
Selective Isolation of Eggerthella lenta from Human Faeces and Characterisation of the Species Prophage Diversity

 ,
,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Growth Requirements
2.2. Antibiotic Resistance Screening
2.3. E. lenta Isolation from Human Faecal Samples
2.4. Bacterial Genomic DNA Extraction and Genome Sequencing
2.5. Bioinformatic Analysis of Bacterial Genomes
2.6. Prophage Features Identification, Phylogenetic and Protein Functional Analysis
2.7. Demonstration of DGR Functionality of Prophage DSM2243phi4
2.8. Detection of Circularised Prophage Genomes among Host Strain Cells
2.9. Detection of Virions in the Supernatant of Strain DSM2243
2.10. Data Processing and Visualisation
2.11. DDBJ/ENA/GenBank Submission Details
3. Results
3.1. Isolation of E. lenta from Human Faecal Samples
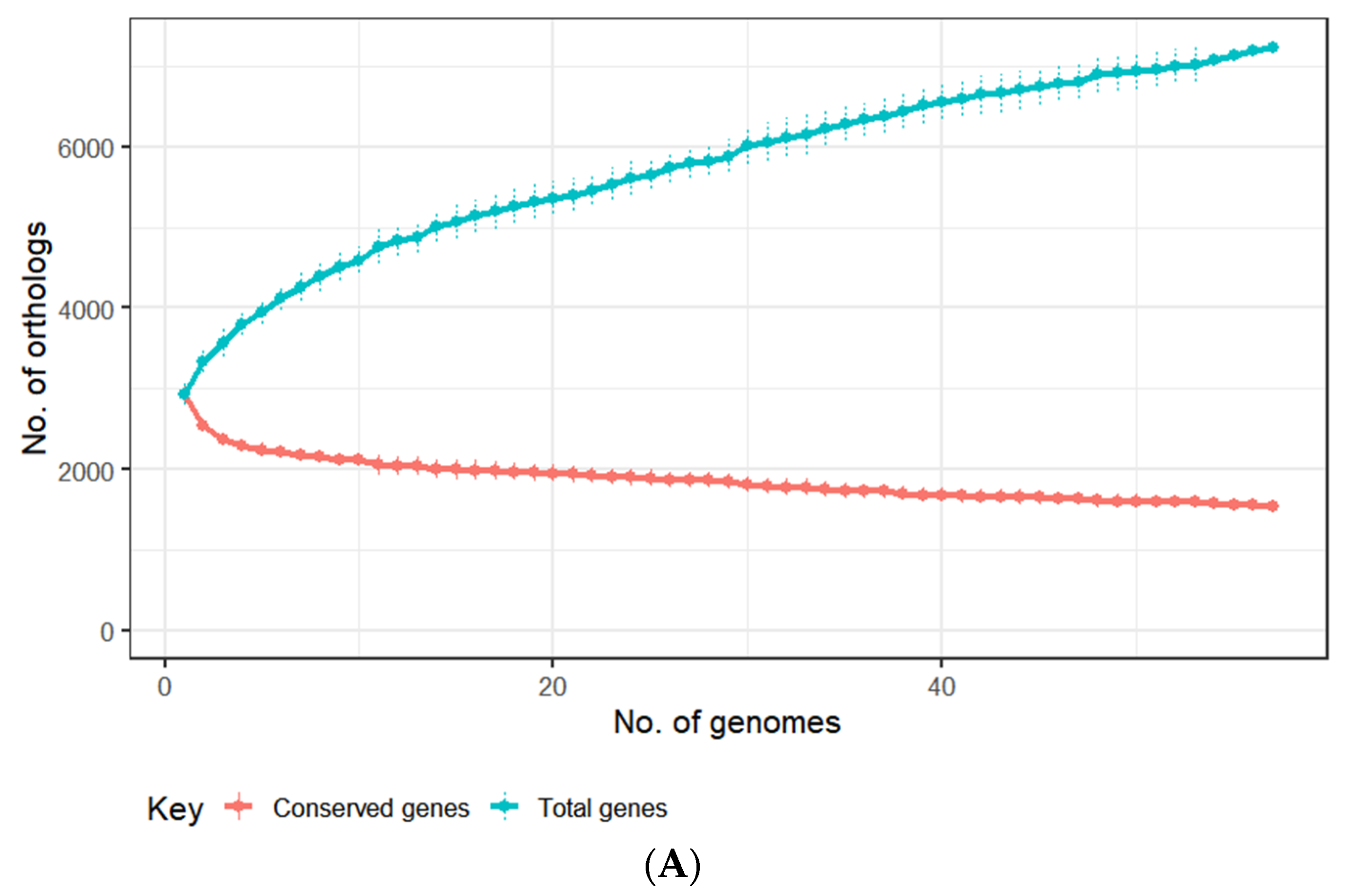
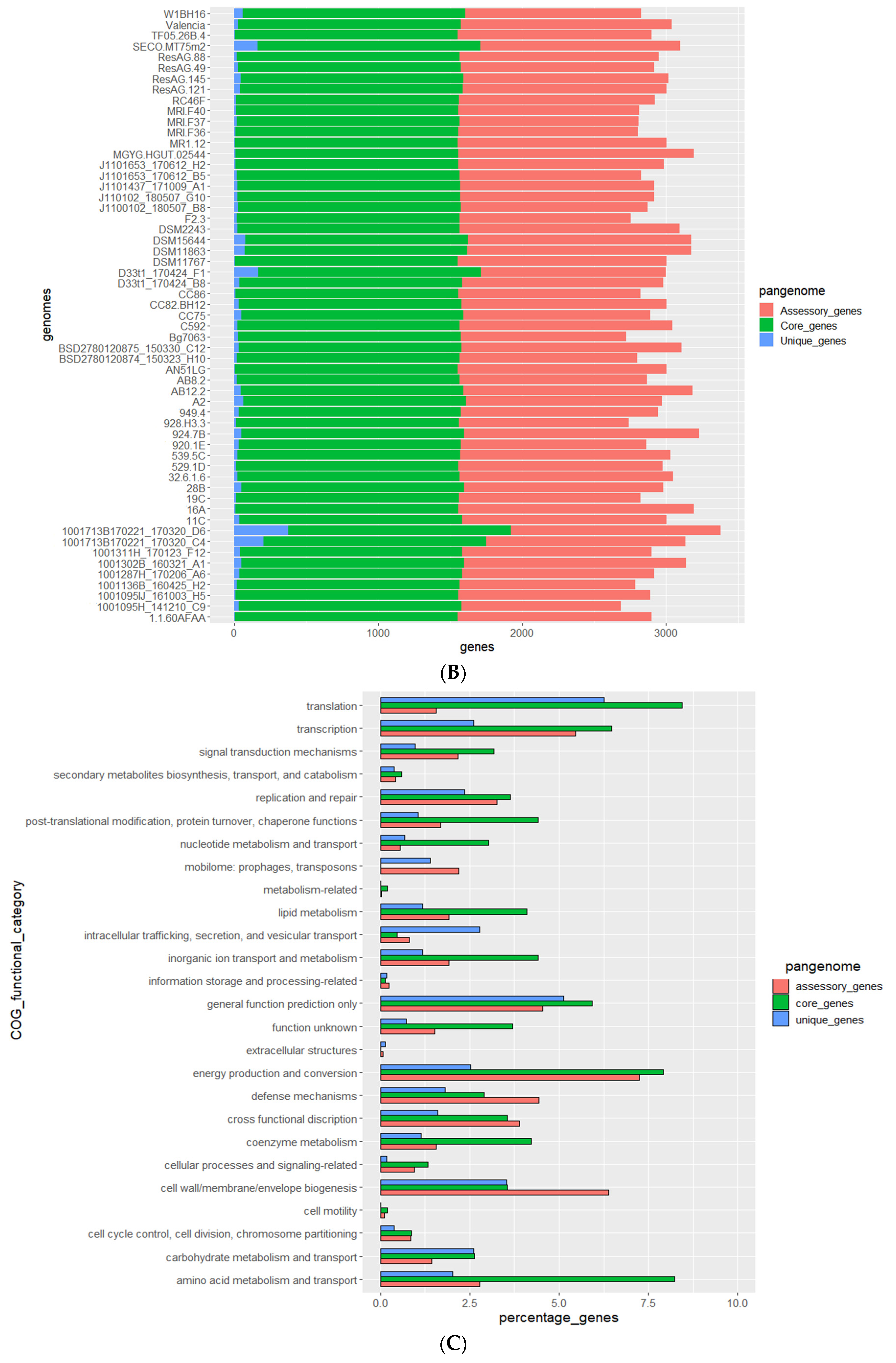
3.2. Genomes of E. lenta Isolates
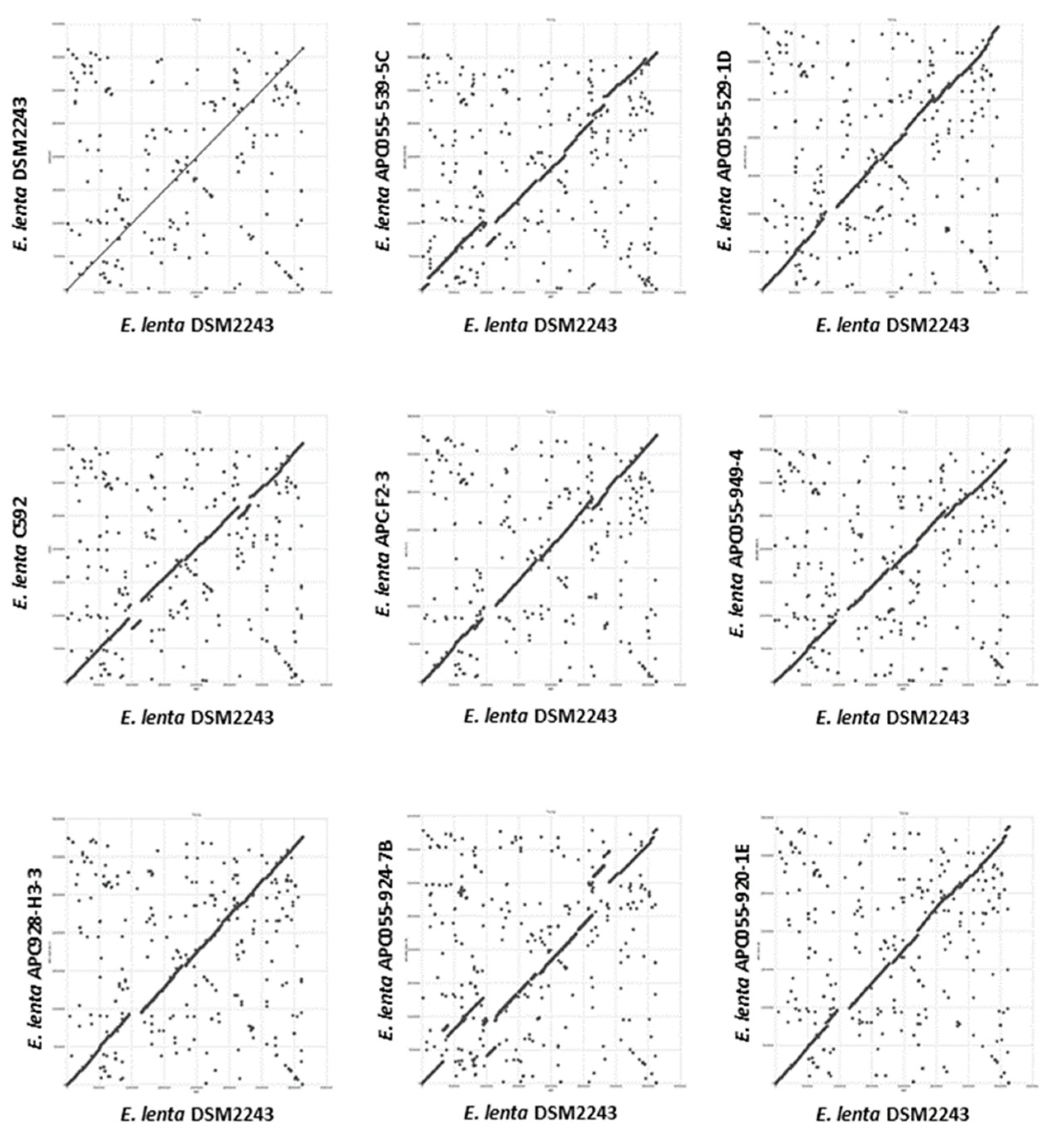
3.3. Comparative Analysis of E. lenta Isolates
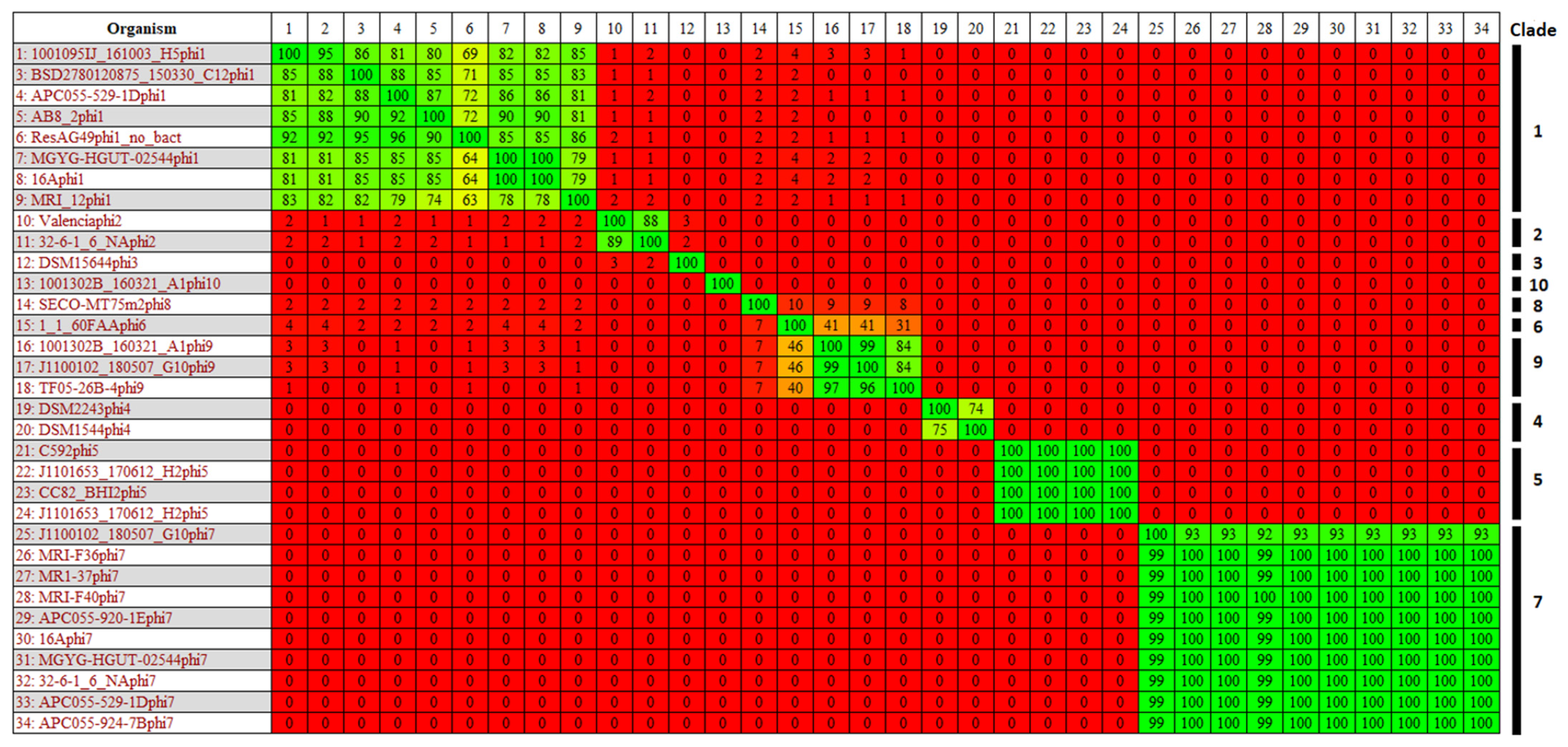
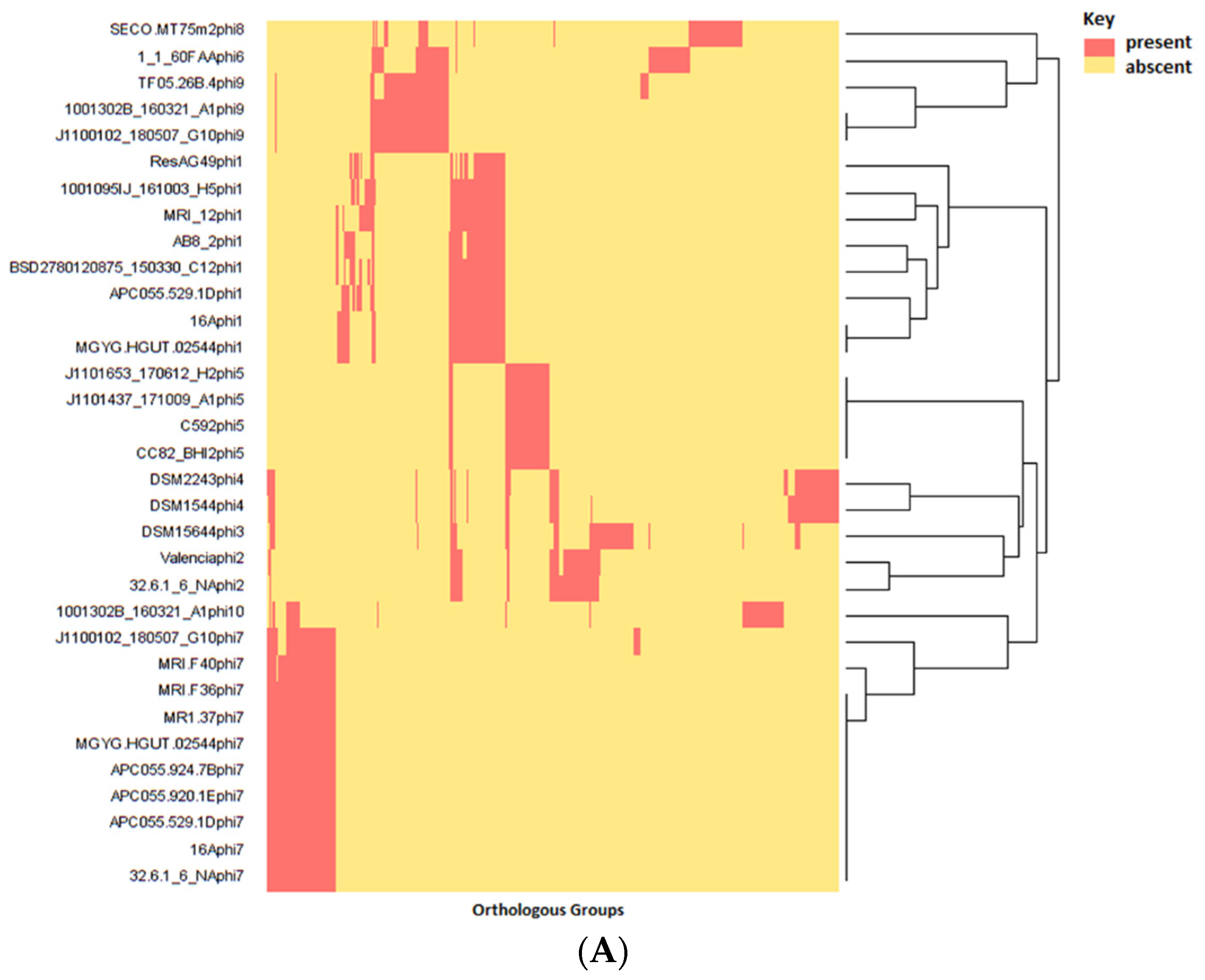
3.4. Identification and Diversity of Prophages
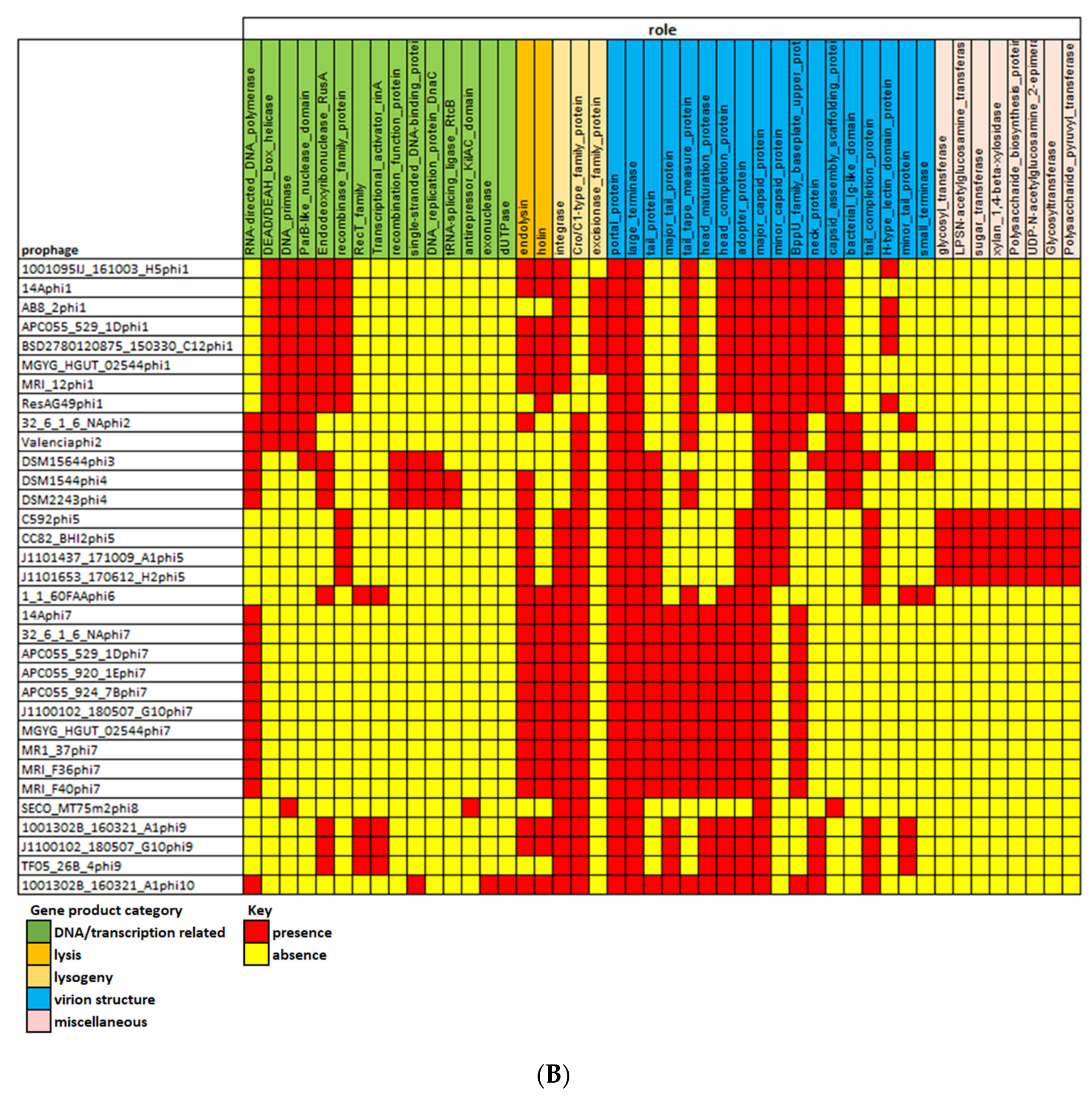
3.5. Gene Content of Prophages and Possible Impact on Host Infection
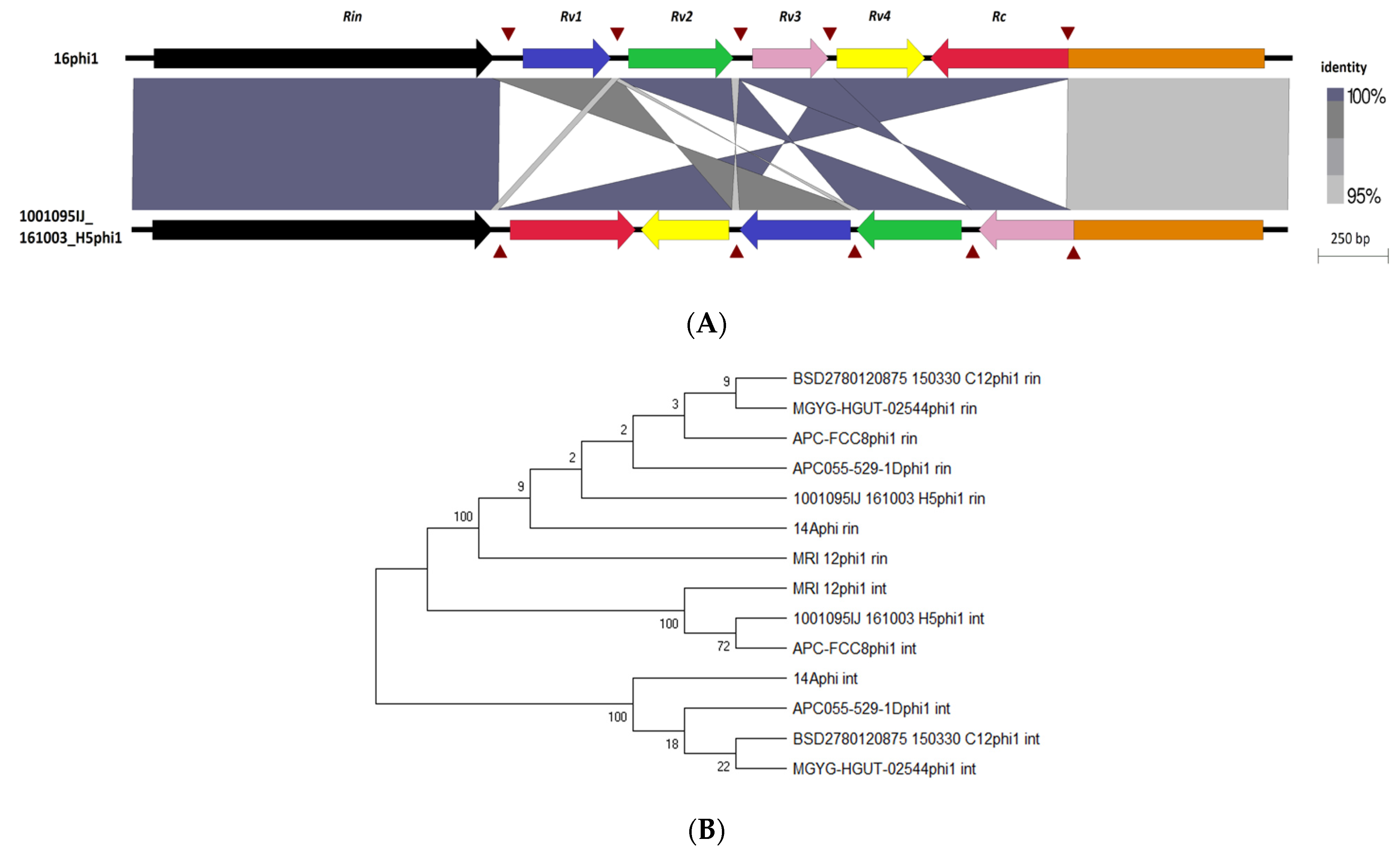
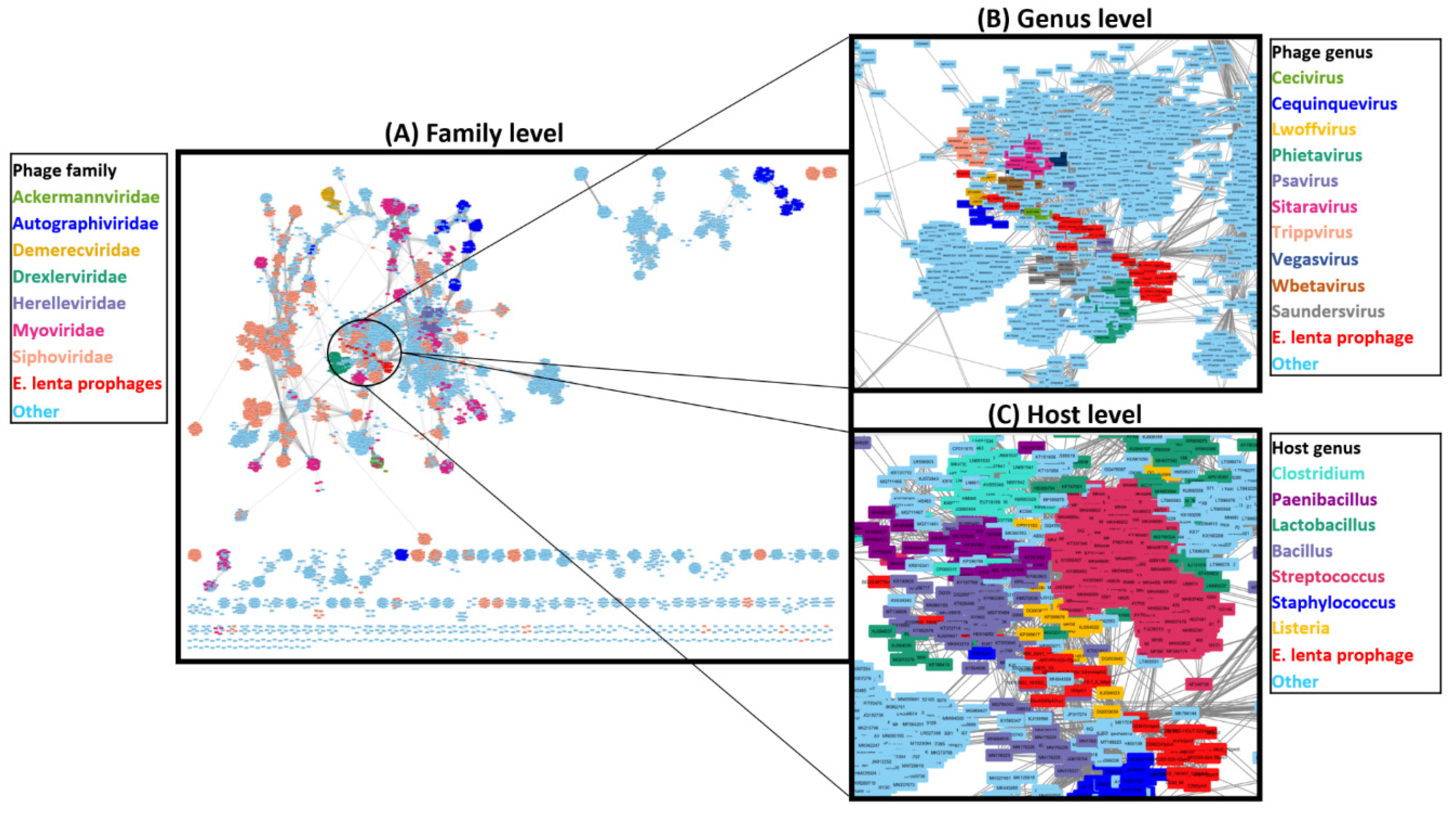
3.6. Taxonomic Placement of Prophages
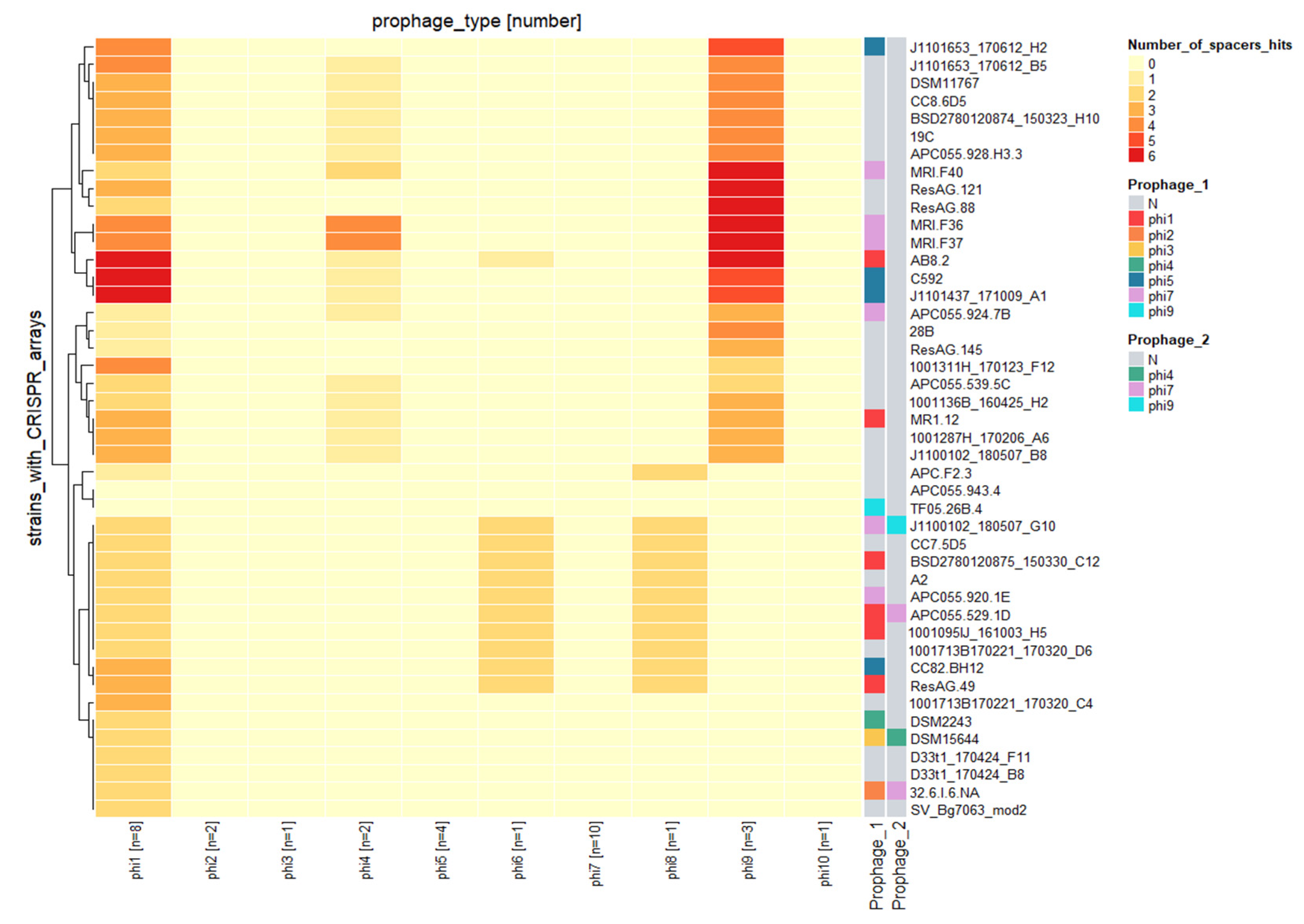
3.7. Prophages and the CRISPR/cas System
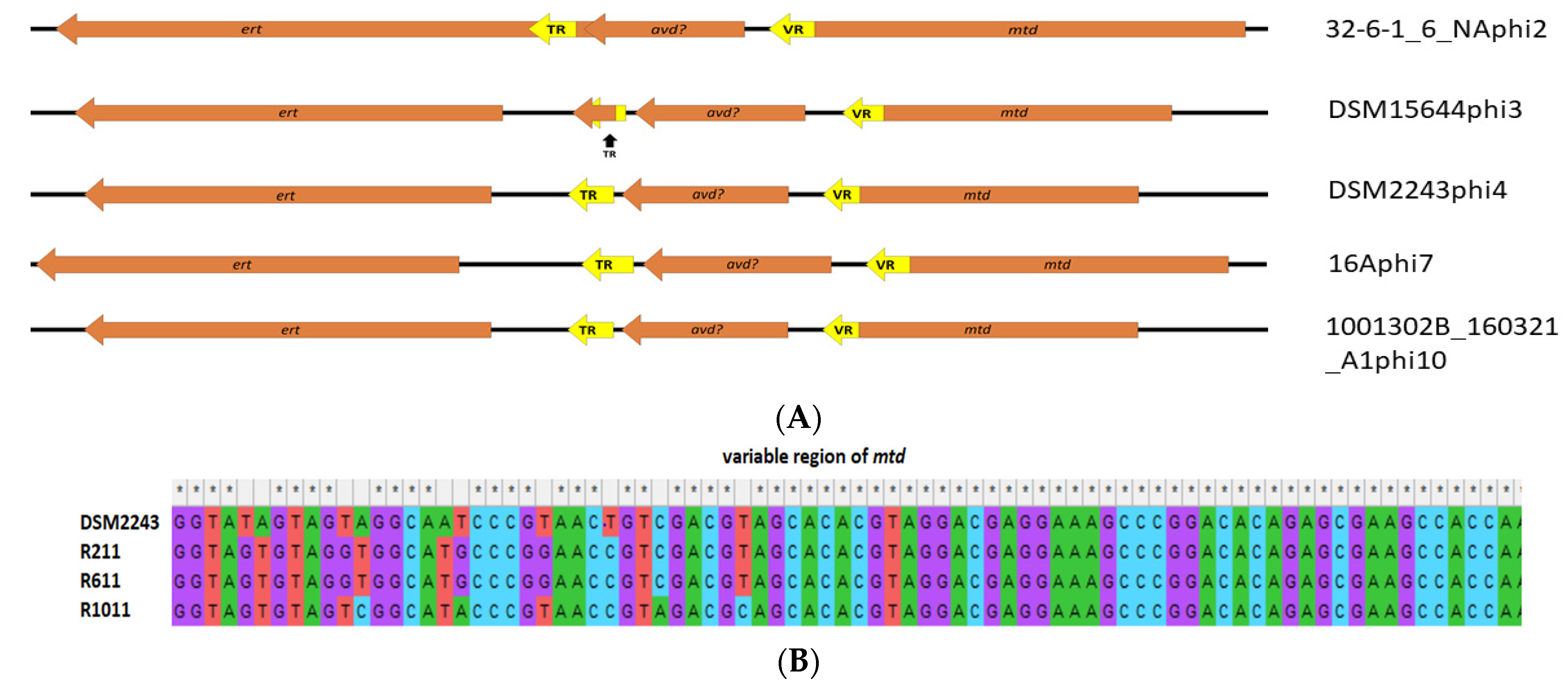
3.8. Evidence That Prophages Are Functional
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppel, N.; Rekdal, V.M.; Balskus, E.P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356, 1246–1257. [Google Scholar] [CrossRef] [PubMed]
- Spanogiannopoulos, P.; Bess, E.N.; Carmody, R.N.; Turnbaugh, P.J. The microbial pharmacists within us: A metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 2016, 14, 273–287. [Google Scholar] [CrossRef]
- Haiser, H.J.; Turnbaugh, P.J. Developing a metagenomic view of xenobiotic metabolism. Pharmacol. Res. 2013, 69, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Wade, W.G.; Downes, J.; Dymock, D.; Hiom, S.J.; Weightman, A.J.; Dewhirst, F.E.; Paster, B.J.; Tzellas, N.; Coleman, B. The family Coriobacteriaceae: Reclassification of Eubacterium exiguum (Poco et al, 1996) and Peptostreptococcus heliotrinreducens (Lanigan 1976) as Slackia exigua gen. nov., comb. nov. and Slackia heliotrinireducens gen. nov., comb. nov., and Eubacterium. Int. J. Syst. Bacteriol. 1999, 49, 595–600. [Google Scholar] [CrossRef]
- Gupta, R.S.; Chen, W.J.; Adeolu, M.; Chai, Y. Molecular signatures for the class Coriobacteriia and its different clades; proposal for division of the class Coriobacteriia into the emended order Coriobacteriales, containing the emended family Coriobacteriaceae and Atopobiaceae fam. nov., and Eggerthe. Int. J. Syst. Evol. Microbiol. 2013, 63, 3379–3397. [Google Scholar] [CrossRef] [Green Version]
