-6, decreased significantly in the livers of diabetic mice infected with XN108 Δ
CcpA, suggesting that the infectivity and pathogenicity of the
CcpA-knockout
S. aureus strain XN108 decreased significantly. These results are in accord with previous reports [
15]. Furthermore, we reported for the first time that compared with mice infected with wild-type strains, the type 1 diabetic mice infected with XN108 Δ
CcpA for 2 weeks showed significantly decreased random blood glucose and increased triglyceride and free fatty acid levels, and they also showed obvious metabolic phenotypic changes.
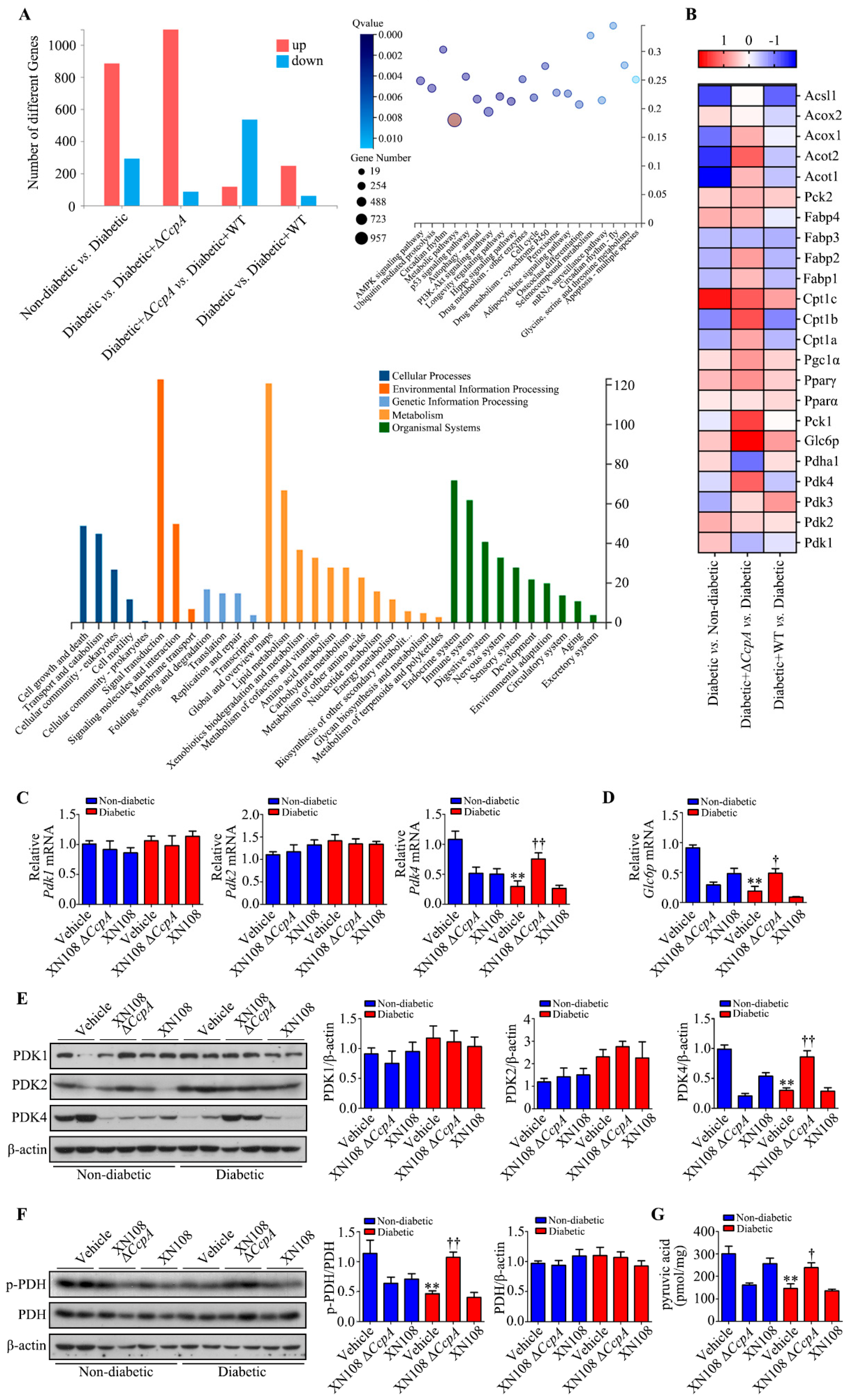
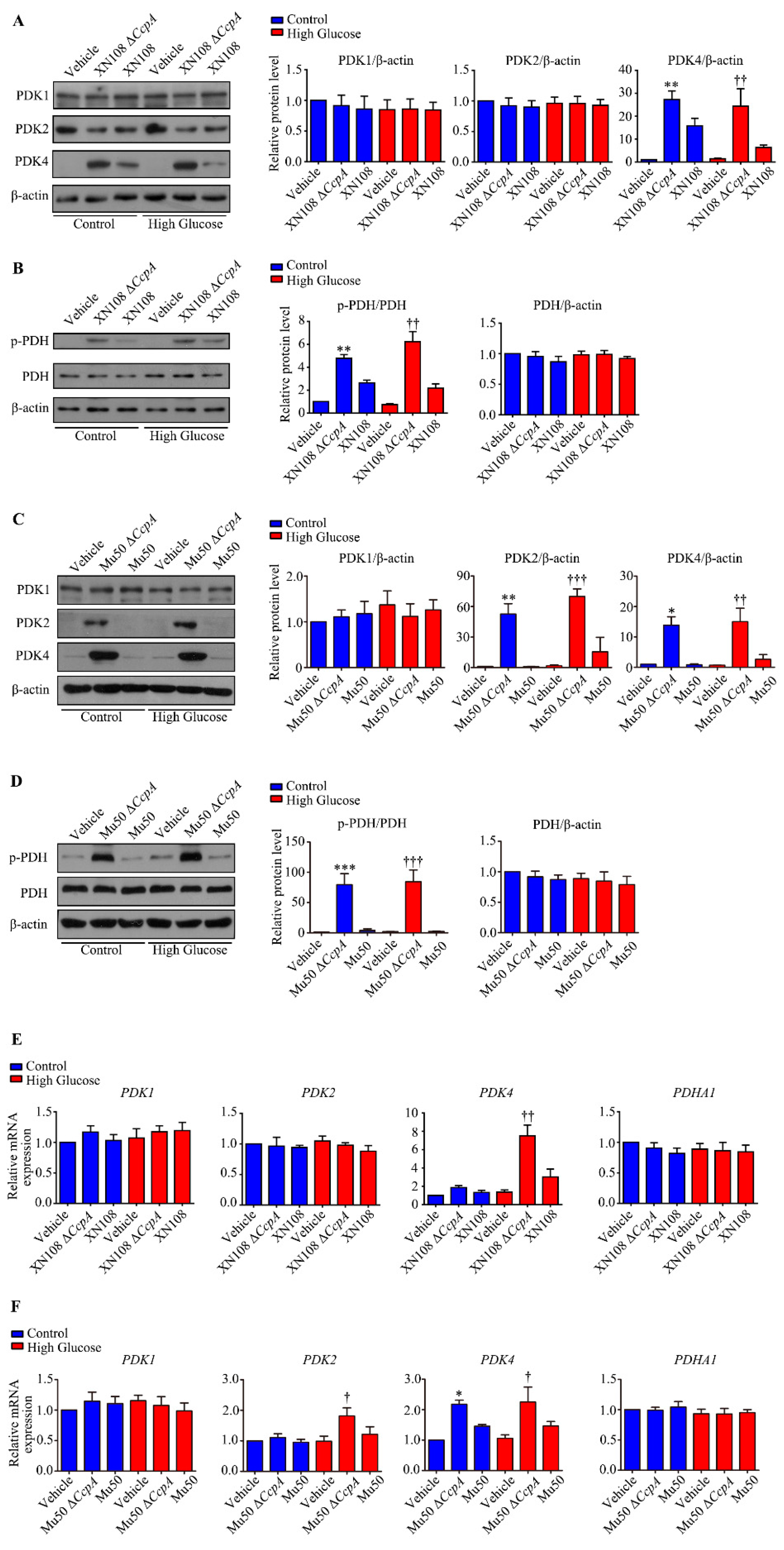
In the present study, RNA was extracted from the liver tissues of each group to investigate the molecular mechanisms behind metabolic phenotypic changes induced by the S. aureus ΔCcpA strain. The RNA-seq technique was used to analyze the differential expression of genes. It was found that, compared with the diabetic group, the diabetic + ΔCcpA group had 1182 differentially expressed genes in the liver, of which 1076 genes were upregulated and 86 genes were downregulated. Our findings suggest that PDK family genes are key target genes for metabolic phenotypic changes. Upon verification in the animal model, we found that infection with the XN108 ΔCcpA strain increased the transcription and translation of PDK4 in the liver of type 1 diabetic mice, increased p-PDH expression, and significantly increased the pyruvate level in the tissues.
