
Physiological and Genomic Characterization of Actinotalea subterranea sp. nov. from Oil-Degrading Methanogenic Enrichment and Reclassification of the Family Actinotaleaceae

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Development of Methanogenic Enrichment Growing on Crude Oil
2.2. DNA Isolation from the Oil-Degrading Enrichment, 16S rRNA Gene Amplification, and Sequencing
2.3. Bacterial and Archaeal Strains
2.4. DNA Extraction from New Strains, 16S rRNA Gene Sequencing, and Phylogenetic Analysis
2.5. Genome Sequencing and Analyses
2.6. Morphological and Physiological Characterization
2.7. Chemotaxonomic Characterization
2.8. Gas Chromatography
2.9. Nucleotide Sequence Accession Number
3. Results
3.1. Methane Production and Phylogenetic Diversity of Prokaryotes in the Anaerobic Oil-Degrading Enrichment
3.2. Isolation of Fermentative and Methanogenic Strains and Analysis of 16S rRNA Genes
3.3. Phenotypic Characterization
3.4. Chemotaxonomic Characterization
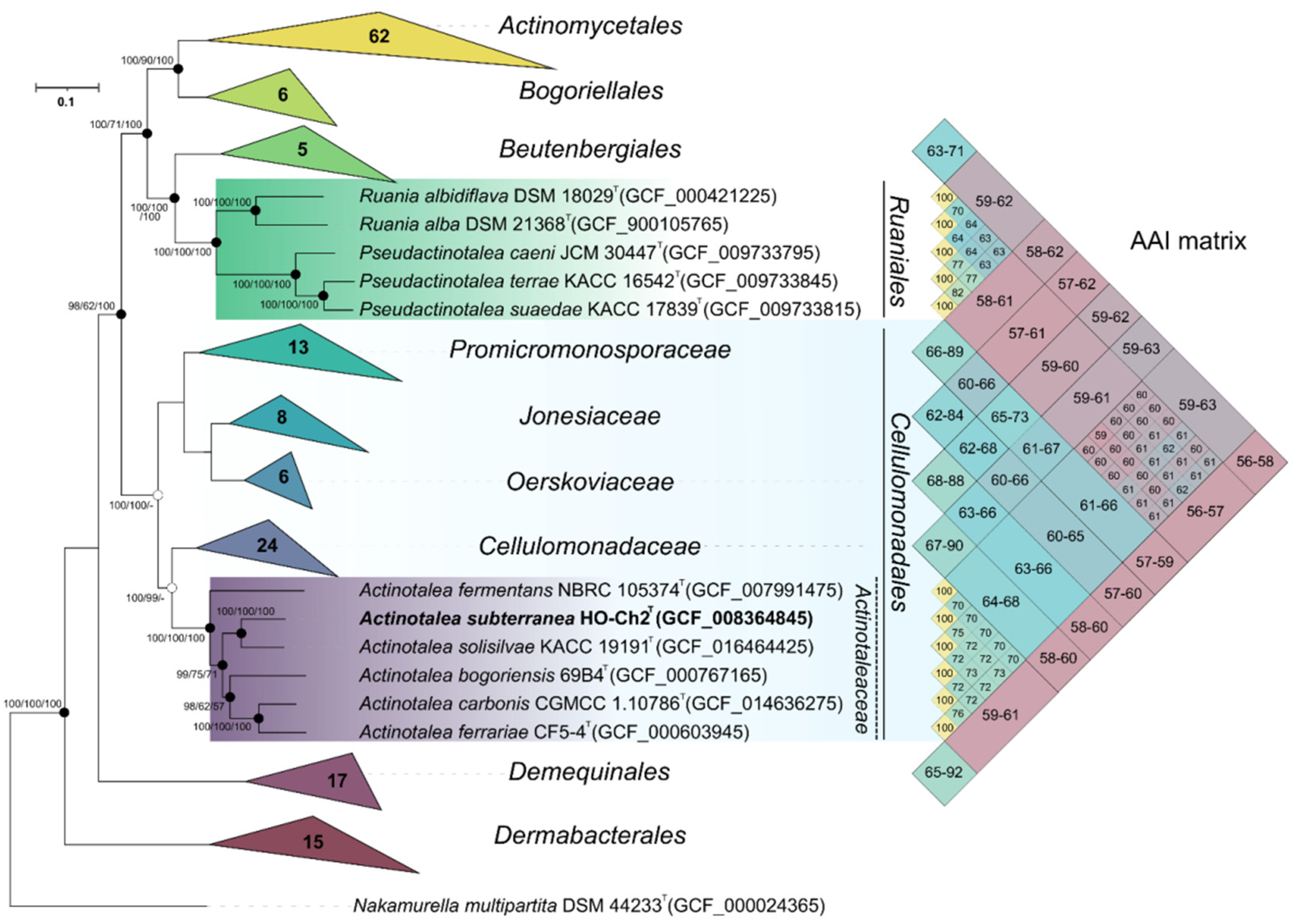
3.5. Whole Genome Sequencing and Phylogenomic Analysis
3.6. Pangenomic Analysis
4. Discussion
5. Conclusions
5.1. Description of Actinotalea subterranea sp. nov.
5.2. Description of Actinotalea carbonis, comb. nov.
5.3. Description of Actinotalea bogoriensis, comb. nov.
5.4. Emended description of the family Actinotaleaceae Salam et al. 2020
5.5. Description of Pseudactinotalea caeni, comb. nov.
5.6. Emended Description of the Genus Pseudactinotalea Cho et al. 2017 Emend. Salam et al. 2020
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Head, I.M.; Gray, N.D.; Larter, S.R. Life in the slow lane; biogeochemistry of biodegraded petroleum containing reservoirs and implications for energy recovery and carbon management. Front. Microbiol. 2014, 5, 566. [Google Scholar] [CrossRef] [PubMed]
- Magot, M.; Ollivier, B.; Patel, B.K.C. Microbiology of petroleum reservoirs. Antonie Van Leeuwenhoek 2000, 77, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.; Elshahed, M.S.; McInerney, M.J. Microbial processes in oil fields: Culprits, problems and opportunities. Adv. Appl. Microbiol. 2009, 66, 141–251. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, S.I. Investigation of the possibility of contemporaneous formation of methane in gas-petroleum formations in the Saratov and Buguruslan regions. Mikrobiologiya 1950, 19, 193–202. (In Russian) [Google Scholar]
- Zengler, K.; Richnow, H.H.; Rossello-Mora, R.; Michaelis, W.; Widdel, F. Methane formation from long-chain alkanes by anaerobic microorganisms. Nature 1999, 401, 266–269. [Google Scholar] [CrossRef]