- Sousa, T.; Paterson, R.; Moore, V.; Carlsson, A.; Abrahamsson, B.; Basit, A.W. The gastrointestinal microbiota as a site for the biotransformation of drugs. Int. J. Pharm. 2008, 363, 1–25. [Google Scholar] [CrossRef]
- Kageyama, A.; Benno, Y.; Nakase, T. Phylogenetic evidence for the transfer of Eubacterium lentum to the genus Eggerthella as Eggerthella lenta gen. nov., comb. nov. Int. J. Syst. Bacteriol. 1999, 49, 1725–1732. [Google Scholar] [CrossRef] [Green Version]
- Koppel, N.; Bisanz, J.E.; Pandelia, M.E.; Turnbaugh, P.J.; Balskus, E.P. Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. Elife 2018, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Haiser, H.J.; Gootenberg, D.B.; Chatman, K.; Sirasani, G.; Balskus, E.P.; Turnbaugh, P.J. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta + NIH Public Access. Science 2013, 341, 295–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenbaum, J.; Rund, D.G.; Butler, V.P.; Tse-Eng, D.; Saha, J.R. Inactivation of Digoxin by the Gut Flora: Reversal by Antibiotic Therapy. N. Engl. J. Med. 1981, 305, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Rekdal, V.M.; Bess, E.N.; Bisanz, J.E.; Turnbaugh, P.J.; Balskus, E.P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 2019, 364, 6445. [Google Scholar]
- Jung, C.M.; Heinze, T.M.; Schnackenberg, L.K.; Mullis, L.B.; Elkins, S.A.; Elkins, C.A.; Steele, R.S.; Sutherland, J.B. Interaction of dietary resveratrol with animal-associated bacteria. FEMS Microbiol. Lett. 2009, 297, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Bess, E.N.; Bisanz, J.E.; Yarza, F.; Bustion, A.; Rich, B.E.; Li, X.; Kitamura, S.; Waligurski, E.; Ang, Q.Y.; Alba, D.L.; et al. Genetic basis for the cooperative bioactivation of plant lignans by Eggerthella lenta and other human gut bacteria. Nat. Microbiol. 2020, 5, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Elias, R.M.; Khoo, S.Y.; Pupaibool, J.; Nienaber, J.H.; Cummins, N.W. Multiple pyogenic liver abscesses caused by Eggerthella lenta treated with ertapenem: A case report. Case Rep. Med. 2012, 2012, 718130. [Google Scholar] [CrossRef]
- Gardiner, B.J.; Tai, A.Y.; Kotsanas, D.; Francis, M.J.; Roberts, S.A.; Ballard, S.A.; Junckerstorff, R.K.; Kormana, T.M. Clinical and microbiological characteristics of Eggerthella lenta bacteremia. J. Clin. Microbiol. 2015, 53, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopal, A.A.; Szpunar, S.; Johnson, L.B. Risk and prognostic factors among patients with bacteremia due to Eggerthella lenta. Anaerobe 2012, 18, 475–478. [Google Scholar] [CrossRef]
