
First Impressions Matter: Immune Imprinting and Antibody Cross-Reactivity in Influenza and SARS-CoV-2
 , , , and
, , , and 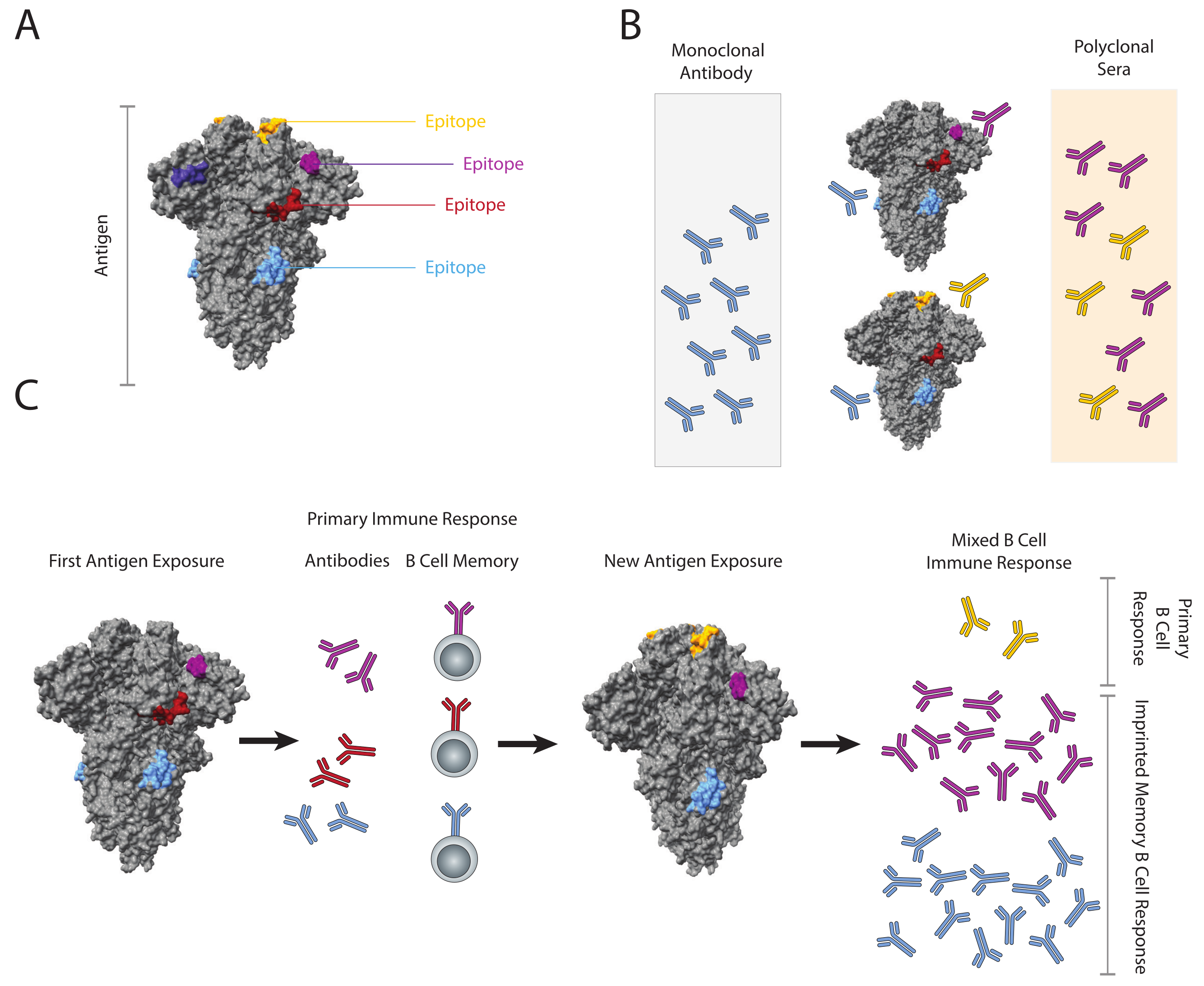{kind=link}
{kind=link}
Abstract
:1. Introduction
2. History of the Concept
3. Immune Imprinting and Influenza

4. The Role of Memory B Cells in Imprinting
5. Measuring Imprinting and Antibody Cross-Reactivity
5.1. Measuring Antibody Cross-Reactivity
5.2. Epitope Mapping Across Variants
5.3. Describing Immunologic Similarity: Antigenic Distance, Cartography, and Landscapes
5.4. Vaccination Strategies and Imprinting
6. Immune Imprinting and SARS-CoV-2
6.1. The Challenge of Emerging SARS-CoV-2 Variants
6.2. Evidence of SARS-CoV-2 Memory B Cell Imprinting
6.3. Antibody Dependent Enhancement (ADE) vs. Imprinting
6.4. Relevance of Imprinting to SARS-CoV-2 Vaccine Development
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ELISA | Enzyme linked immunosorbant assay |
| CoV | Coronavirus |
| HA | Hemagglutinin |
| HAI | Hemagglutinin inhibition assay |
| HCoV | Human coronavirus |
| HLA | Human leukocyte antigen |
| IAV | Influenza A virus |
| Ig | Immunoglobulin |
| MBC | Memory B cell |
| MDA | Multidimensional assay |
| OAS | Original antigenic sin |
| RBD | Receptor binding domain |
| SARS | Severe acute respiratory syndrome |
References
- Dörner, T.; Radbruch, A. Antibodies and B Cell Memory in Viral Immunity. Immunity 2007, 27, 384–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, A.E.; Henry, C. Remembrance of Things Past: Long-Term B Cell Memory After Infection and Vaccination. Front. Immunol. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangster, M.Y.; Nguyen, P.Q.T.; Topham, D.J. Role of Memory B Cells in Hemagglutinin-Specific Antibody Production Following Human Influenza A Virus Infection. Pathogens 2019, 8, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodda, L.B.; Morawski, P.A.; Pruner, K.B.; Fahning, M.L.; Howard, C.A.; Franko, N.; Logue, J.; Eggenberger, J.; Stokes, C.; Golez, I.; et al. Imprinted SARS-CoV-2-specific memory lymphocytes define hybrid immunity. Cell 2022, 185, 1588–1601.e14. [Google Scholar] [CrossRef]
- Muecksch, F.; Wang, Z.; Cho, A.; Gaebler, C.; Ben Tanfous, T.; DaSilva, J.; Bednarski, E.; Ramos, V.; Zong, S.; Johnson, B.; et al. Increased Memory B Cell Potency and Breadth After a SARS-CoV-2 mRNA Boost. Nature 2022, 607, 128–134. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Santra, D.; Maiti, S. Energetics and IC50 based epitope screening in SARS-CoV-2 (COVID-19) spike protein by immunoinformatic analysis implicating for a suitable vaccine development. J. Transl. Med. 2020, 18, 281. [Google Scholar] [CrossRef]
- Kirkpatrick, E.; Qiu, X.; Wilson, P.C.; Bahl, J.; Krammer, F. The influenza virus hemagglutinin head evolves faster than the stalk domain. Sci. Rep. 2018, 8, 10432. [Google Scholar] [CrossRef] [Green Version]
- Hoehn, K.B.; Turner, J.S.; Miller, F.I.; Jiang, R.; Pybus, O.G.; Ellebedy, A.H.; Kleinstein, S.H. Human B cell lineages associated with germinal centers following influenza vaccination are measurably evolving. eLife 2021, 10, e70873. [Google Scholar] [CrossRef]
- Kaku, C.I.; Starr, T.N.; Zhou, P.; Dugan, H.L.; Khalifé, P.; Song, G.; Champney, E.R.; Mielcarz, D.W.; Geoghegan, J.C.; Burton, D.R.; et al. Evolution of antibody immunity following omicron BA.1 breakthrough infection. bioRxiv 2022. Available online: http://xxx.lanl.gov/abs/https://www.biorxiv.org/content/early/2022/09/22/2022.09.21.508922.full.pdf (accessed on 14 December 2022). [CrossRef]
