
The Gill Microbiota of Argopecten purpuratus Scallop Is Dominated by Symbiotic Campylobacterota and Upwelling Intensification Differentially Affects Their Abundance

 , , and
, , and 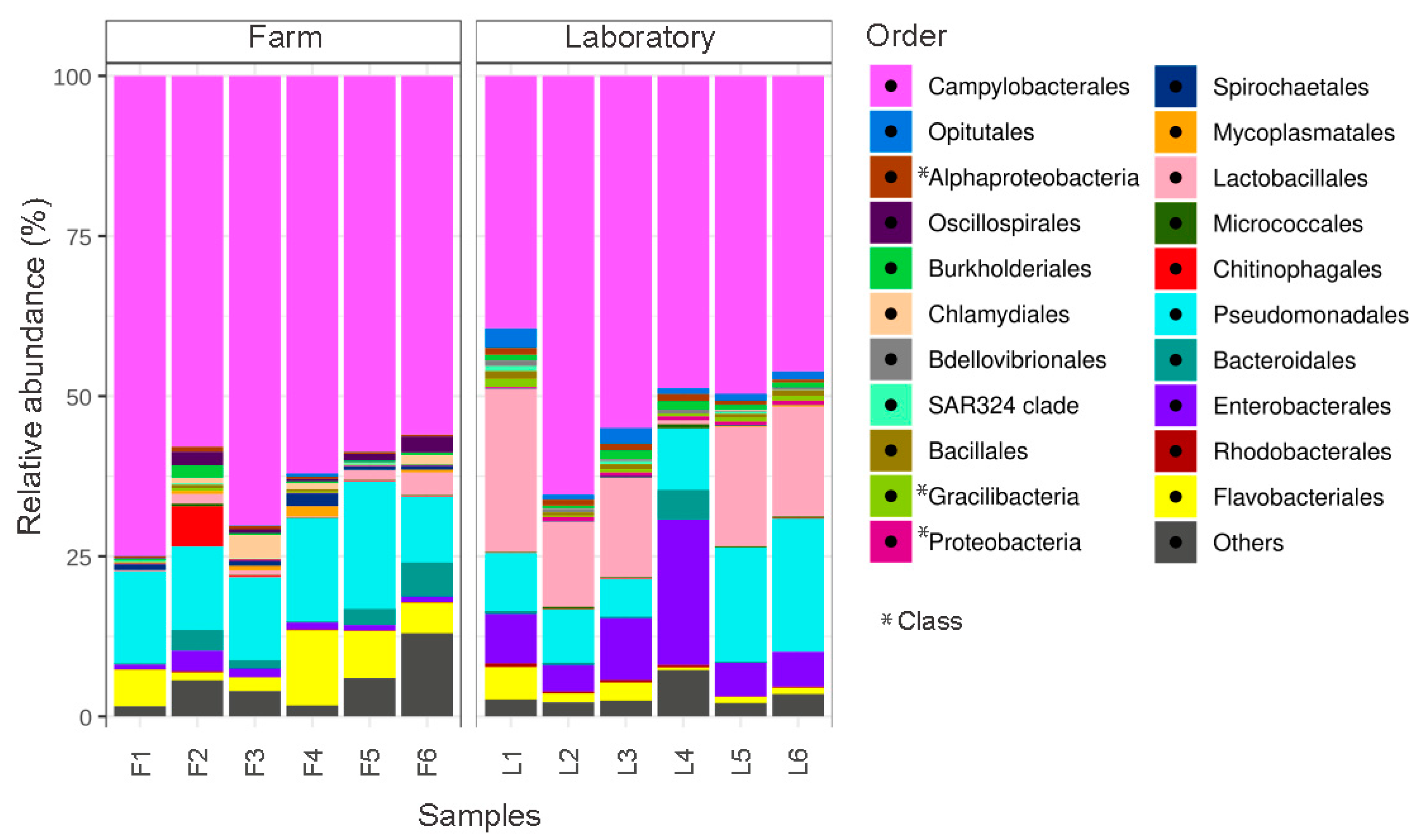{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Scallop Maintenance, Experimental Procedure and Sample Collection
2.2. Genomic DNA Extraction and Deep Amplicon Sequencing of the 16S rRNA Gene
2.3. 16S rRNA Deep Amplicon Sequencing Analysis
2.4. Phylogenetic Inference for the 16S rRNA Sequences from Campylobacterota
2.5. In Situ Hybridization (ISH)
2.6. Scanning Electron Microscopy (SEM)
2.7. Absolute Quantification of Bacterial Symbionts by qPCR
3. Results and Discussion
3.1. The Gill Microbiota of A. purpuratus Is Dominated by the Phylum Campylobacterota in the Farm and under Controlled Laboratory Conditions
3.2. The Gill Microbiota of A. purpuratus Have a Potential Chemoautotrophic Function That May Contribute to Its Nutrition
3.3. The Campylobacterota from Scallops Are Part of a Widely Distributed Family of Symbionts of Bivalves That Inhabit Extreme and Challenging Environments
3.4. Campylobacterota Are Located in the Surface of Scallop Gills
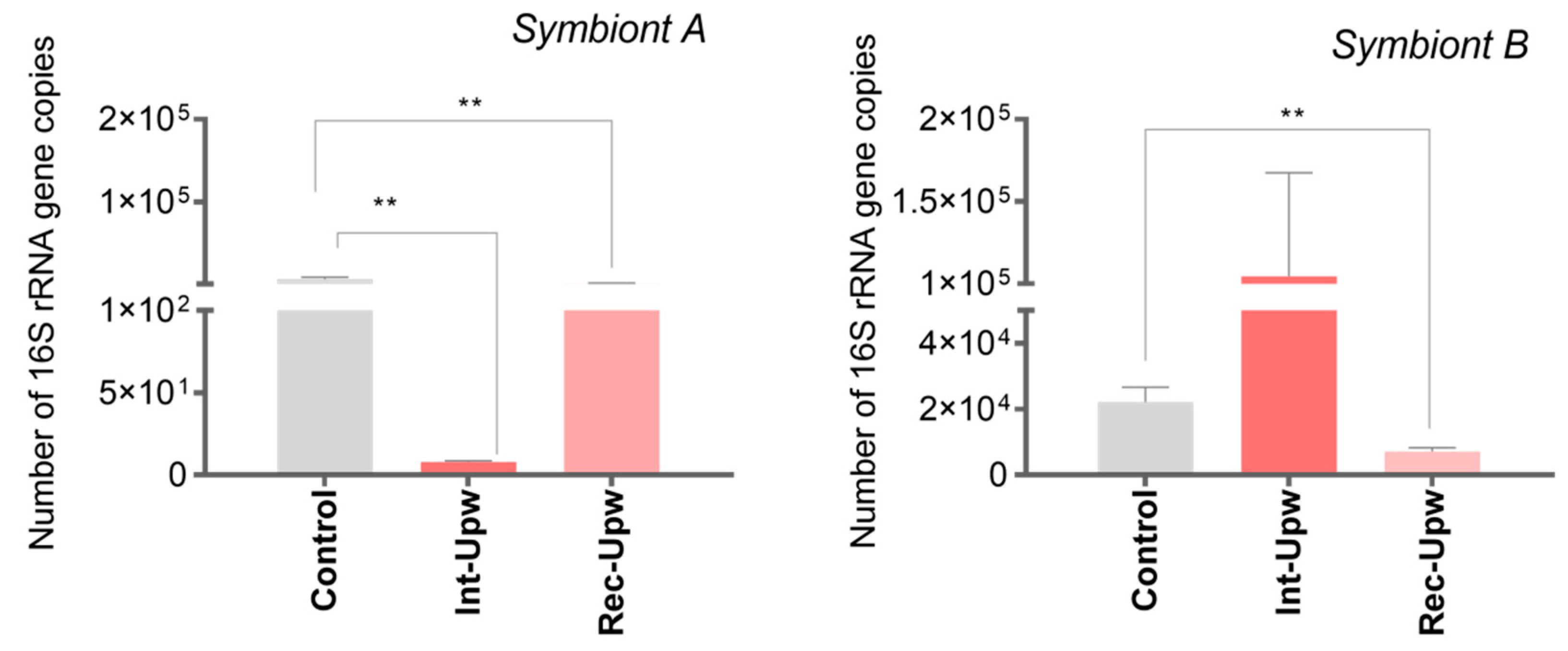
3.5. Upwelling Intensification Differentially Affects the Abundance of the Most Relevant Campylobacterota
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dubé, C.E.; Ky, C.-L.; Planes, S. Microbiome of the Black-Lipped Pearl Oyster Pinctada margaritifera, a Multi-Tissue Description With Functional Profiling. Front. Microbiol. 2019, 10, 1548. [Google Scholar] [CrossRef] [PubMed]
- Evariste, L.; Barret, M.; Mottier, A.; Mouchet, F.; Gauthier, L.; Pinelli, E. Gut microbiota of aquatic organisms: A key endpoint for ecotoxicological studies. Environ. Pollut. 2019, 248, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Woodhams, D.C.; Bletz, M.C.; Becker, C.G.; Bender, H.A.; Buitrago-Rosas, D.; Diebboll, H.; Huynh, R.; Kearns, P.J.; Kueneman, J.; Kurosawa, E.; et al. Host-associated microbiomes are predicted by immune system complexity and climate. Genome Biol. 2020, 21, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, C.M.; Aneja, K.K.; Beyhan, S.; Cho, C.E.; Woloszynek, S.; Convertino, M.; McCoy, S.J.; Zhang, Y.; Anderson, M.Z.; Alvarez-Ponce, D.; et al. Emerging Priorities for Microbiome Research. Front. Microbiol. 