
Rokubacteria in Northern Peatlands: Habitat Preferences and Diversity Patterns

 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Analysis of Microbial Diversity in Boreal Wetlands
2.2. Peat Sampling for Incubation Studies
2.3. Methane Incubation Experiments
2.4. DNA Extraction and 16S rRNA Gene Sequencing
2.5. Bioinformatic Analyses of Microbial Community Structure
2.6. Correlations between Microbial Groups and Chemical Properties of Peat
2.7. Analyses of the Publicly Available Rokubacterial Metagenomes
3. Results and Discussion
3.1. Microbial Community Composition in Boreal Wetlands
3.2. Diversity and Most Abundant OTUs of Methylomirabilota
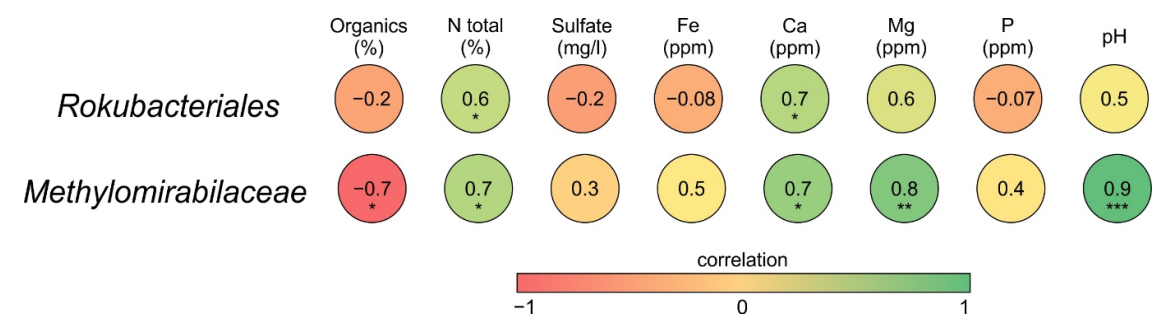
3.3. Correlations between Chemical Properties of Peat and the Abundance of Methylomirabilota
3.4. Microbial Community Response to Incubation with Methane
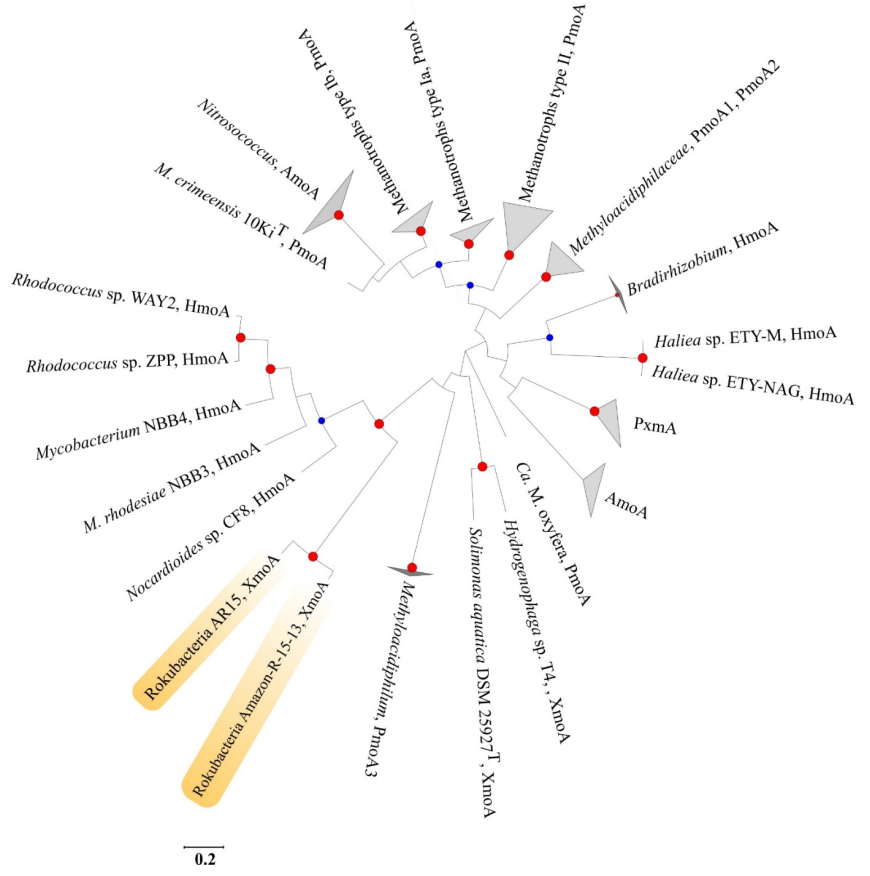
3.5. Search for the Presence of Genomic Determinants of Methanotrophy in Rokubacterial MAGs
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Anantharaman, K.; Brown, C.T.; Hug, L.A.; Sharon, I.; Castelle, C.J.; Probst, A.J.; Thomas, B.C.; Singh, A.; Wilkins, M.J.; Karaoz, U.; et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 2016, 7, 13219. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Thomas, B.C.; Sharon, I.; Brown, C.T.; Sharma, R.; Hettich, R.L.; Wilkins, M.J.; Williams, K.H.; Singh, A.; Banfield, J.F. Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ. Microbiol. 2016, 18, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, M.E.; Delmont, T.O.; Eren, A.M.; Meyer, K.M.; Guo, J.; Khan, K.; Rodrigues, J.L.M.; Bohannan, B.J.M.; Tringe, S.G.; Borges, C.D.; et al. New biological insights into how deforestation in amazonia affects soil microbial communities using metagenomics and metagenome-assembled genomes. Front. Microbiol. 2018, 9, 1635. [Google Scholar] [CrossRef]
- Lipson, D.A.; Schmidt, S.K. Seasonal changes in an alpine soil bacterial community in the Colorado Rocky Mountains. Appl. Environ. Microbiol. 2004, 70, 2867–2879. [Google Scholar] [CrossRef] [Green Version]
- Becraft, E.D.; Woyke, T.; Jarett, J.; Ivanova, N.; Godoy-Vitorino, F.; Poulton, N.; Brown, J.M.; Brown, J.; Lau, M.C.Y.; Onstott, T.; et al. Rokubacteria: Genomic giants among the uncultured bacterial phyla. Front. Microbiol. 2017, 8, 2264. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.-A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2021, gkab776. [Google Scholar] [CrossRef]