- Li, G.M.; Chiu, C.; Wrammert, J.; McCausland, M.; Andrews, S.F.; Zheng, N.Y.; Lee, J.H.; Huang, M.; Qu, X.; Edupuganti, S.; et al. Pandemic H1N1 influenza vaccine induces a recall response in humans that favors broadly cross-reactive memory B cells. Proc. Natl. Acad. Sci. USA 2012, 109, 9047–9052. [Google Scholar] [CrossRef] [Green Version]
- Francis, T. On the Doctrine of Original Antigenic Sin. Proc. Am. Philos. Soc. 1960, 106, 7. [Google Scholar]
- Lessler, J.; Riley, S.; Read, J.M.; Wang, S.; Zhu, H.; Smith, G.J.; Guan, Y.; Jiang, C.Q.; Cummings, D.A. Evidence for antigenic seniority in influenza A (H3N2) antibody responses in southern China. PLoS Pathog. 2012, 8, e1002802. [Google Scholar] [CrossRef]
- Fonville, J.M.; Wilks, S.H.; James, S.L.; Fox, A.; Ventresca, M.; Aban, M.; Xue, L.; Jones, T.C.; Le, N.M.H.; Pham, Q.T.; et al. Antibody landscapes after influenza virus infection or vaccination. Science 2014, 346, 996–1000. [Google Scholar] [CrossRef] [Green Version]
- Kohler, H. Novel vaccine concept based on back-boost effect in viral infection. Vaccine 2015, 33, 3274–3275. [Google Scholar] [CrossRef]
- Lv, H.; So, R.T.Y.; Teo, Q.W.; Yuan, M.; Liu, H.; Lee, C.C.D.; Yip, G.K.; Ng, W.W.; Wilson, I.A.; Peiris, M.; et al. Neutralizing Antibody Response to Sarbecovirus Is Delayed in Sequential Heterologous Immunization. Viruses 2022, 14, 1382. [Google Scholar] [CrossRef]
- Francis, T.J.; Davenport, F.M.; Hennessy, A.V. A serological recapitulation of human infection with different strains of influenza virus. Trans. Assoc. Am. Physicians 1953, 66, 231–239. [Google Scholar]
- Davenport, F.M.; Hennessy, A.V.; Francis, T.J. Epidemiologic and immunologic significance of age distribution of antibody to antigenic variants of influenza virus. J. Exp. Med. 1953, 98, 641–656. [Google Scholar] [CrossRef]
- Köhler, H.; Müller, S.; Nara, P.L. Deceptive imprinting in the immune response against HIV-1. Immunol. Today 1994, 15, 475–478. [Google Scholar] [CrossRef]
- Abadie, V.; Bonduelle, O.; Duffy, D.; Parizot, C.; Verrier, B.; Combadiere, B. Original encounter with antigen determines antigen-presenting cell imprinting of the quality of the immune response in mice. PLoS ONE 2009, 4, e8159. [Google Scholar] [CrossRef]
- de St. Groth, S.F.; Webster, R.G. Disquisitions On Original Antigenic Sin: II. Proof In Lower Creatures. J. Exp. Med. 1966, 124, 347–361. [Google Scholar] [CrossRef]
- Skarlupka, A.L.; Ross, T.M. Immune Imprinting in the Influenza Ferret Model. Vaccines 2020, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.R.; Von Holle, T.A.; Sutherland, L.L.; Oguin, T.H., III; Sempowski, G.D.; Harrison, S.C.; Moody, M.A. Differential immune imprinting by influenza virus vaccination and infection in nonhuman primates. Proc. Natl. Acad. Sci. USA 2021, 118, e2026752118. [Google Scholar] [CrossRef] [PubMed]
- Monto, A.S.; Malosh, R.E.; Petrie, J.G.; Martin, E.T. The Doctrine of Original Antigenic Sin: Separating Good From Evil. J. Infect. Dis. 2017, 215, 1782–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Dushoff, J.; Earn, D.J. Age-specific mortality risk from pandemic influenza. J. Theor. Biol. 2011, 288, 29–34. [Google Scholar] [CrossRef]
- Morens, D.M.; Burke, D.S.; Halstead, S.B. The Wages of Original Antigenic Sin. Emerg. Infect. Dis. J. 2010, 16, 1023. [Google Scholar] [CrossRef]