2020, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- Paillard, C.; Gueguen, Y.; Wegner, K.M.; Bass, D.; Pallavicini, A.; Vezzulli, L.; Arzul, I. Recent advances in bivalve-microbiota interactions for disease prevention in aquaculture. Curr. Opin. Biotechnol. 2021, 73, 225–232. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.M.; Fung, J.M.; Sharp, K.H.; Benner, J.S.; McClung, C.; Cushing, S.; Lamkin, E.R.; Fomenkov, A.I.; Henrissat, B.; Londer, Y.Y.; et al. Gill bacteria enable a novel digestive strategy in a wood-feeding mollusk. Proc. Natl. Acad. Sci. USA 2014, 111, E5096–E5104. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.D.; Glover, E.A. Chemosymbiotic Bivalves. In The Vent and Seep Biota: Aspects from Microbes to Eco-Systems; Kiel, S., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 107–135. [Google Scholar]
- King, W.L.; Siboni, N.; Williams, N.L.R.; Kahlke, T.; Nguyen, K.V.; Jenkins, C.; Dove, M.; O’Connor, W.; Seymour, J.; Labbate, M. Variability in the Composition of Pacific Oyster Microbiomes Across Oyster Families Exhibiting Different Levels of Susceptibility to OsHV-1 μvar Disease. Front. Microbiol. 2019, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Longford, S.R.; Campbell, A.H.; Nielsen, S.; Case, R.; Kjelleberg, S.; Steinberg, P.D. Interactions within the microbiome alter microbial interactions with host chemical defences and affect disease in a marine holobiont. Sci. Rep. 2019, 9, 1363. [Google Scholar] [CrossRef] [Green Version]
- Ros, M.; Suggett, D.J.; Edmondson, J.; Haydon, T.; Hughes, D.J.; Kim, M.; Guagliardo, P.; Bougoure, J.; Pernice, M.; Raina, J.-B.; et al. Symbiont shuffling across environmental gradients aligns with changes in carbon uptake and translocation in the reef-building coral Pocillopora acuta. Coral Reefs 2021, 40, 595–607. [Google Scholar] [CrossRef]
- Apprill, A. Marine Animal Microbiomes: Toward Understanding Host–Microbiome Interactions in a Changing Ocean. Front. Mar. Sci. 2017, 4, 222. [Google Scholar] [CrossRef] [Green Version]
- Allam, B.; Espinosa, E.P. Bivalve immunity and response to infections: Are we looking at the right place? Fish Shellfish Immunol. 2016, 53, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, E.T.F.; Moreira, A.M.S.; de Paula, R.S.; Andrade, G.R.; de Carvalho, M.D.; Assis, P.S.; Jorge, E.C.; Cardoso, A.V. Ultra-structure of the gill ciliary epithelium of Limnoperna fortunei (Dunker 1857), the invasive golden mussel. BMC Zool. 2022, 7, 6. [Google Scholar] [CrossRef]