- Ettwig, K.F.; Butler, M.K.; Le Paslier, D.; Pelletier, E.; Mangenot, S.; Kuypers, M.M.M.; Schreiber, F.; Dutilh, B.E.; Zedelius, J.; De Beer, D.; et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 2010, 464, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Hansel, C.M.; Fendorf, S.; Jardine, P.M.; Francis, C.A. Changes in bacterial and archaeal community structure and functional diversity along a geochemically variable soil profile. Appl. Environ. Microbiol. 2008, 74, 1620–1633. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Hu, Y.; Yin, Y.; Yang, X.; Xiang, C.; Wang, B.; Chen, Y.; Yang, F.; Lei, F.; Wu, N.; et al. Functional screening of antibiotic resistance genes from human gut microbiota reveals a novel gene fusion. FEMS Microbiol. Lett. 2012, 336, 11–16. [Google Scholar] [CrossRef]
- Figuerola, E.L.M.; Guerrero, L.D.; Türkowsky, D.; Wall, L.G.; Erijman, L. Crop monoculture rather than agriculture reduces the spatial turnover of soil bacterial communities at a regional scale. Environ. Microbiol. 2015, 17, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, V.D.; Torres, T.T.; Ottoboni, L.M.M. Bacterial diversity assessment in soil of an active Brazilian copper mine using high-throughput sequencing of 16S rDNA amplicons. Antonie van Leeuwenhoek 2014, 106, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Green, S.; Tfaily, M.M.; Prakash, O.; Konstantinidis, K.T.; Corbett, J.E.; Chanton, J.P.; Cooper, W.T.; Kostka, J.E. Microbial community structure and activity linked to contrasting biogeochemical gradients in bog and fen environments of the Glacial Lake Agassiz Peatland. Appl. Environ. Microbiol. 2012, 78, 7023–7031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernov, T.I.; Zhelezova, A.D.; Tkhakakhova, A.K.; Bgazhba, N.A.; Zverev, A.O. Microbiomes of virgin soils of Southern Vietnam tropical forests. Microbiology 2019, 88, 489–498. [Google Scholar] [CrossRef]
- Wang, Y.; Li, T.; Li, C.; Song, F. Differences in microbial community and metabolites in litter layer of plantation and original Korean Pine forests in North temperate zone. Microorganisms 2020, 8, 2023. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.D.; Frank, J.; Mitchell, H.; Markillie, L.M.; Wilkins, M.J. Wetland sediments host diverse microbial taxa capable of cycling alcohols. Appl. Environ. Microbiol. 2019, 85, e00189-19. [Google Scholar]
- Ivanova, A.A.; Beletsky, A.V.; Rakitin, A.L.; Kadnikov, V.V.; Philippov, D.A.; Mardanov, A.V.; Ravin, N.V.; Dedysh, S.N. Closely located but totally distinct: Highly contrasting prokaryotic diversity patterns in raised bogs and eutrophic fens. Microorganisms 2020, 8, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anantharaman, K.; Hausmann, B.; Jungbluth, S.P.; Kantor, R.S.; Lavy, A.; Warren, L.A.; Rappé, M.S.; Pester, M.; Loy, A.; Thomas, B.C.; et al. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 2018, 12, 1715–1728. [Google Scholar] [CrossRef] [Green Version]