- Kucharski, A.J.; Gog, J.R. Age profile of immunity to influenza: Effect of original antigenic sin. Theor. Popul. Biol. 2012, 81, 102–112. [Google Scholar] [CrossRef]
- Knight, M.; Changrob, S.; Li, L.; Wilson, P.C. Imprinting, immunodominance, and other impediments to generating broad influenza immunity. Immunol. Rev. 2020, 296, 191–204. [Google Scholar] [CrossRef]
- Dukor, P.; Dietrich, F.M. The immune response to heterologous red cells in mice. V. The effect of cyclophosphamide and cortisone on antigenic competition. J. Immunol. 1970, 105, 118–125. [Google Scholar] [CrossRef]
- Smith, D.J.; Forrest, S.; Ackley, D.H.; Perelson, A.S. Variable efficacy of repeated annual influenza vaccination. Proc. Natl. Acad. Sci. USA 1999, 96, 14001–14006. [Google Scholar] [CrossRef] [Green Version]
- Mesin, L.; Ersching, J.; Victora, G.D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gostic, K.M.; Ambrose, M.; Worobey, M.; Lloyd-Smith, J.O. Potent protection against H5N1 and H7N9 influenza via childhood hemagglutinin imprinting. Science 2016, 354, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Cobey, S.; Hensley, S.E. Immune history and influenza virus susceptibility. Curr. Opin. Virol. 2017, 22, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggink, D.; Goff, P.H.; Palese, P. Guiding the immune response against influenza virus hemagglutinin toward the conserved stalk domain by hyperglycosylation of the globular head domain. J. Virol. 2014, 88, 699–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chen, S.; Jiang, Y.; Huang, K.; Huang, J.; Yang, D.; Zhu, J.; Zhu, Y.; Shi, S.; Peng, D.; et al. Hemagglutinin glycosylation modulates the pathogenicity and antigenicity of the H5N1 avian influenza virus. Vet. Microbiol. 2015, 175, 244–256. [Google Scholar] [CrossRef]
- Wang, J.; Li, D.; Perry, S.; Hilchey, S.P.; Wiltse, A.; Treanor, J.J.; Sangster, M.Y.; Zand, M.S. Broadly Reactive IgG Responses to Heterologous H5 Prime-Boost Influenza Vaccination Are Shaped by Antigenic Relatedness to Priming Strains. mBio 2021, 12, e0044921. [Google Scholar] [CrossRef]
- Cheung, T.K.; Poon, L.L. Biology of influenza a virus. Ann. N. Y. Acad. Sci. 2007, 1102, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Carreno, J.M.; Strohmeier, S.; Kirkpatrick Roubidoux, E.; Hai, R.; Palese, P.; Krammer, F. H1 Hemagglutinin Priming Provides Long-Lasting Heterosubtypic Immunity against H5N1 Challenge in the Mouse Model. mBio 2020, 11, e02090-20. [Google Scholar] [CrossRef]
- Marchi, S.; Manini, I.; Kistner, O.; Piu, P.; Remarque, E.J.; Manenti, A.; Biuso, F.; Carli, T.; Lazzeri, G.; Montomoli, E.; et al. Serologically-Based Evaluation of Cross-Protection Antibody Responses among Different A(H1N1) Influenza Strains. Vaccines 2020, 8, 656. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The Adaptive Immune System. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; Book Section 24. [Google Scholar]
- Quast, I.; Tarlinton, D. B cell memory: Understanding COVID-19. Immunity 2021, 54, 205–210. [Google Scholar] [CrossRef]
- Andrews, S.F.; Chambers, M.J.; Schramm, C.A.; Plyler, J.; Raab, J.E.; Kanekiyo, M.; Gillespie, R.A.; Ransier, A.; Darko, S.; Hu, J.; et al. Activation Dynamics and Immunoglobulin Evolution of Pre-existing and Newly Generated Human Memory B cell Responses to Influenza Hemagglutinin. Immunity 2019, 51, 398–410.e5. [Google Scholar] [CrossRef]