- Townsend, T.G.; Prince, R.C.; Suflita, J.M. Anaerobic oxidation of crude oil hydrocarbons by the resident microorganisms of a contaminated anoxic aquifer. Environ. Sci. Technol. 2003, 37, 5213–5218. [Google Scholar] [CrossRef]
- Jones, D.M.; Head, I.M.; Gray, N.D.; Adams, J.J.; Rowan, A.K.; Aitken, C.M.; Bennett, B.; Huang, H.; Brown, A.; Bowler, B.F.J.; et al. Crude-oil biodegradation via methanogenesis in subsurface petroleum reservoirs. Nature 2008, 451, 176–180. [Google Scholar] [CrossRef]
- Wang, L.-Y.; Gao, C.-X.; Mbadinga, S.M.; Zhou, L.; Liu, J.-F.; Gu, J.-D.; Mu, B.-Z. Characterization of an alkane-degrading methanogenic enrichment culture from production water of an oil reservoir after 274 days of incubation. Int. Biodeterior. Biodegrad. 2011, 65, 444–450. [Google Scholar] [CrossRef]
- Gray, N.D.; Sherry, A.; Hubert, C.; Dolfing, J.; Head, I.M. Methanogenic degradation of petroleum hydrocarbons in subsurface environments remediation, heavy oil formation, and energy recovery. Adv. Appl. Microbiol. 2010, 72, 137–161. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Y.F.; Zhou, L.; Irfan, M.; Hou, Z.-W.; Li, W.; Mbadinga, S.M.; Liu, J.F.; Yang, S.Z.; Wu, X.-L.; et al. Long-chain n-alkane biodegradation coupling to methane production in an enriched culture from production water of a high-temperature oil reservoir. AMB Expr. 2020, 10, 63. [Google Scholar] [CrossRef]
- Gray, N.D.; Sherry, A.; Grant, R.J.; Rowan, A.K.; Hubert, C.R.J.; Callbeck, C.M.; Aitken, C.M.; Jones, D.M.; Adams, J.J.; Larter, S.R.; et al. The quantitative significance of Syntrophaceae and syntrophic partnerships in methanogenic degradation of crude oil alkanes. Environ. Microbiol. 2011, 13, 2957–2975. [Google Scholar] [CrossRef] [PubMed]
- Mbadinga, S.M.; Li, K.; Zhou, L.; Wang, L.-Y.; Yang, S.-Z.; Liu, J.-F.; Gu, J.-D.; Mu, B.-Z. Analysis of alkane-dependent methanogenic community derived from production water of a high-temperature petroleum reservoir. Appl. Microbiol. Biotechnol. 2012, 96, 531–542. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Brown, D.; Chatterjee, I.; Dong, X.; Ramos-Padron, E.; Wilson, S.; Bordenave, S.; Caffrey, S.M.; Gieg, L.M.; Sensen, C.W.; et al. Microbial community and potential functional gene diversity involved in anaerobic hydrocarbon degradation and methanogenesis in an oil sands tailings pond. Genome 2013, 56, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Berdugo-Clavijo, C.; Gieg, L.M. Conversion of crude oil to methane by a microbial consortium enriched from oil reservoir production waters. Front. Microbiol. 2014, 5, 197. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Wang, L.-Y.; Mbadinga, S.M.; Liu, J.-F.; Yang, S.-Z.; Gu, J.-D.; Mu, B.-Z. Anaerolineaceae and Methanosaeta turned to be the dominant microorganisms in alkanes-dependent methanogenic culture after long-term of incubation. AMB Expr. 2015, 5, 37. [Google Scholar] [CrossRef]