- Dedysh, S.N.; Beletsky, A.V.; Ivanova, A.A.; Kulichevskaya, I.S.; Suzina, N.E.; Philippov, D.A.; Rakitin, A.L.; Mardanov, A.V.; Ravin, N.V. Wide distribution of Phycisphaera-like planctomycetes from WD2101 soil group in peatlands and genome analysis of the first cultivated representative. Environ. Microbiol. 2020, 23, 1510–1526. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Rime, T.; Phillips, M.; Stierli, B.; Hajdas, I.; Widmer, F.; Hartmann, M. Microbial diversity in European alpine permafrost and active layers. FEMS Microbiol. Ecol. 2016, 92, fiw018. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.; Kuczynski, J.; Stombaugh, J. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Kennedy, D.; Fredrickson, J.; Bjornstad, B.; Konopka, A. Vertical stratification of subsurface microbial community composition across geological formations at the Hanford Site. Environ. Microbiol. 2012, 14, 414–425. [Google Scholar] [CrossRef]
- Butterfield, C.N.; Li, Z.; Andeer, P.F.; Spaulding, S.; Thomas, B.C.; Singh, A.; Hettich, R.L.; Suttle, K.B.; Probst, A.J.; Tringe, S.G.; et al. Proteogenomic analyses indicate bacterial methylotrophy and archaeal heterotrophy are prevalent below the grass root zone. PeerJ 2016, 4, e2687. [Google Scholar] [CrossRef] [Green Version]
- Semrau, J.D.; Chistoserdov, A.; Lebron, J.; Costello, A.; Davagnino, J.; Kenna, E.; Holmes, A.J.; Finch, R.; Murrell, J.C.; Lidstrom, M.E. Particulate methane monooxygenase genes in methanotrophs. J. Bacteriol. 1995, 177, 3071–3079. [Google Scholar] [CrossRef] [Green Version]
- Hakemian, A.S.; Rosenzweig, A.C. The Biochemistry of methane oxidation. Annu. Rev. Biochem. 2007, 76, 223–241. [Google Scholar] [CrossRef]
- Tavormina, P.L.; Orphan, V.J.; Kalyuzhnaya, M.G.; Jetten, M.S.M.; Klotz, M.G. A novel family of functional operons encoding methane/ammonia monooxygenase-related proteins in gammaproteobacterial methanotrophs. Environ. Microbiol. Rep. 2011, 3, 91–100. [Google Scholar] [CrossRef]
- Khadka, R.; Clothier, L.; Wang, L.; Lim, C.K.; Klotz, M.G.; Dunfield, P.F. Evolutionary history of copper membrane monooxygenases. Front. Microbiol. 2018, 9, 2493. [Google Scholar] [CrossRef] [PubMed]
- Dedysh, S.N.; Knief, C. Diversity and phylogeny of described aerobic methanotrophs. In Methane Biocatalysis: Paving the Way to Sustainability; Springer: Cham, Switzerland, 2018; pp. 17–42. [Google Scholar]