- Creanga, A.; Gillespie, R.A.; Fisher, B.E.; Andrews, S.F.; Lederhofer, J.; Yap, C.; Hatch, L.; Stephens, T.; Tsybovsky, Y.; Crank, M.C.; et al. A comprehensive influenza reporter virus panel for high-throughput deep profiling of neutralizing antibodies. Nat. Commun. 2021, 12, 1722. [Google Scholar] [CrossRef]
- Doria-Rose, N.A.; Joyce, M.G. Strategies to guide the antibody affinity maturation process. Curr. Opin. Virol. 2015, 11, 137–147. [Google Scholar] [CrossRef] [Green Version]
- King, H.W.; Orban, N.; Riches, J.C.; Clear, A.J.; Warnes, G.; Teichmann, S.A.; James, L.K. Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci. Immunol. 2021, 6, eabe6291. [Google Scholar] [CrossRef]
- Roco, J.A.; Mesin, L.; Binder, S.C.; Nefzger, C.; Gonzalez-Figueroa, P.; Canete, P.F.; Ellyard, J.; Shen, Q.; Robert, P.A.; Cappello, J.; et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 2019, 51, 337–350.e7. [Google Scholar] [CrossRef]
- Bannard, O.; Cyster, J.G. Germinal centers: Programmed for affinity maturation and antibody diversification. Curr. Opin. Immunol. 2017, 45, 21–30. [Google Scholar] [CrossRef]
- Lambert, P.H.; Liu, M.; Siegrist, C.A. Can successful vaccines teach us how to induce efficient protective immune responses? Nat. Med. 2005, 11, S54–S62. [Google Scholar] [CrossRef]
- Winarski, K.L.; Tang, J.; Klenow, L.; Lee, J.; Coyle, E.M.; Manischewitz, J.; Turner, H.L.; Takeda, K.; Ward, A.B.; Golding, H.; et al. Antibody-dependent enhancement of influenza disease promoted by increase in hemagglutinin stem flexibility and virus fusion kinetics. Proc. Natl. Acad. Sci. USA 2019, 116, 15194–15199. [Google Scholar] [CrossRef] [Green Version]
- Yip, M.S.; Leung, H.L.; Li, P.H.; Cheung, C.Y.; Dutry, I.; Li, D.; Daeron, M.; Bruzzone, R.; Peiris, J.S.; Jaume, M. Antibody-dependent enhancement of SARS coronavirus infection and its role in the pathogenesis of SARS. Hong Kong Med. J. 2016, 22, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Tetro, J.A. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect. 2020, 22, 72–73. [Google Scholar] [CrossRef]
- Henry, C.; Palm, A.E.; Krammer, F.; Wilson, P.C. From Original Antigenic Sin to the Universal Influenza Virus Vaccine. Trends Immunol. 2018, 39, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Ogawa, H. Hemagglutination (inhibition) assay. Methods Mol. Biol. 2014, 1200, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Elshal, M.F.; McCoy, J.P. Multiplex bead array assays: Performance evaluation and comparison of sensitivity to ELISA. Methods 2006, 38, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duschl, C.; Hall, E.A. Adsorption and complex formation of immunoglobulins on silicon wafers, studied by interference-enhanced reflectometry. J. Colloid Interface Sci. 1991, 144, 368–380. [Google Scholar] [CrossRef]
- Trombetta, C.M.; Remarque, E.J.; Mortier, D.; Montomoli, E. Comparison of hemagglutination inhibition, single radial hemolysis, virus neutralization assays, and ELISA to detect antibody levels against seasonal influenza viruses. Influenza Other Respir. Viruses 2018, 12, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Watson, D.S.; Reddy, S.M.; Brahmakshatriya, V.; Lupiani, B. A multiplexed immunoassay for detection of antibodies against avian influenza virus. J. Immunol. Methods 2009, 340, 123–131. [Google Scholar] [CrossRef]