- Hsu, B.B.; Gibson, T.E.; Yeliseyev, V.; Liu, Q.; Lyon, L.; Bry, L.; Silver, P.A.; Gerber, G.K. Dynamic Modulation of the Gut Microbiota and Metabolome by Bacteriophages in a Mouse Model. Cell Host Microbe 2019, 25, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Manrique, P.; Dills, M.; Young, M.J. The Human Gut Phage Community and Its Implications for Health and Disease. Viruses 2017, 9, 141. [Google Scholar] [CrossRef] [Green Version]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumby, N.; Davidson, A.R.; Maxwell, K.L. The moron comes of age. Bacteriophage 2012, 2, e23146. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Denomy, J.; Davidson, A.R. When a Virus is not a Parasite: The Beneficial Effects of Prophages on Bacterial Fitness. J. Microbiol. 2014, 52, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Canchaya, C.; Fournous, G.; Chibani-Chennoufi, S.; Dillmann, M.L.; Brüssow, H. Phage as agents of lateral gene transfer. Curr. Opin. Microbiol. 2003, 6, 417–424. [Google Scholar] [CrossRef]
- Soto-Perez, P.; Bisanz, J.E.; Berry, J.D.; Lam, K.N.; Bondy-Denomy, J.; Turnbaugh, P.J. CRISPR-Cas System of a Prevalent Human Gut Bacterium Reveals Hyper-targeting against Phages in a Human Virome Catalog. Cell Host Microbe 2019, 26, 325–335.e5. [Google Scholar] [CrossRef]
- Bisanz, J.E.; Soto-Perez, P.; Lam, K.N.; Bess, E.N.; Haiser, H.J.; Allen-Vercoe, E.; Rekdal, V.M.; Balskus, E.P.; Turnbaugh, P.J. Illuminating the microbiome’s dark matter: A functional genomic toolkit for the study of human gut Actinobacteria. BioRxiv 2018, 11, 304840. [Google Scholar]
- Bolger, A.M.M.; Lohse, M.; Usadel, B. Genome analysis Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.A.; Dvorkin, M.; Kulikov, A.S.S.; Lesin, V.M.M.; Nikolenko, S.I.I.; Pham, S.; Prjibelski, A.D.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 4245. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.P.F.; Prohaska, S.J.S.J. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Jia, X.; Yang, J.; Ling, Y.; Zhang, Z.; Yu, J.; Wu, J.; Xiao, J. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 2014, 30, 1297–1299. [Google Scholar] [CrossRef] [Green Version]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Vera Alvarez, R.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef]
- Wu, S.; Zhu, Z.; Fu, L.; Niu, B.; Li, W. WebMGA: A customizable web server for fast metagenomic sequence analysis. BMC Genom. 2011, 12, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 2014, 1079, 155–170. [Google Scholar]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Ågren, J.; Sundström, A.; Håfström, T.; Segerman, B. Gegenees: Fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS ONE 2012, 7, e39107. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S.D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic virus orthologous groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.H.Y.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.; Tavares, P.; Petit, M.A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef] [Green Version]