- Assié, A.; Borowski, C.; van der Heijden, K.; Raggi, L.; Geier, B.; Leisch, N.; Schimak, M.P.; Dubilier, N.; Petersen, J.M. A specific and widespread association between deep-sea Bathymodiolus mussels and a novel family of Epsilonproteobacteria. Environ. Microbiol. Rep. 2016, 8, 805–813. [Google Scholar] [CrossRef] [PubMed]
- González, R.; Gonçalves, A.T.; Rojas, R.; Brokordt, K.; Rosa, R.D.; Schmitt, P. Host Defense Effectors Expressed by Hemocytes Shape the Bacterial Microbiota From the Scallop Hemolymph. Front. Immunol. 2020, 11, 599625. [Google Scholar] [CrossRef]
- von Brand, E.; Merino, G.E.; Abarca, A.; Stotz, W. Chapter 27 Scallop fishery and aquaculture in Chile. In Developments in Aquaculture and Fisheries Science; Shumway, S.E., Parsons, G.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 1293–1314. [Google Scholar]
- Messié, M.; Chavez, F.P. Seasonal regulation of primary production in eastern boundary upwelling systems. Prog. Oceanogr. 2015, 134, 1–18. [Google Scholar] [CrossRef]
- García-Reyes, M.; Sydeman, W.J.; Schoeman, D.; Rykaczewski, R.R.; Black, B.A.; Smit, A.J.; Bograd, S.J. Under Pressure: Climate Change, Upwelling, and Eastern Boundary Upwelling Ecosystems. Front. Mar. Sci. 2015, 2, 109. [Google Scholar] [CrossRef] [Green Version]
- Grez, P.W.; Aguirre, C.; Farías, L.; Contreras-López, M.; Masotti, Í. Evidence of climate-driven changes on atmospheric, hydrological, and oceanographic variables along the Chilean coastal zone. Clim. Chang. 2020, 163, 633–652. [Google Scholar] [CrossRef]
- Ramajo, L.; Valladares, M.; Astudillo, O.; Fernández, C.; Rodríguez-Navarro, A.B.; Watt-Arévalo, P.; Núñez, M.; Grenier, C.; Román, R.; Aguayo, P.; et al. Upwelling intensity modulates the fitness and physiological performance of coastal species: Implications for the aquaculture of the scallop Argopecten purpuratus in the Humboldt Current System. Sci. Total Environ. 2020, 745, 140949. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, C.; Rojas, M.; Garreaud, R.D.; Rahn, D. Role of synoptic activity on projected changes in upwelling-favourable winds at the ocean’s eastern boundaries. NPJ Clim. Atmos. Sci. 2019, 2, 44. [Google Scholar] [CrossRef]