- Wang, J.; Wiltse, A.; Zand, M.S. A Complex Dance: Measuring the Multidimensional Worlds of Influenza Virus Evolution and Anti-Influenza Immune Responses. Pathogens 2019, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hilchey, S.P.; Hyrien, O.; Huertas, N.; Perry, S.; Ramanunninair, M.; Bucher, D.; Zand, M.S. Multi-Dimensional Measurement of Antibody-Mediated Heterosubtypic Immunity to Influenza. PLoS ONE 2015, 10, e0129858. [Google Scholar] [CrossRef] [Green Version]
- Price, J.V.; Jarrell, J.A.; Furman, D.; Kattah, N.H.; Newell, E.; Dekker, C.L.; Davis, M.M.; Utz, P.J. Characterization of influenza vaccine immunogenicity using influenza antigen microarrays. PLoS ONE 2013, 8, e64555. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, R.; Supnet, M.; Jasinskas, A.; Jain, A.; Taghavian, O.; Obiero, J.; Milton, D.K.; Chen, W.H.; Grantham, M.; Webby, R.; et al. Protein Microarray Analysis of the Specificity and Cross-Reactivity of Influenza Virus Hemagglutinin-Specific Antibodies. mSphere 2018, 3, e00592-18. [Google Scholar] [CrossRef] [Green Version]
- Nachbagauer, R.; Feser, J.; Naficy, A.; Bernstein, D.I.; Guptill, J.; Walter, E.B.; Berlanda-Scorza, F.; Stadlbauer, D.; Wilson, P.C.; Aydillo, T.; et al. A chimeric hemagglutinin-based universal influenza virus vaccine approach induces broad and long-lasting immunity in a randomized, placebo-controlled phase I trial. Nat. Med. 2021, 27, 106–114. [Google Scholar] [CrossRef]
- Potocnakova, L.; Bhide, M.; Pulzova, L.B. An Introduction to B-Cell Epitope Mapping and In Silico Epitope Prediction. J. Immunol. Res. 2016, 2016, 6760830. [Google Scholar] [CrossRef]
- Hou, J.; Wu, T.; Cao, R.; Cheng, J. Protein tertiary structure modeling driven by deep learning and contact distance prediction in CASP13. Proteins 2019, 87, 1165–1178. [Google Scholar] [CrossRef] [Green Version]
- Tambur, A.R.; Claas, F.H. HLA epitopes as viewed by antibodies: What is it all about? Am. J. Transpl. 2015, 15, 1148–1154. [Google Scholar] [CrossRef]
- Duquesnoy, R.J. HLA matching at the epitope level: The way to go. Clin. Transpl. 2013, 53, 441–451. [Google Scholar]
- Duquesnoy, R.J.; Mostecki, J.; Hariharan, J.; Balazs, I. Structurally based epitope analysis of major histocompatibility complex class I-related chain A (MICA) antibody specificity patterns. Hum. Immunol. 2008, 69, 826–832. [Google Scholar] [CrossRef] [Green Version]
- Duquesnoy, R.J. A structurally based approach to determine HLA compatibility at the humoral immune level. Hum. Immunol. 2006, 67, 847–862. [Google Scholar] [CrossRef] [Green Version]
- Duquesnoy, R.J.; Marrari, M.; Jelenik, L.; Zeevi, A.; Claas, F.H.; Mulder, A. Structural aspects of HLA class I epitopes reacting with human monoclonal antibodies in Ig-binding, C1q-binding and lymphocytotoxicity assays. Hum. Immunol. 2013, 74, 1271–1279. [Google Scholar] [CrossRef]
- Fidler, S.; D’Orsogna, L.; Irish, A.B.; Lewis, J.R.; Wong, G.; Lim, W.H. Correlation and agreement between eplet mismatches calculated using serological, low-intermediate and high resolution molecular human leukocyte antigen typing methods. Oncotarget 2018, 9, 13116–13124. [Google Scholar] [CrossRef] [Green Version]
- Usureau, C.; Jacob, V.; Dubois, V.; Masson, D.; Jollet, I.; Desoutter, J.; Taupin, J.L.; Guillaume, N. HLA graph, a free and ready-to-use bioinformatics tool to explore anti-HLA eplets reactivity pattern. HLA 2022, 100, 244–253. [Google Scholar] [CrossRef]
- Engen, R.M.; Tambur, A.R. Accurate eplet identification is necessary for accurate risk assessment. Am. J. Transpl. 2021, 21, 3504. [Google Scholar] [CrossRef] [PubMed]