- Söding, J.; Biegert, A.; Lupas, A.N.; Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Clavel, T.; Lepage, P.; Charrier, C. The family Coriobacteriaceae. In The Prokaryotes: Actinobacteria; Springer: Berlin/Heidelberg, Germany, 2014; pp. 201–238. ISBN 9783642301384. [Google Scholar]
- Danylec, N.; Stoll, D.A.; Göbl, A.; Huch, M. Draft Genome Sequences of 13 Isolates of Adlercreutzia equolifaciens, Eggerthella lenta, and Gordonibacter urolithinfaciens, Isolated from Human Fecal Samples in Karlsruhe, Germany. Microbiol. Resour. Announc. 2020, 9, e00017–e00020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobkin, J.F.; Saha; J.R.; Butler, V.P.; Neu, H.C.; Lindenbaum, J. Digoxin-inactivating bacteria: Identification in human gut flora. Science 1983, 220, 325–327. [Google Scholar]
- Clavel, T.; Henderson, G.; Alpert, C.A.; Philippe, C.; Rigottier-Gois, L.; Doré, J.; Blaut, M. Intestinal bacterial communities that produce active estrogen-like compounds enterodiol and enterolactone in humans. Appl. Environ. Microbiol. 2005, 71, 6077–6085. [Google Scholar] [CrossRef] [Green Version]
- Liderot, K.; Larsson, M.; Boräng, S.; Özenci, V. Polymicrobial bloodstream infection with Eggerthella lenta and Desulfovibrio desulfuricans. J. Clin. Microbiol. 2010, 48, 3810–3812. [Google Scholar] [CrossRef] [Green Version]
- Bisanz, J.E.; Soto-Perez, P.; Noecker, C.; Aksenov, A.A.; Lam, K.N.; Kenney, G.E.; Bess, E.N.; Haiser, H.J.; Kyaw, T.S.; Yu, F.B.; et al. A Genomic Toolkit for the Mechanistic Dissection of Intractable Human Gut Bacteria. Cell Host Microbe 2020, 27, 1001–1013.e9. [Google Scholar] [CrossRef] [PubMed]
- Soto Perez, P.A. Plasmids, Immunity, and Phages of Gut Bacterium Eggerthella Lenta; UCSF: San Francisco, CA, USA, 2021. [Google Scholar]
- Rasko, D.A.; Rosovitz, M.J.; Myers, G.S.A.; Mongodin, E.F.; Fricke, W.F.; Gajer, P.; Crabtree, J.; Sebaihia, M.; Thomson, N.R.; Chaudhuri, R.; et al. The pangenome structure of Escherichia coli: Comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 2008, 190, 6881–6893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arboleya, S.; Bottacini, F.; O’Connell-Motherway, M.; Ryan, C.A.; Ross, R.P.; van Sinderen, D.; Stanton, C. Gene-trait matching across the Bifidobacterium longum pan-genome reveals considerable diversity in carbohydrate catabolism among human infant strains. BMC Genom. 2018, 19, 245. [Google Scholar] [CrossRef] [PubMed]
- Declerck, B.; Van der Beken, Y.; De Geyter, D.; Piérard, D.; Wybo, I. Antimicrobial susceptibility testing of Eggerthella lenta blood culture isolates at a university hospital in Belgium from 2004 to 2018. Anaerobe 2021, 69, 102348. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Casey, E.; Oliveira, J.; Bottacini, F.; James, K.; Franz, C.M.A.P.; Lugli, G.A.; Neve, H.; Ventura, M.; Hatfull, G.F.; et al. Characterization and induction of prophages in human gut-associated Bifidobacterium hosts. Sci. Rep. 2018, 8, 117. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Turroni, F.; Tremblay, D.; Ferrario, C.; Mancabelli, L.; Duranti, S.; Ward, D.V.; Ossiprandi, M.C.; Moineau, S.; et al. Prophages of the genus Bifidobacterium as modulating agents of the infant gut microbiota. Environ. Microbiol. 2016, 18, 2196–2213. [Google Scholar] [CrossRef]