- Tan, B.; Fowler, S.J.; Abu Laban, N.; Dong, X.; Sensen, C.W.; Foght, J.M.; Gieg, L.M. Comparative analysis of metagenomes from three methanogenic hydrocarbon-degrading enrichment cultures with 41 environmental samples. ISME J. 2015, 9, 2028–2045. [Google Scholar] [CrossRef]
- Fowler, S.J.; Toth, C.R.A.; Gieg, L.M. Community structure in methanogenic enrichments provides insight into syntrophic interactions in hydrocarbon-impacted environments. Front. Microbiol. 2016, 7, 562. [Google Scholar] [CrossRef]
- Davidova, I.A.; Marks, C.R.; Suflita, J.M. Anaerobic hydrocarbon-degrading Deltaproteobacteria. In Taxonomy, Genomics and Ecophysiology of Hydrocarbon-Degrading Microbes, Handbook of Hydrocarbon and Lipid Microbiology; McGenity, J., Ed.; Springer International Publishing AG: Berlin/Heidelberg, Germany, 2018; pp. 1–38. [Google Scholar] [CrossRef]
- Liu, Y.F.; Qi, Z.Z.; Shou, L.B.; Liu, J.-F.; Yang, S.-Z.; Gu, J.-D.; Mu, B.-Z. Anaerobic hydrocarbon degradation in candidate phylum ‘Atribacteria’ (JS1) inferred from genomics. ISME J. 2019, 13, 2377–2390. [Google Scholar] [CrossRef]
- Borrel, G.; Adam, P.S.; McKay, L.J.; Chen, L.-X.; Sierra-García, I.N.; Sieber, C.M.K.; Letourneur, Q.; Ghozlane, A.; Andersen, G.L.; Li, W.J.; et al. Wide diversity of methane and short-chain alkane metabolisms in uncultured archaea. Nat. Microbiol. 2019, 4, 603–613. [Google Scholar] [CrossRef]
- Laso-Pérez, R.; Hahn, C.; Van Vliet, D.M.; Tegetmeyer, H.E.; Schubotz, F.; Smit, N.T.; Pape, T.; Sahling, H.; Bohrmann, G.; Boetius, A.; et al. Anaerobic degradation of non-methane alkanes by ‘Candidatus Methanoliparia’ in hydrocarbon seeps of the Gulf of Mexico. mBio 2019, 10, e01814–e01819. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, C.-J.; Liu, P.-F.; Laso-Pérez, R.; Yang, L.; Bai, L.P.; Li, J.; Yang, M.; Lin, J.-Z.; Wang, W.-D.; et al. Non-syntrophic methanogenic hydrocarbon degradation by an archaeal species. Nature 2022, 601, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Chen, J.; Liu, Z.L.; Shou, L.B.; Lin, D.D.; Zhou, L.; Yang, S.Z.; Liu, J.F.; Li, W.; Gu, J.D.; et al. Anaerobic degradation of paraffins by thermophilic Actinobacteria under methanogenic conditions. Environ. Sci. Technol. 2020, 54, 10610–10620. [Google Scholar] [CrossRef] [PubMed]
- Nazina, T.N.; Grigoryan, A.A.; Xue, Y.-F.; Sokolova, D.S.H.; Novikova, E.V.; Tourova, T.P.; Poltaraus, A.B.; Belyaev, S.S.; Ivanov, M.V. Phylogenetic diversity of aerobic saprotrophic bacteria isolated from the Daqing Oil Field. Microbiology 2002, 71, 91–97. [Google Scholar] [CrossRef]