PDC is a key regulatory enzyme in the second stage of aerobic glucose oxidation and is also known as the hub of aerobic sugar oxidation [
16]. It catalyzes the irreversible oxidation of pyruvate to acetyl CoA. PDH is the core structure of PDC. The activity of PDC is regulated by PDK and PDP [
17,
32]. In the oxidation process, PDH regulates the decarboxylation of pyruvate to hydroxyethyl-TPP. PDK4 phosphorylates and inactivates PDH [
24]. Thus, PDK4 inhibits the oxidative decarboxylation of pyruvate in the mitochondria [
33]. Consequently, glucose oxidation is inhibited, which leads to glucose utilization disorder. It was interesting to observe that
CcpA-knockout
S. aureus infection upregulated PDK4 transcription and translation in the livers of diabetic mice. As a result, PDH phosphorylation was increased, and this led to PDC inactivation. The inactivation of PDC affects the aerobic oxidation of glucose. The reaction process of the oxidative decarboxylation of pyruvate into acetyl CoA is thus limited and results in the accumulation of pyruvate produced by glycolysis, which is consistent with our experimental results.
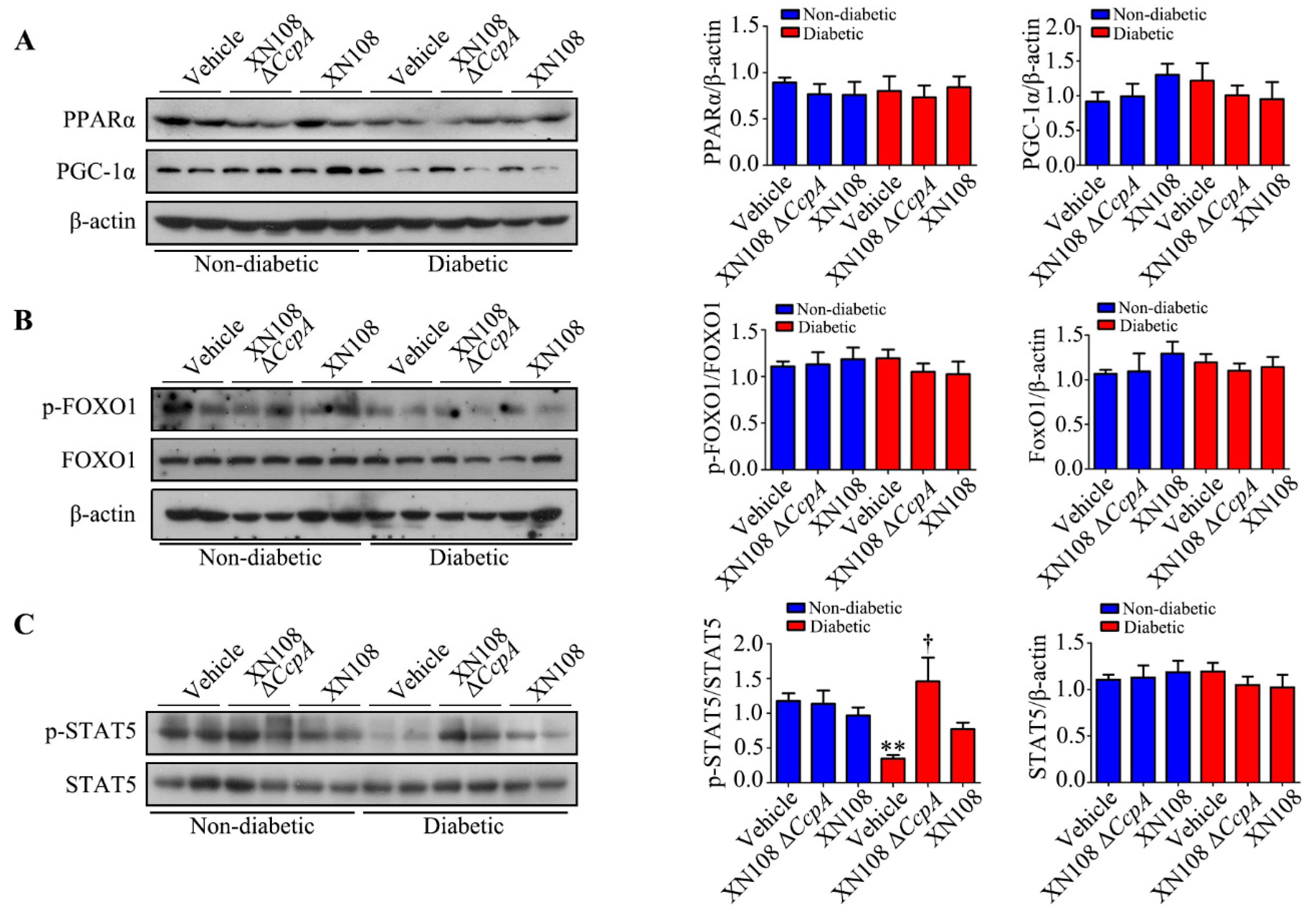
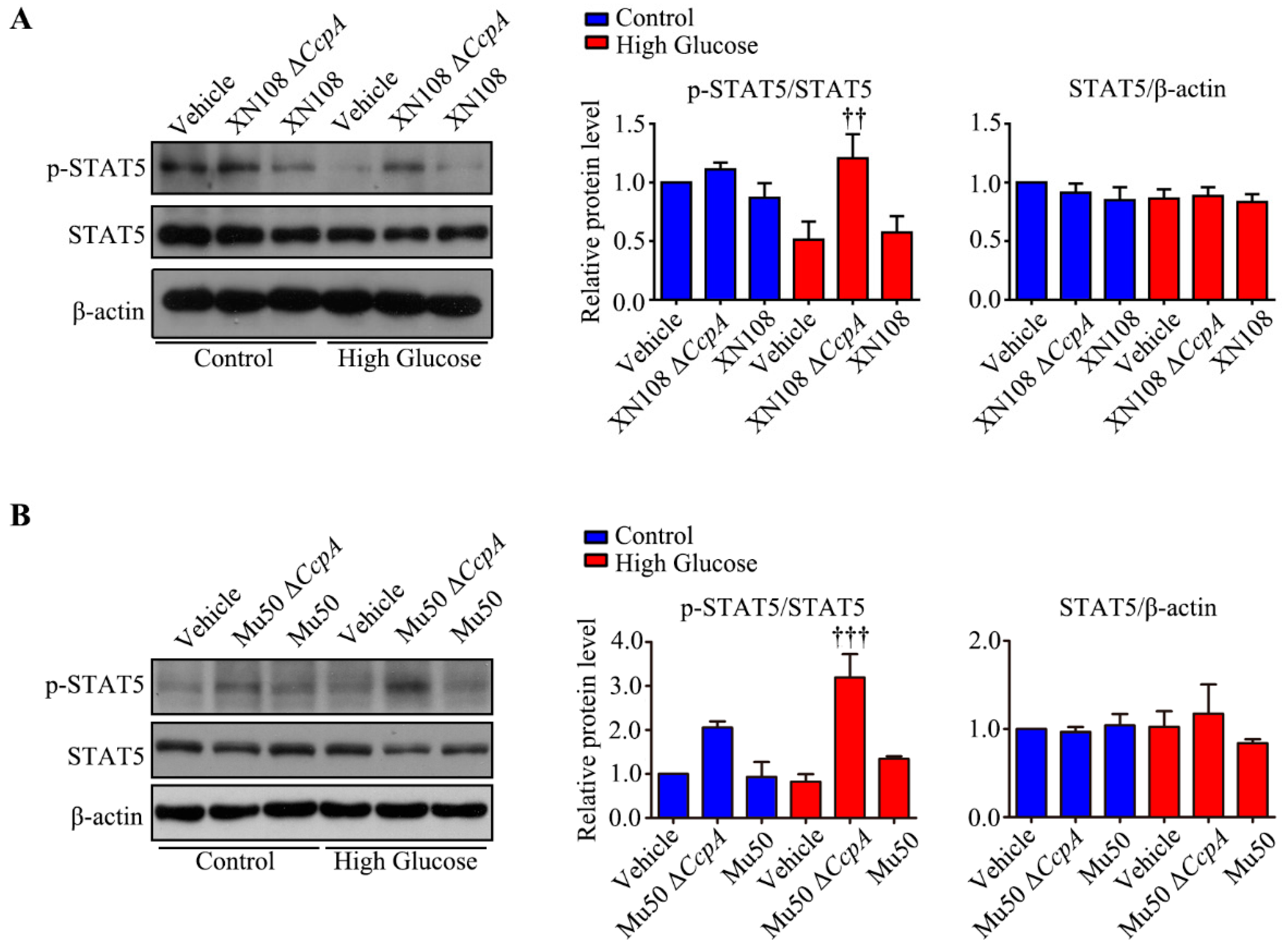