- Pope, W.H.; Jacobs-Sera, D.; Russell, D.A.; Peebles, C.L.; Al-Atrache, Z.; Alcoser, T.A.; Alexander, L.M.; Alfano, M.B.; Alford, S.T.; Amy, N.E.; et al. Expanding the Diversity of Mycobacteriophages: Insights into Genome Architecture and Evolution. PLoS ONE 2011, 6, e16329. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, L.; Tavares, P.; Alonso, J.C. Headful DNA packaging: Bacteriophage SPP1 as a model system. Virus Res. 2013, 173, 247–259. [Google Scholar] [CrossRef]
- Ellis, D.M.; Dean, D.H. Nucleotide sequence of the cohesive single-stranded ends of Bacillus subtilis temperate bacteriophage phi 105. J. Virol. 1985, 55, 1212. [Google Scholar] [CrossRef] [Green Version]
- Lillehaug, D.; Lindqvist, B.H.; Birkeland, N.K. Characterization of φLC3, a Lactococcus lactis subsp. cremoris temperate bacteriophage with cohesive single-stranded DNA ends. Appl. Environ. Microbiol. 1991, 57, 3206–3211. [Google Scholar] [CrossRef] [Green Version]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.L.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.; et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 10771–10776. [Google Scholar] [CrossRef] [Green Version]
- Cho, G.-S.; Ritzmann, F.; Eckstein, M.; Huch, M.; Briviba, K.; Behsnilian, D.; Neve, H.; Franz, C.M.A.P. Quantification of Slackia and Eggerthella spp. in Human Feces and Adhesion of Representatives Strains to Caco-2 Cells. Front. Microbiol. 2016, 7, 658. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Deora, R.; Doulatov, S.R.; Gingery, M.; Eiserling, F.A.; Preston, A.; Maskell, D.J.; Simons, R.W.; Cotter, P.A.; Parkhill, J.; et al. Reverse Transcriptase-Mediated Tropism Switching in Bordetella Bacteriophage. Science 2016, 295, 2091–2094. [Google Scholar] [CrossRef]
- Alayyoubi, M.; Guo, H.; Dey, S.; Golnazarian, T.; Brooks, G.A.; Rong, A.; Miller, J.F.; Ghosh, P. Structure of the essential diversity-generating retroelement protein bAvd and its functionally important interaction with reverse transcriptase. Structure 2013, 21, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Arambula, D.; Ghosh, P.; Miller, J.F. Diversity-generating Retroelements in Phage and Bacterial Genomes. In Mobile DNA III; American Society of Microbiology: Washington, DC, USA, 2015; Volume 2, pp. 1237–1252. [Google Scholar]
- Dai, W.; Hodes, A.; Hui, W.H.; Gingery, M.; Miller, J.F.; Zhou, Z.H. Three-dimensional structure of tropism-switching Bordetella bacteriophage. Proc. Natl. Acad. Sci. USA 2010, 107, 4347–4352. [Google Scholar] [CrossRef] [Green Version]