- Bonino, G.; Di Lorenzo, E.; Masina, S.; Iovino, D. Interannual to decadal variability within and across the major Eastern Boundary Upwelling Systems. Sci. Rep. 2019, 9, 19949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakun, A.; Black, B.A.; Bograd, S.J.; García-Reyes, M.; Miller, A.J.; Rykaczewski, R.R.; Sydeman, W.J. Anticipated Effects of Climate Change on Coastal Upwelling Ecosystems. Curr. Clim. Chang. Rep. 2015, 1, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Scanes, E.; Parker, L.M.; Seymour, J.R.; Siboni, N.; King, W.L.; Wegner, K.M.; Dove, M.C.; O’Connor, W.A.; Ross, P.M. Microbiome response differs among selected lines of Sydney rock oysters to ocean warming and acidification. FEMS Microbiol. Ecol. 2021, 97, fiab099. [Google Scholar] [CrossRef] [PubMed]
- Newell, R.I.E.; Fisher, T.R.; Holyoke, R.R.; Cornwell, J.C. Influence of Eastern Oysters on Nitrogen and Phosphorus Regeneration in Chesapeake Bay, USA. In The Comparative Roles of Suspension-Feeders in Ecosystems; NATO Science Series IV: Earth and Environmental Series; Springer: Dordrecht, The Netherlands, 2005; pp. 93–120. [Google Scholar] [CrossRef]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [Green Version]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534, Correction in Mol. Biol. Evol. 2020, 37, 2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, I.; van Aerle, R.; Ross, S.; Verner-Jeffreys, D.W.; Paley, R.K.; Rimmer, G.S.E.; Ryder, D.; Hooper, P.; Stone, D.; Feist, S.W. Molecular Characterization of an Endozoicomonas-Like Organism Causing Infection in the King Scallop (Pecten maximus L.). Appl. Environ. Microbiol. 2018, 84, e00952-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDowell, E.M.; Trump, B.F. Histologic fixatives suitable for diagnostic light and electron microscopy. Arch. Pathol. Lab. Med. 1976, 100, 405–414. [Google Scholar]
- Ma, Y.; Li, M.; Sun, J.; Hao, Z.; Liang, J.; Zhao, X. Characterization of Bacterial Community Associated with Four Organs of the Yesso Scallop (Patinopecten yessoensis) by High-Throughput Sequencing. J. Ocean Univ. China 2019, 18, 493–500. [Google Scholar] [CrossRef]
- Baccouri, O.; Boukerb, A.M.; Farhat, L.B.; Zébré, A.; Zimmermann, K.; Domann, E.; Cambronel, M.; Barreau, M.; Maillot, O.; Rincé, I.; et al. Probiotic Potential and Safety Evaluation of Enterococcus faecalis OB14 and OB15, Isolated From Traditional Tunisian Testouri Cheese and Rigouta, Using Physiological and Genomic Analysis. Front. Microbiol. 2019, 10, 881. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Albores, A.; Lopez-Santamarina, A.; Rodriguez, J.A.; Ibarra, I.S.; Del Carmen Mondragón, A.; Miranda, J.M.; Lamas, A.; Cepeda, A. Complementary Methods to Improve the Depuration of Bivalves: A Review. Foods 2020, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Du, S.; Chen, W.; Yao, Z.; Huang, X.; Chen, C.; Guo, H.; Zhang, D. Enterococcus faecium are associated with the modification of gut microbiota and shrimp post-larvae survival. Anim. Microbiome 2021, 3, 88. [Google Scholar] [CrossRef]