Furthermore, we studied the upstream mechanism of PDK4 activation induced by XN108 Δ
CcpA in the livers of diabetic mice. The expression of PDK4 is mainly regulated by the PGC-1α, FOXO1, and JAK2/STAT5 pathways [
1,
25,
34]. Our results showed that the expression of p-STAT5 was significantly upregulated due to XN108 Δ
CcpA in the livers of diabetic mice. Moreover, the trend of the changes was consistent with that of PDK4, suggesting that XN108 Δ
CcpA upregulated the expression of PDK4 by activating the STAT5 pathway.
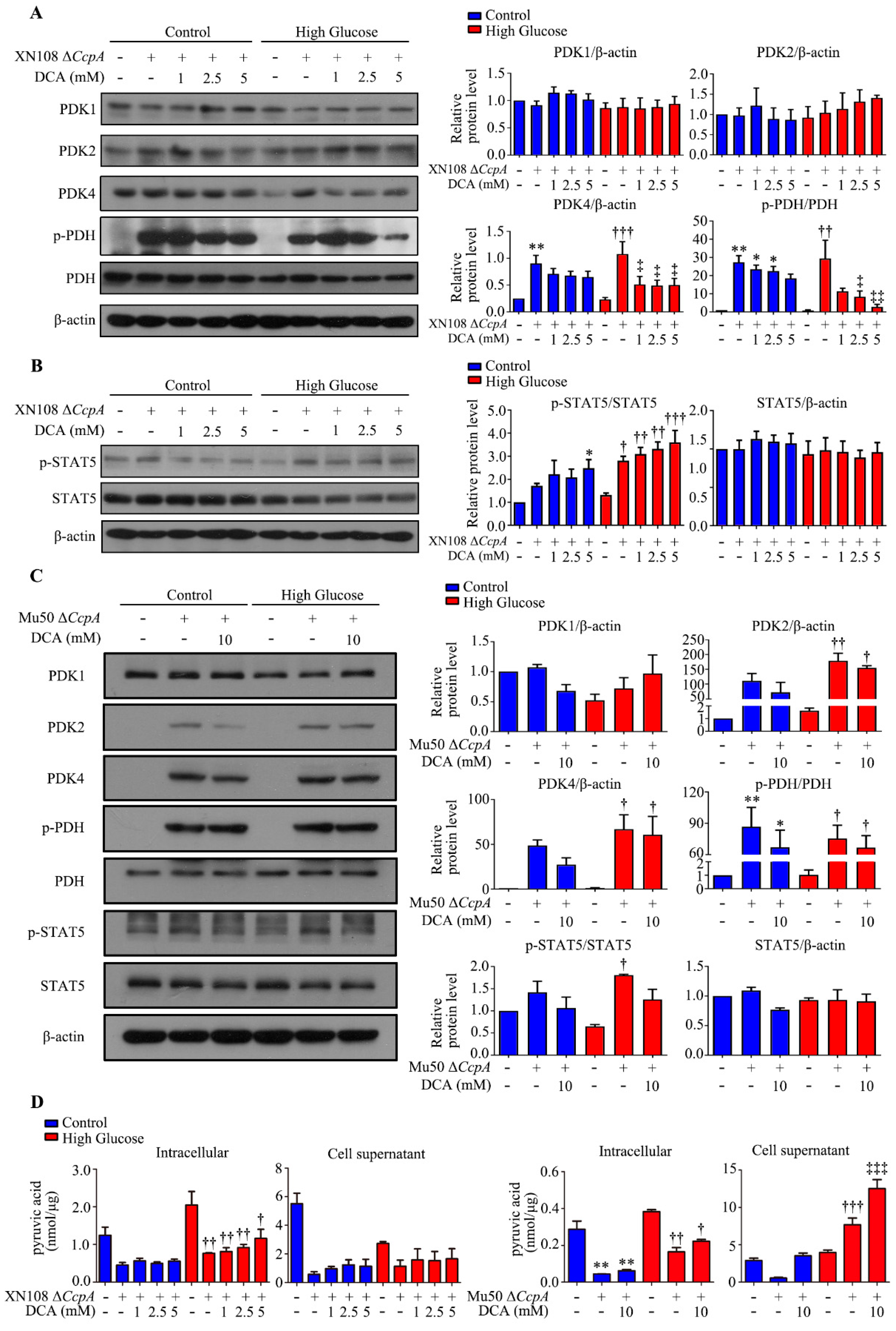
We also used two
S. aureus strains from different sources and different genetic backgrounds (XN108 and Mu50) to infect the human liver cell line WRL68 induced by high glucose and high glucose combined with the PDK4 inhibitor DCA. This helped further clarify the effect and molecular mechanism of Δ
CcpA S. aureus on the metabolic phenotype of diabetes and excluded the influence of the genetic background. The results showed that
S. aureus Δ
CcpA activated the p-STAT5/PDK4 pathway and caused an increase in PDH phosphorylation and the inactivation of PDH. Conversely, DCA significantly inhibited PDK4, and the phosphorylation of PDH also decreased. The results indicated that p-PDH was increased by PDK4. The significant change in triglycerides caused by infection with