- Sapir-Pichhadze, R.; Zhang, X.; Ferradji, A.; Madbouly, A.; Tinckam, K.J.; Gebel, H.M.; Blum, D.; Marrari, M.; Kim, S.J.; Fingerson, S.; et al. Epitopes as characterized by antibody-verified eplet mismatches determine risk of kidney transplant loss. Kidney Int. 2020, 97, 778–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, K.; Subieta, K.C.; Deem, M.W. A novel sequence-based antigenic distance measure for H1N1, with application to vaccine effectiveness and the selection of vaccine strains. Protein Eng. Des. Sel. 2011, 24, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.S.; Sangster, M.Y.; Yang, H.; Mariani, T.J.; Chaudhury, S.; Topham, D.J. Implementing sequence-based antigenic distance calculation into immunological shape space model. BMC Bioinform. 2020, 21, 256. [Google Scholar] [CrossRef]
- Smith, D.J.; Lapedes, A.S.; de Jong, J.C.; Bestebroer, T.M.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. Mapping the antigenic and genetic evolution of influenza virus. Science 2004, 305, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Zhang, T.; Wan, X.F. Antigenic distance measurements for seasonal influenza vaccine selection. Vaccine 2012, 30, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Gostic, K.M.; Bridge, R.; Brady, S.; Viboud, C.; Worobey, M.; Lloyd-Smith, J.O. Childhood immune imprinting to influenza A shapes birth year-specific risk during seasonal H1N1 and H3N2 epidemics. PLoS Pathog. 2019, 15, e1008109. [Google Scholar] [CrossRef] [Green Version]
- Tesini, B.L.; Kanagaiah, P.; Wang, J.; Hahn, M.; Halliley, J.L.; Chaves, F.A.; Nguyen, P.Q.T.; Nogales, A.; DeDiego, M.L.; Anderson, C.S.; et al. Broad Hemagglutinin-Specific Memory B Cell Expansion by Seasonal Influenza Virus Infection Reflects Early-Life Imprinting and Adaptation to the Infecting Virus. J. Virol. 2019, 93, e00169-19. [Google Scholar] [CrossRef] [Green Version]
- Sherman, A.C.; Lai, L.; Bower, M.; Natrajan, M.S.; Huerta, C.; Karmali, V.; Kleinhenz, J.; Xu, Y.; Rouphael, N.; Mulligan, M.J. The Effects of Imprinting and Repeated Seasonal Influenza Vaccination on Adaptive Immunity after Influenza Vaccination. Vaccines 2020, 8, 663. [Google Scholar] [CrossRef]
- Kim, J.H.; Liepkalns, J.; Reber, A.J.; Lu, X.; Music, N.; Jacob, J.; Sambhara, S. Prior infection with influenza virus but not vaccination leaves a long-term immunological imprint that intensifies the protective efficacy of antigenically drifted vaccine strains. Vaccine 2016, 34, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, A.K.; Fox, A.; Tan, H.X.; Juno, J.A.; Davenport, M.P.; Subbarao, K.; Kent, S.J. Immune imprinting and SARS-CoV-2 vaccine design. Trends Immunol. 2021, 42, 956–959. [Google Scholar] [CrossRef]
- Monto, A.S.; Cowling, B.J.; Peiris, J.S.M. Coronaviruses. In Viral Infections of Humans: Epidemiology and Control; Kaslow, R.A., Stanberry, L.R., Le Duc, J.W., Eds.; Springer: Boston, MA, USA, 2014; pp. 199–223. [Google Scholar] [CrossRef] [Green Version]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef]
- Huang, A.T.; Garcia-Carreras, B.; Hitchings, M.D.T.; Yang, B.; Katzelnick, L.C.; Rattigan, S.M.; Borgert, B.A.; Moreno, C.A.; Solomon, B.D.; Trimmer-Smith, L.; et al. A systematic review of antibody mediated immunity to coronaviruses: Kinetics, correlates of protection, and association with severity. Nat. Commun. 2020, 11, 4704. [Google Scholar] [CrossRef]
- Wang, J.; Li, D.; Cameron, A.; Zhou, Q.; Wiltse, A.; Nayak, J.; Pecora, N.D.; Zand, M.S. IgG Against Human beta-Coronavirus Spike Proteins Correlates with SARS-CoV-2 anti-Spike IgG Responses and COVID-19 Disease Severity. J. Infect. Dis. 2022, 226, 474–484. [Google Scholar] [CrossRef]