- Yu, M.; Wang, X.; Yan, A. Microbial Profiles of Retail Pacific Oysters (Crassostrea gigas) From Guangdong Province, China. Front. Microbiol. 2021, 12, 689520. [Google Scholar] [CrossRef]
- Gao, Y.-M.; Zou, K.-S.; Zhou, L.; Huang, X.-D.; Li, Y.-Y.; Gao, X.-Y.; Chen, X.; Zhang, X.-Y. Deep Insights into Gut Microbiota in Four Carnivorous Coral Reef Fishes from the South China Sea. Microorganisms 2020, 8, 426. [Google Scholar] [CrossRef]
- Franco, A.; Busse, H.-J.; Schubert, P.; Wilke, T.; Kampfer, P.; Glaeser, S.P. Winogradskyella pocilloporae sp. nov. isolated from healthy tissue of the coral Pocillopora damicornis. Int. J. Syst. Evol. Microbiol. 2018, 68, 1689–1696. [Google Scholar] [CrossRef]
- Schellenberg, J.; Busse, H.-J.; Hardt, M.; Schubert, P.; Wilke, T.; Kämpfer, P.; Glaeser, S.P. Winogradskyella haliclonae sp. nov., isolated from a marine sponge of the genus Haliclona. Int. J. Syst. Evol. Microbiol. 2017, 67, 4902–4910. [Google Scholar] [CrossRef] [PubMed]
- Bass, D.; Stentiford, G.D.; Wang, H.-C.; Koskella, B.; Tyler, C.R. The Pathobiome in Animal and Plant Diseases. Trends Ecol. Evol. 2019, 34, 996–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassone-Corsi, M.; Raffatellu, M. No vacancy: How beneficial microbes cooperate with immunity to provide colonization resistance to pathogens. J. Immunol. 2015, 194, 4081–4087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leggett, H.C.; Buckling, A.; Long, G.H.; Boots, M. Generalism and the evolution of parasite virulence. Trends Ecol. Evol. 2013, 28, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Assié, A.; Leisch, N.; Meier, D.V.; Gruber-Vodicka, H.; Tegetmeyer, H.E.; Meyerdierks, A.; Kleiner, M.; Hinzke, T.; Joye, S.; Saxton, M.; et al. Horizontal acquisition of a patchwork Calvin cycle by symbiotic and free-living Campylobacterota (formerly Epsilonproteobacteria). ISME J. 2020, 14, 104–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bloa, S.; Boidin-Wichlacz, C.; Cueff-Gauchard, V.; Rosa, R.D.; Cuvillier, V.; Durand, L.; Methou, P.; Pradillon, F.; Cambon-Bonavita, M.-A.; Tasiemski, A. Antimicrobial Peptides and Ectosymbiotic Relationships: Involvement of a Novel Type IIa Crustin in the Life Cycle of a Deep-Sea Vent Shrimp. Front. Immunol. 2020, 11, 1511. [Google Scholar] [CrossRef]