CcpA-knockout
S. aureus may be attributed to the accumulation of pyruvate, which promotes the citric acid–pyruvate cycle. Pyruvate can be directly used to synthesize several amino acids. It can also be metabolized into lactic acid or ethyl acetate or converted into acetyl CoA. Acetyl CoA can be metabolized into acetate and introduced into the tricarboxylic acid cycle. It can also be used for the synthesis of fatty acids. Thus, the triglyceride content is increased [
35]. However, the results of the cell experiments showed that the Δ
CcpA strain reduced the concentration of intracellular pyruvate. Moreover, the concentration of pyruvate in the livers of the diabetic + XN108 Δ
CcpA group was higher than that in the livers of the diabetic + vehicle group. The results of these two parts of the experiment were not consistent. The differences could be attributed to the different strains used in the experiment. The mechanism needs to be further explored for a better understanding.
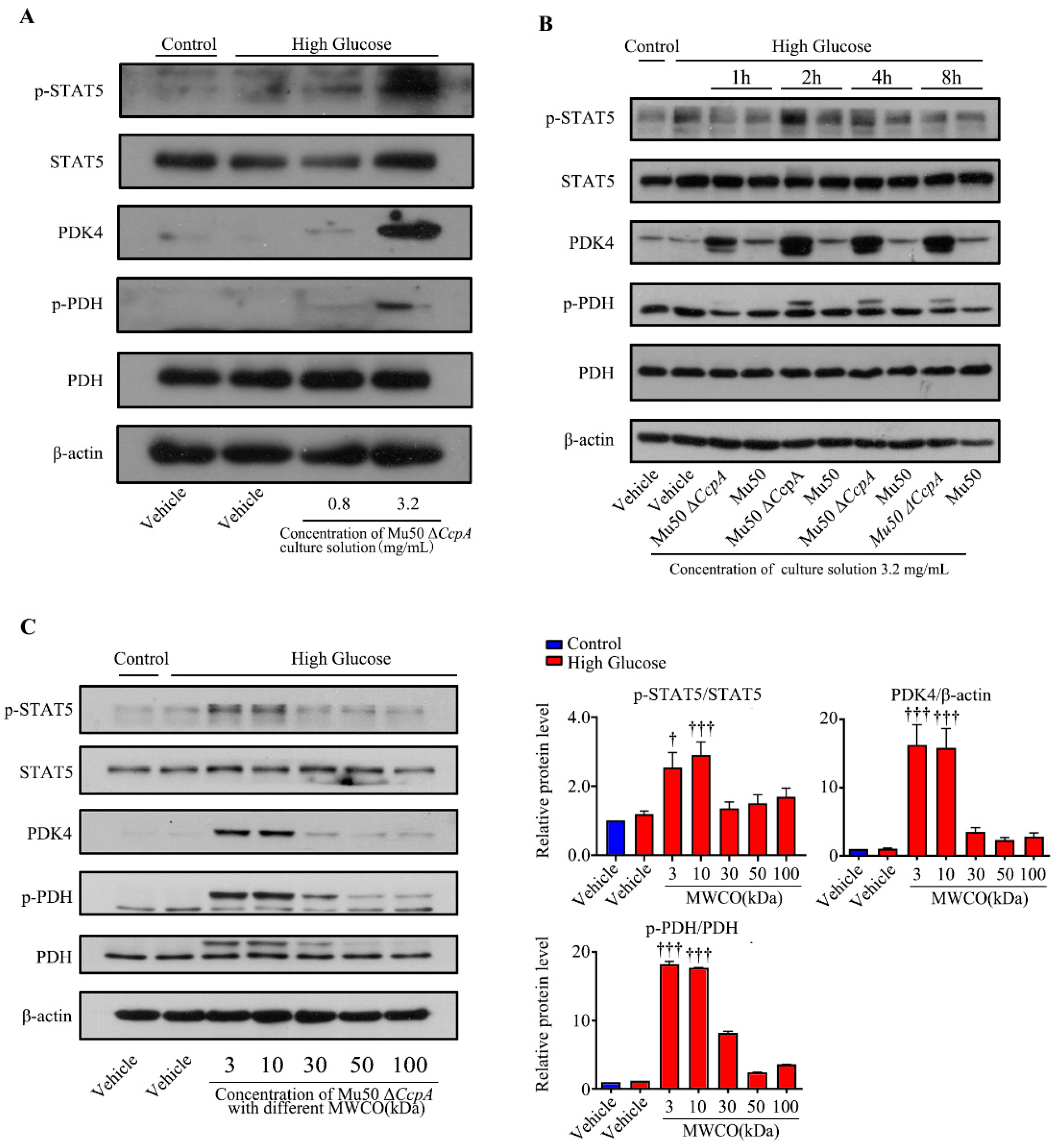