- Op den Camp, H.J.M.; Islam, T.; Stott, M.B.; Harhangi, H.R.; Hynes, A.; Schouten, S.; Jetten, M.S.M.; Birkeland, N.K.; Pol, A.; Dunfield, P.F. Environmental, genomic and taxonomic perspectives on methanotrophic Verrucomicrobia. Environ. Microbiol. Rep. 2009, 1, 293–306. [Google Scholar] [CrossRef]
- Sayavedra-Soto, L.A.; Hamamura, N.; Liu, C.W.; Kimbrel, J.A.; Chang, J.H.; Arp, D.J. The membrane-associated monooxygenase in the butane-oxidizing Gram-positive bacterium Nocardioides sp. strain CF8 is a novel member of the AMO/PMO family. Environ. Microbiol. Rep. 2011, 3, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.V.; Le, N.B.; Ly, M.A.; Ogawa, H.E.; McCarl, V.; Wilson, N.L.; Holmes, A.J. Hydrocarbon monooxygenase in Mycobacterium: Recombinant expression of a member of the ammonia monooxygenase superfamily. ISME J. 2012, 6, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, B.; Huang, Y.; Zhang, P.P.; Ding, X.M.; den Camp, H.J.M.O.; Quan, Z.X. Horizontal gene transfer of genes encoding copper-containing membrane-bound monooxygenase (CuMMO) and soluble diiron monooxygenase (SDIMO) in ethane and propane-oxidizing Rhodococcus bacteria. Appl. Environ. Microbiol. 2021, 87, e00227-21. [Google Scholar] [CrossRef] [PubMed]
- Burrows, K.J.; Cornish, A.; Scott, D.; Higgins, I.J. Substrate specificities of the soluble and particulate methane mono-oxygenases of Methylosinus trichosporium OB3b. J. Gen. Microbiol. 1984, 130, 3327–3333. [Google Scholar] [CrossRef] [Green Version]
- Moreno, R.; Rojo, F. Enzymes for aerobic degradation of alkanes in bacteria. In Aerobic Utilization of Hydrocarbons, Oils and Lipids; Rojo, F., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–25. ISBN 978-3-319-39782-5. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mires | Characteristics | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Coordinates | pH | TOC (%) | N Total (%) | Sulfate (mg/L) | Fe (ppm) | Ca (ppm) | Mg (ppm) | P (ppm) | ||
| RAISED BOGS * | 1 | 59°56′56″ N 41°16′59″ E | 4.3 | 88.5 | 0.605 | 172 | 343 | 3522 | 634 | 614 |
| 2 | 60°46′29″ N 36°49′35″ E | 3.7 | 85.1 | 0.923 | 220 | 1347 | 4190 | 682 | 791 | |
| 3 | 59°27′10″ N 40°30′45″ E | 4.3 | 88 | 0.685 | 211 | 662 | 4191 | 905 | 721 | |
| 4 | 59°22′33″ N 39°59′26″ E | 4.1 | 81.5 | 1.16 | 200 | 5306 | 3765 | 816 | 1020 | |
| FENS ** | 1 | 59°56′31″ N 41°15′53″ E | 7.4 | 73.6 | 2.31 | 202 | 9387 | 29,834 | 2575 | 1179 |
| 2 | 60°46′08″ N 36°49′30″ E | 6.9 | 71.6 | 1.65 | 222 | 16,344 | 27,373 | 1078 | 1305 | |
| 3 | 59°47′08″ N 37°52′08″ E | 7.6 | 41.8 | 1.06 | 186 | 106,966 | 32,196 | 1599 | 8920 | |
| 4 | 61°08′18″ N 36°33′27″ E | 6.9 | 83.2 | 2.55 | 230 | 3455 | 15,968 | 2583 | 1049 | |