- Srain, B.M.; Sobarzo, M.; Daneri, G.; González, H.E.; Testa, G.; Farías, L.; Schwarz, A.; Pérez, N.; Pantoja-Gutiérrez, S. Fermentation and Anaerobic Oxidation of Organic Carbon in the Oxygen Minimum Zone of the Upwelling Ecosystem Off Concepción, in Central Chile. Front. Mar. Sci. 2020, 7, 533. [Google Scholar] [CrossRef]
- Zbinden, M.; Marqué, L.; Gaudron, S.M.; Ravaux, J.; Léger, N.; Duperron, S. Epsilonproteobacteria as gill epibionts of the hydrothermal vent gastropod Cyathermia naticoides (North East-Pacific Rise). Mar. Biol 2015, 162, 435–448. [Google Scholar] [CrossRef]
- Yáñez, E.; Lagos, N.A.; Norambuena, R.; Silva, C.; Letelier, J.; Muck, K.-P.; Martin, G.S.; Benítez, S.; Broitman, B.R.; Contreras, H.; et al. Impacts of Climate Change on Marine Fisheries and Aquaculture in Chile. In Climate Change Impacts on Fisheries and Aquaculture; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2017; pp. 239–332. [Google Scholar]
- Goris, T.; Schubert, T.; Gadkari, J.; Wubet, T.; Tarkka, M.; Buscot, F.; Adrian, L.; Diekert, G. Insights into organohalide respiration and the versatile catabolism of Sulfurospirillum multivorans gained from comparative genomics and physiological studies. Environ. Microbiol. 2014, 16, 3562–3580. [Google Scholar] [CrossRef]
- Dahle, H.; Roalkvam, I.; Thorseth, I.H.; Pedersen, R.B.; Steen, I.H. The versatile in situ gene expression of an Epsilonproteobacteria-dominated biofilm from a hydrothermal chimney. Environ. Microbiol. Rep. 2013, 5, 282–290. [Google Scholar] [CrossRef] [PubMed]
- van der Stel, A.-X.; Wösten, M.M.S.M. Regulation of Respiratory Pathways in Campylobacterota: A Review. Front. Microbiol. 2019, 10, 1719. [Google Scholar] [CrossRef] [Green Version]
- Foster, K.R.; Schluter, J.; Coyte, K.Z.; Rakoff-Nahoum, S. The evolution of the host microbiome as an ecosystem on a leash. Nature 2017, 548, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, R.; González, D.; Stambuk, F.; Ramírez, F.; Guzmán, F.; Mercado, L.; Rojas, R.; Henríquez, C.; Brokordt, K.; Schmitt, P. A g-type lysozyme from the scallop Argopecten purpuratus participates in the immune response and in the stability of the hemolymph microbiota. Fish Shellfish Immunol. 2022, 123, 324–334. [Google Scholar] [CrossRef]