Taken together, the findings of the cell experiments and the animal experiments may verify that
CcpA-knockout
S. aureus infection activates the p-STAT5/PDK4 pathway in the liver. However, how this strain activates p-STAT5 remains unknown. Subsequently, we isolated the cells and culture medium of Mu50 Δ
CcpA. The bacterial culture medium collected was concentrated and utilized to stimulate WRL cells induced by high glucose. We found that the concentrated culture also activated the STAT5/PDK4 pathway in a concentration- and time-dependent manner. Then, the culture medium was concentrated using a centrifuge filtration device with different interception sizes before stimulating the cells. It was found that secretory proteins with masses 3 kDa and 10 kDa effectively increased the expression of PDK4 in hepatocytes under high-glucose conditions. Therefore, we speculated that Mu50 Δ
CcpA may activate the STAT5/PDK4 pathway by releasing secretory proteins with a mass of around 30 kDa. Kim et al. reported that growth hormone significantly induced PDK4 expression in the liver through the JAK2/STAT5 signaling pathway. It was reported that growth hormone-mediated PDK4 expression may induce PDC phosphorylation and inhibit pyruvate utilization in the livers of wild-type mice [
27]. When glucose levels are low, growth hormone may act as a reverse regulatory hormone of insulin and stimulate adipose tissue to release free fatty acids into the blood and activate the liver JAK2/STAT5 signaling pathway to promote the utilization of free fatty acids in the liver and the surrounding tissues [
36]. In this study, we found that the increase in triglyceride and free fatty acid levels in the serum of XN108 Δ
CcpA-infected diabetic mice was similar to that influenced by growth hormone. We thus speculate that the XN108 Δ
CcpA strain may activate the STAT5/PDK4 pathway by producing growth hormone-like proteins. The specific types of secreted proteins need to be further identified using protein spectrum analysis and metabolomics. The experimental results may provide new insights for understanding the molecular mechanism underlying glucose and lipid metabolism disorders in patients with diabetes complicated with
S. aureus infection. The findings of the present study provide vital information for the development of new antimicrobial agents targeting
CcpA and its use in patients with diabetes.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}