- Dziewit, L.; Jazurek, M.; Drewniak, L.; Baj, J.; Bartosik, D. The SXT conjugative element and linear prophage N15 encode toxin-antitoxin-stabilizing systems homologous to the tad-ata module of the Paracoccus aminophilus plasmid pAMI2. J. Bacteriol. 2007, 189, 1983–1997. [Google Scholar] [CrossRef] [Green Version]
- Adriaenssens, E.M.; Rodney Brister, J. How to name and classify your phage: An informal guide. Viruses 2017, 9, 4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riipinen, K.A.; Forsman, P.; Alatossava, T. The genomes and comparative genomics of Lactobacillus delbrueckii phages. Arch. Virol. 2011, 156, 1217–1233. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R. Resistance to β-lactam antibiotics in bacteroides spp. J. Med. Microbiol. 1997, 46, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Hedberg, M.; Lindqvist, L.; Bergman, T.; Nord, C.E. Purification and characterization of a new β-lactamase from Bacteroides uniformis. Antimicrob. Agents Chemother. 1995, 39, 1458–1461. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Liu, Q.; Pei, Z.; Wang, L.; Tian, P.; Liu, Z.; Zhao, J.; Zhang, H.; Chen, W. The Diversity of the CRISPR-Cas System and Prophages Present in the Genome Reveals the Co-evolution of Bifidobacterium pseudocatenulatum and Phages. Front. Microbiol. 2020, 11, 1088. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Yu, X.; Duan, Z.; Lu, J.; Jia, B.; Qiao, Y.; Sun, C.; Wei, C. Discovery and characterization of the evolution, variation and functions of diversity-generating retroelements using thousands of genomes and metagenomes. BMC Genom. 2019, 20, 685. [Google Scholar] [CrossRef] [PubMed]
- Cornuault, J.K.; Petit, M.-A.; Mariadassou, M.; Benevides, L.; Moncaut, E.; Langella, P.; Sokol, H.; De Paepe, M. Phages infecting Faecalibacterium prausnitzii belong to novel viral genera that help to decipher intestinal viromes. Microbiome 2018, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Benler, S.; Cobián-Güemes, A.G.; McNair, K.; Hung, S.H.; Levi, K.; Edwards, R.; Rohwer, F. A diversity-generating retroelement encoded by a globally ubiquitous Bacteroides phage 06 Biological Sciences 0605 Microbiology. Microbiome 2018, 6, 243. [Google Scholar]
- Bondy-Denomy, J.; Pawluk, A.; Maxwell, K.L.; Davidson, A.R. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 2013, 493, 429–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, S.Y.; Maxwell, K.L. Phage-Encoded Anti-CRISPR Defenses. Annu. Rev. Genet. 2018, 11, 861. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Isolate | Accession (Genome; Plasmids) |
|---|---|---|
| 1 | APC055-529-1D | CP089331; CP089332 |
| 2 | APC055-539-5C | CP089333 |
| 3 | APC055-920-1E | CP089334 |
| 4 | APC055-924-7B | CP089335 |
| 5 | APC055-928-H3-3 | CP089336 |
| 6 | APC055-943-4 | CP089337 |
| 7 | APC-F2-3 | CP089338; CP089339, CP089340 |
| 8 | DSM2243_R211 | JAJQIW000000000 |