- Hernroth, B.E.; Baden, S.P. Alteration of host-pathogen interactions in the wake of climate change—Increasing risk for shellfish associated infections? Environ. Res. 2018, 161, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Gebrayel, P.; Nicco, C.; al Khodor, S.; Bilinski, J.; Caselli, E.; Comelli, E.M.; Egert, M.; Giaroni, C.; Karpinski, T.M.; Loniewski, I.; et al. Microbiota medicine: Towards clinical revolution. J. Transl. Med. 2022, 20, 111. [Google Scholar] [CrossRef] [PubMed]
- de Lorgeril, J.; Escoubas, J.M.; Loubiere, V.; Pernet, F.; le Gall, P.; Vergnes, A.; Aujoulat, F.; Jeannot, J.L.; Jumas-Bilak, E.; Got, P.; et al. Inefficient immune response is associated with microbial permissiveness in juvenile oysters affected by mass mortalities on field. Fish Shellfish Immunol. 2018, 77, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Clerissi, C.; De Lorgeril, J.; Petton, B.; Lucasson, A.; Escoubas, J.-M.; Gueguen, Y.; Dégremont, L.; Mitta, G.; Toulza, E. Microbiota Composition and Evenness Predict Survival Rate of Oysters Confronted to Pacific Oyster Mortality Syndrome. Front. Microbiol. 2020, 11, 311. [Google Scholar] [CrossRef] [Green Version]
- Egan, S.; Gardiner, M. Microbial Dysbiosis: Rethinking Disease in Marine Ecosystems. Front. Microbiol. 2016, 7, 991. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, R.; Henríquez-Castillo, C.; Lohrmann, K.B.; Romero, M.S.; Ramajo, L.; Schmitt, P.; Brokordt, K. The Gill Microbiota of Argopecten purpuratus Scallop Is Dominated by Symbiotic Campylobacterota and Upwelling Intensification Differentially Affects Their Abundance. Microorganisms 2022, 10, 2330. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10122330
González R, Henríquez-Castillo C, Lohrmann KB, Romero MS, Ramajo L, Schmitt P, Brokordt K. The Gill Microbiota of Argopecten purpuratus Scallop Is Dominated by Symbiotic Campylobacterota and Upwelling Intensification Differentially Affects Their Abundance. Microorganisms. 2022; 10(12):2330. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10122330
Chicago/Turabian StyleGonzález, Roxana, Carlos Henríquez-Castillo, Karin B. Lohrmann, María Soledad Romero, Laura Ramajo, Paulina Schmitt, and Katherina Brokordt. 2022. "The Gill Microbiota of Argopecten purpuratus Scallop Is Dominated by Symbiotic Campylobacterota and Upwelling Intensification Differentially Affects Their Abundance" Microorganisms 10, no. 12: 2330. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10122330