- Aydillo, T.; Rombauts, A.; Stadlbauer, D.; Aslam, S.; Abelenda-Alonso, G.; Escalera, A.; Amanat, F.; Jiang, K.; Krammer, F.; Carratala, J.; et al. Immunological imprinting of the antibody response in COVID-19 patients. Nat. Commun. 2021, 12, 3781. [Google Scholar] [CrossRef]
- Guo, L.; Wang, Y.; Kang, L.; Hu, Y.; Wang, L.; Zhong, J.; Chen, H.; Ren, L.; Gu, X.; Wang, G.; et al. Cross-reactive antibody against human coronavirus OC43 spike protein correlates with disease severity in COVID-19 patients: A retrospective study. Emerg. Microbes Infect. 2021, 10, 664–676. [Google Scholar] [CrossRef]
- Altmann, D.M.; Reynolds, C.J.; Boyton, R.J. SARS-CoV-2 variants: Subversion of antibody response and predicted impact on T cell recognition. Cell Rep. Med. 2021, 2, 100286. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, D.; Nutalai, R.; Duyvesteyn, H.M.; Tuekprakhon, A.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Mentzer, A.J.; Wang, B.; et al. The antibody response to SARS-CoV-2 Beta underscores the antigenic distance to other variants. Cell Host Microbe 2022, 30, 53–68.e12. [Google Scholar] [CrossRef]
- Roltgen, K.; Nielsen, S.C.A.; Silva, O.; Younes, S.F.; Zaslavsky, M.; Costales, C.; Yang, F.; Wirz, O.F.; Solis, D.; Hoh, R.A.; et al. Immune imprinting, breadth of variant recognition, and germinal center response in human SARS-CoV-2 infection and vaccination. Cell 2022, 185, 1025–1040.e14. [Google Scholar] [CrossRef]
- Alsoussi, W.B.; Malladi, S.K.; Zhou, J.Q.; Liu, Z.; Ying, B.; Kim, W.; Schmitz, A.J.; Lei, T.; Horvath, S.C.; Sturtz, A.J.; et al. SARS-CoV-2 Omicron boosting induces de novo B cell response in humans. bioRxiv 2022. Available online: http://xxx.lanl.gov/abs/https://www.biorxiv.org/content/early/2022/09/22/2022.09.22.509040.full.pdf (accessed on 14 December 2022). [CrossRef]
- McNaughton, A.L.; Paton, R.S.; Edmans, M.; Youngs, J.; Wellens, J.; Phalora, P.; Fyfe, A.; Belij-Rammerstorfer, S.; Bolton, J.S.; Ball, J.; et al. Fatal COVID-19 outcomes are associated with an antibody response targeting epitopes shared with endemic coronaviruses. JCI Insight 2022, 7, e156372. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zand, M.S. The potential for antibody-dependent enhancement of SARS-CoV-2 infection: Translational implications for vaccine development. J. Clin. Transl. Sci. 2021, 5, e2. [Google Scholar] [CrossRef]
- Huisman, W.; Martina, B.E.; Rimmelzwaan, G.F.; Gruters, R.A.; Osterhaus, A.D. Vaccine-induced enhancement of viral infections. Vaccine 2009, 27, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Wheatley, A.K.; Kent, S.J.; DeKosky, B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 2020, 5, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B.; Katzelnick, L. COVID-19 Vaccines: Should We Fear ADE? J. Infect. Dis. 2020, 222, 1946–1950. [Google Scholar] [CrossRef]
- Guzman, M.G.; Vazquez, S. The complexity of antibody-dependent enhancement of dengue virus infection. Viruses 2010, 2, 2649–2662. [Google Scholar] [CrossRef] [Green Version]
- Chareonsirisuthigul, T.; Kalayanarooj, S.; Ubol, S. Dengue virus (DENV) antibody-dependent enhancement of infection upregulates the production of anti-inflammatory cytokines, but suppresses anti-DENV free radical and pro-inflammatory cytokine production, in THP-1 cells. J. Gen. Virol. 2007, 88, 365–375. [Google Scholar] [CrossRef]