| 9 | DSM2243_R611 | JAJQIX000000000 |
| 10 | DSM2243_R1011 | JAJQKW000000000 |
| No. | Prophage | Host (Contig Accession) | Left Boundary | Right Boundary | Size (bp) | GC% | Integration Locus (Locus Tag) Relative to Strains DSM2243 or C529 | Clade |
|---|---|---|---|---|---|---|---|---|
| 1 | 14Aphi1 | 14A (NZ_PPUR01000006.1) | 29,937 | 71,301 | 41,365 | 67 | tRNA-Leu (ELEN_RS15020) | 1 |
| 2 | MGYG-HGUT-02544phi1 | MGYG-HGUT-02544 (NZ_CABMOO010000006.1) | 29,956 | 71,302 | 41,347 | 67 | tRNA-Leu (ELEN_RS15020) | 1 |
| 3 | AB8_2phi1 | AB8 #2 (NZ_PPUJ01000004.1) | 191,735 | end of contig | >38,510 | 67 | tRNA-Leu (ELEN_RS15020) | 1 |
| 4 | APC055-529-1Dphi1 | APC055-529-1D (CP089331) | 3,240,976 | 3,282,082 | 41,106 | 67 | tRNA-Leu (ELEN_RS15020) | 1 |
| 5 | ResAG49phi1 | ResAG49 (NZ_WPON01000034.1) | full contig | full contig | >30,988 | 67 | unknown | 1 |
| 6 | MR1_12phi1 | MR1_#12 (NZ_PPTX01000022.1) | 3020 | 45,178 | 42,159 | 67 | tRNA-Leu (ELEN_RS02880) | 1 |
| 7 | 1001095IJ_161003_H5phi1 | 1001095IJ_161003_H5 (JADMUV010000007.1) | 137,112 | 178,204 | 41,092 | 67 | tRNA-Leu (ELEN_RS02880) | 1 |
| 8 | BSD2780120875_150330_C12phi1 | BSD2780120875_150330_C12 (JADMOT010000003.1) | 209,825 | 251,107 | 41,282 | 66 | tRNA-Leu (ELEN_RS15020) | 1 |
| 9 | Valenciaphi2 | Valencia (NZ_PPTV01000006.1) | 55,915 | 93,457 | 37,543 | 69 | tRNA-Ser (ELEN_RS00500) | 2 |
| 10 | 32-6-I_6_NAphi2 | 32-6-I_6_NA (NZ_PPUM01000011.1) | 26,284 | 63,686 | 37,403 | 69 | tRNA-Ser (ELEN_RS00500) | 2 |
| 11 | DSM15644phi3 | DSM15644 (NZ_PPUB01000019.1) | 32,924 | 69,743 | 36,820 | 67 | tRNA-Ala (ELEN_RS00055) | 3 |
| 12 | DSM15644phi4 | DSM15644 (NZ_PPUB01000037.1) | full contig | full contig | >36,317 | 63 | unknown | 4 |
| 13 | DSM2243phi4 | DSM2243 (NC_013204) | 3,031,719 | 3,068,586 | 36,618 | 63 | tRNA-Ala (C592_00654) | 4 |
| 14 | CC82_BHI2phi5 | CC82_BHI2 (NZ_PPUF01000005.1) | 22,245 | 56,042 | 33,798 | 65 | hypothetical protein (ELEN_RS14245) | 5 |
| 15 | C592phi5 | C592 (NZ_CP021140) | 500,864 | 534,671 | 33,808 | 65 | hypothetical protein (ELEN_RS14245) | 5 |
| 16 | J1101437_171009_A1phi5 | J1101437_171009_A1 (JADNJK010000003.1) | 299,175 | 333,022 | 33,847 | 65 | hypothetical protein (ELEN_RS14245) | 5 |
| 17 | J1101653_170612_H2phi5 | J1101653_170612_H2 (JADPDY010000015.1) | 27,299 | 61,125 | 33,826 | 65 | hypothetical protein (ELEN_RS14245) | 5 |
| 18 | 1-1-60AFAAphi6 | 1-1-60AFAA (NZ_KN214093.1) | 487,345 | 528,047 | 40,703 | 64 | unknown | 6 |
| 19 | APC055-529-1Dphi7 | APC055-529-1D (CP089331) | 3,337,687 | 3,371,357 | 33,670 | 59 | tRNA-Arg (ELEN_RS15245) | 7 |
| 20 | APC055-924-7Bphi7 | APC055-924-7B (CP089335) | 3,608,848 | 3,642,543 | 33,695 | 59 | tRNA-Arg (ELEN_RS15245) | 7 |
| 21 | 14Aphi7 | 14A (NZ_PPUR01000011.1) | 37,521 | 71,198 | 33,678 | 59 | tRNA-Arg (ELEN_RS15245) | 7 |
| 22 | MGYG-HGUT-02544phi7 | MGYG-HGUT-02544 (NZ_CABMOO010000011.1) | 37,518 | 71,195 | 33,678 | 59 | tRNA-Arg (ELEN_RS15245) | 7 |