| 5 | 61°07′16″ N 36°33′21″ E | 6.5 | 48.6 | 1.51 | 607 | 19,264 | 8494 | 2665 | 1192 | |
| 6 | 60°30′42″ N 38°38′59″ E | 7.1 | 66.2 | 2.4 | 188 | 5333 | 31,193 | 2695 | 985 | |
| OTU | FENS (Relative Abundance %) | Taxon | Close Match | Habitat | Similarity (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||||
| 1 | 1.31 | 0.00 | 0.12 | 0.00 | 0.00 | 1.50 | Rokubacteriales | EU135330 | soil grass | 97.0 |
| 2 | 0.54 | 0.00 | 0.02 | 0.00 | 0.00 | 0.22 | Rokubacteriales | JF265688 | mat lava tube | 93.5 |
| 3 | 0.35 | 0.26 | 0.03 | 0.00 | 0.00 | 1.01 | Rokubacteriales | DQ067010 | sediment lake | 98.8 |
| 4 | 0.24 | 0.00 | 0.00 | 0.00 | 0.00 | 0.22 | Rokubacteriales | JX025748 | polluted soil | 99.8 |
| 5 | 0.22 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | Rokubacteriales | EU135329 | soil grass | 96.5 |
| 6 | 0.17 | 0.00 | 0.00 | 0.00 | 0.00 | 0.07 | Rokubacteriales | EU135345 | soil grass | 92.8 |
| 7 | 0.14 | 0.00 | 0.13 | 0.00 | 0.00 | 0.31 | Rokubacteriales | JF266550 | mat lava tube | 99.3 |
| 8 | 0.12 | 0.04 | 0.25 | 0.25 | 0.23 | 0.17 | Methylomirabilaceae | FQ658910 | polluted soil | 96.8 |
| 9 | 0.09 | 0.02 | 0.00 | 0.00 | 0.00 | 0.03 | Rokubacteriales_WX65 | EU135337 | soil grass | 86.8 |
| 10 | 0.07 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | Rokubacteriales | KM205483 | rice rhizosphere | 97.5 |
| 11 | 0.00 | 0.11 | 0.02 | 0.00 | 0.02 | 0.00 | Methylomirabilaceae | AB426228 | anaerobic field soil | 95.5 |
| 12 | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 | 0.12 | Rokubacteriales | JF265688 | mat lava tube | 92.0 |
| 13 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 | 0.11 | Methylomirabilaceae | KX504792 | sediment lake | 96.8 |
| 14 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.07 | Rokubacteriales | EU135331 | soil grass | 96.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, A.A.; Oshkin, I.Y.; Danilova, O.V.; Philippov, D.A.; Ravin, N.V.; Dedysh, S.N. Rokubacteria in Northern Peatlands: Habitat Preferences and Diversity Patterns. Microorganisms 2022, 10, 11. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10010011
Ivanova AA, Oshkin IY, Danilova OV, Philippov DA, Ravin NV, Dedysh SN. Rokubacteria in Northern Peatlands: Habitat Preferences and Diversity Patterns. Microorganisms. 2022; 10(1):11. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10010011
Chicago/Turabian StyleIvanova, Anastasia A., Igor Y. Oshkin, Olga V. Danilova, Dmitriy A. Philippov, Nikolai V. Ravin, and Svetlana N. Dedysh. 2022. "Rokubacteria in Northern Peatlands: Habitat Preferences and Diversity Patterns" Microorganisms 10, no. 1: 11. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10010011