- Paul, L.M.; Carlin, E.R.; Jenkins, M.M.; Tan, A.L.; Barcellona, C.M.; Nicholson, C.O.; Michael, S.F.; Isern, S. Dengue virus antibodies enhance Zika virus infection. Clin. Transl. Immunol. 2016, 5, e117. [Google Scholar] [CrossRef]
- Shukla, R.; Ramasamy, V.; Shanmugam, R.K.; Ahuja, R.; Khanna, N. Antibody-Dependent Enhancement: A Challenge for Developing a Safe Dengue Vaccine. Front. Cell Infect. Microbiol. 2020, 10, 572681. [Google Scholar] [CrossRef]
- Bigay, J.; Le Grand, R.; Martinon, F.; Maisonnasse, P. Vaccine-associated enhanced disease in humans and animal models: Lessons and challenges for vaccine development. Front. Microbiol. 2022, 13, 932408. [Google Scholar] [CrossRef]
- Rijkers, G.T.; van Overveld, F.J. The “original antigenic sin” and its relevance for SARS-CoV-2 (COVID-19) vaccination. Clin. Immunol. Commun. 2021, 1, 13–16. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Sun, S.; Tai, W.; Chen, J.; Geng, Q.; He, L.; Chen, Y.; Wu, J.; Shi, Z.; et al. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J. Virol. 2020, 94, e02015-19. [Google Scholar] [CrossRef]
- van der Straten, K.; Guerra, D.; van Gils, M.J.; Bontjer, I.; Caniels, T.G.; van Willigen, H.D.; Wynberg, E.; Poniman, M.; Burger, J.A.; Bouhuijs, J.H.; et al. Mapping the antigenic diversification of SARS-CoV-2. medRxiv 2022. Available online: http://xxx.lanl.gov/abs/https://www.medrxiv.org/content/early/2022/01/03/2022.01.03.21268582.full.pdf (accessed on 1 December 2022). [CrossRef]
- Wu, J.; Nie, J.; Zhang, L.; Song, H.; An, Y.; Liang, Z.; Yang, J.; Ding, R.; Liu, S.; Li, Q.; et al. The antigenicity of SARS-CoV-2 Delta variants aggregated 10 high-frequency mutations in RBD has not changed sufficiently to replace the current vaccine strain. Signal Transduct. Target. Ther. 2022, 7, 18. [Google Scholar] [CrossRef]
- Offit, P.A. Bivalent Covid-19 Vaccines—A Cautionary Tale. N. Engl. J. Med. 2023. Available online: https://www.nejm.org/doi/full/10.1056/NEJMp2215780 (accessed on 12 January 2023). [CrossRef]
- Winokur, P.; Gayed, J.; Fitz-Patrick, D.; Thomas, S.J.; Diya, O.; Lockhart, S.; Xu, X.; Zhang, Y.; Bangad, V.; Schwartz, H.I.; et al. Bivalent Omicron BA.1–Adapted BNT162b2 Booster in Adults Older than 55 Years. N. Engl. J. Med. 2023, 388, 214–227. [Google Scholar] [CrossRef]
- Wang, Q.; Bowen, A.; Valdez, R.; Gherasim, C.; Gordon, A.; Liu, L.; Ho, D.D. Antibody responses to Omicron BA.4/BA.5 bivalent mRNA vaccine booster shot. bioRxiv 2022. Available online: https://www.biorxiv.org/content/10.1101/2022.10.22.513349v1 (accessed on 12 January 2023). [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, S.M.; Bryan, S.P.; Hilchey, S.P.; Wang, J.; Zand, M.S. First Impressions Matter: Immune Imprinting and Antibody Cross-Reactivity in Influenza and SARS-CoV-2. Pathogens 2023, 12, 169. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens12020169
King SM, Bryan SP, Hilchey SP, Wang J, Zand MS. First Impressions Matter: Immune Imprinting and Antibody Cross-Reactivity in Influenza and SARS-CoV-2. Pathogens. 2023; 12(2):169. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens12020169
Chicago/Turabian StyleKing, Samantha M., Shane P. Bryan, Shannon P. Hilchey, Jiong Wang, and Martin S. Zand. 2023. "First Impressions Matter: Immune Imprinting and Antibody Cross-Reactivity in Influenza and SARS-CoV-2" Pathogens 12, no. 2: 169. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens12020169