| 23 | APC055-920-1Ephi7 | APC055-920-1E (CP089334) | 3,189,344 | 3,223,015 | 33,671 | 59 | tRNA-Arg (ELEN_RS15245) | 7 |
| 24 | MR1-F37phi7 | MRI-F37 (NZ_WPOI01000001.1) | 69,802 | 103,502 | 33,701 | 59 | tRNA-Arg (ELEN_RS15245) | 7 |
| 25 | MRI-F36phi7 | MRI-F36 (NZ_WPOJ01000009.1) | 49,797 | 83,505 | 33,709 | 59 | tRNA-Arg (ELEN_RS15245) | 7 |
| 26 | 32-6-1-6_NAphi7 | 32-6-I_6_NA (NZ_PPUM01000009.1) | 46,614 | 46,614 | 33,684 | 59 | tRNA-Arg (ELEN_RS15245) | 7 |
| 27 | MRI-F40phi7 | MRI-F40 (NZ_WPOH01000001.1) | 69,804 | 103,501 | 33,448 | 59 | tRNA-Arg (ELEN_RS15245) | 7 |
| 28 | J1100102_180507_G10phi7 | J1100102_180507_G10 (JADOZP010000001.1) | 109,609 | 145,440 | 35,831 | 58 | tRNA-Arg (ELEN_RS15245) | 7 |
| 29 | SECO-MT75m2phi8 | SECO-MT75m2 (NZ_VEVP01000036.1) | 3653 | end of contig | >28,901 | 63 | unknown | 8 |
| 30 | TF05-26B-4phi9 | TF05-26B-4 (NZ_QSSL01000026.1) | 16,687 | end of contig | >31,589 | 64 | unknown | 9 |
| 31 | J1100102_180507_G10phi9 | J1100102_180507_G10 (JADOZP010000013.1) | 10,288 | 46,514 | 36,226 | 65 | unknown | 9 |
| 32 | 1001302B_160321_A1phi9 | 1001302B_160321_A1 (JADNIO010000007.1) | 165,588 | 202,064 | 36,476 | 65 | unknown | 9 |
| 33 | 1001302B_160321_A1phi10 | 1001302B_160321_A1 (JADNIO010000005.1) | 220,405 | 252,147 | 31,742 | 58 | unknown | 10 |
| Prophage | Predicted attP-Site | attB Relative to DSM2243 (Locus) |
|---|---|---|
| 14Aphi1 | CAACCCCATGGAGGTTCAAGTCCTCTCGCCCGCACCATCTGAA | tRNA-Leu (ELEN_RS15020) |
| MGYG-HGUT-02544phi1 | AACCCCATGGAGGTTCAAGTCCTCTCGCCCGCACCATCTGAA | tRNA-Leu (ELEN_RS15020) |
| APC055-529-1Dphi1 | TTCAGATGGTGCGGGCGAGAGGACTTGAACCTCCATGGGGTT | tRNA-Leu (ELEN_RS15020) |
| 1001095IJ_161003_H5phi1 | ACTTAAAATCTTCCGGCTTCGGCCTTGCGGGTTCGAGTCCCGCCGCCCCTACCA | tRNA-Leu (ELEN_RS02880) |
| BSD2780120875_150330_C12phi1 | TTCAGATGGTGCGGGCGAGAGGACTTGAACCTCCATGGGGTT | tRNA-Leu (ELEN_RS15020) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buttimer, C.; Bottacini, F.; Shkoporov, A.N.; Draper, L.A.; Ross, P.; Hill, C. Selective Isolation of Eggerthella lenta from Human Faeces and Characterisation of the Species Prophage Diversity. Microorganisms 2022, 10, 195. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10010195
Buttimer C, Bottacini F, Shkoporov AN, Draper LA, Ross P, Hill C. Selective Isolation of Eggerthella lenta from Human Faeces and Characterisation of the Species Prophage Diversity. Microorganisms. 2022; 10(1):195. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10010195
Chicago/Turabian StyleButtimer, Colin, Francesca Bottacini, Andrey N. Shkoporov, Lorraine A. Draper, Paul Ross, and Colin Hill. 2022. "Selective Isolation of Eggerthella lenta from Human Faeces and Characterisation of the Species Prophage Diversity" Microorganisms 10, no. 1: 195. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10010195