- An, B.A.; Shen, Y.; Voordouw, G. Control of sulfide production in high salinity Bakken Shale oil reservoirs by halophilic bacteria reducing nitrate to nitrite. Front. Microbiol. 2017, 8, 1164. [Google Scholar] [CrossRef]
- Nazina, T.N.; Sokolova, D.S.H.; Babich, T.L.; Semenova, E.M.; Ershov, A.P.; Bidzhieva, S.K.; Borzenkov, I.A.; Poltaraus, A.B.; Khisametdinov, M.R.; Tourova, T.P. Microorganisms of low-temperature heavy oil reservoirs (Russia) and their possible application for enhanced oil recovery. Microbiology 2017, 86, 773–785. [Google Scholar] [CrossRef]
- Pfennig, N.; Lippert, K.D. Über das vitamin B12—Bedürfnis phototropher Schweferelbakterien. Arch. Microbiol. 1966, 55, 245–256. [Google Scholar]
- Bergmann, G.T.; Bates, S.T.; Eilers, K.G.; Lauber, C.L.; Caparoso, J.G.; Walters, W.A.; Knight, R.; Fierer, N. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol. Biochem. 2011, 43, 1450–1455. [Google Scholar] [CrossRef]
- Nazina, T.N.; Bidzhieva, S.K.; Grouzdev, D.S.; Sokolova, D.S.; Tourova, T.P.; Parshina, S.N.; Avtukh, A.N.; Poltaraus, A.B.; Talybly, A.K. Soehngenia longivitae sp. nov., a fermenting bacterium isolated from a petroleum reservoir in Azerbaijan, and emended description of the genus Soehngenia. Microorganisms 2020, 8, 1967. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv 2016, 15, 081257. [Google Scholar] [CrossRef]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Reasoner, D.J.; Geldreich, E.E. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 1985, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K. Preparation of genomic DNA from bacteria. Curr. Protoc. Mol. Biol. 2001, 56, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley& Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Hoang, D.T.; Vinh, L.S.; Flouri, T.; Stamatakis, A.; von Haeseler, A.; Minh, B.Q. MPBoot: Fast phylogenetic maximum parsimony tree inference and bootstrap approximation. BMC Evol. Biol. 2018, 18, 11. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 2013, 14, 60. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Delmont, T.O.; Eren, E.M. Linking pangenomes and metagenomes: The Prochlorococcus metapangenome. PeerJ 2018, 6, e4320. [Google Scholar] [CrossRef] [PubMed]
- Eren, A.M.; Esen, O.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ’omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef] [PubMed]
- Trüper, H.G.; Schlegel, H.G. Sulfur metabolism in Thiorhodaceae. I. Quantitative measurements on growing cells of Chromatium okenii. Antonie van Leeuwenhoek 1964, 30, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.M.; Grouzdev, D.S.; Tourova, T.P.; Nazina, T.N. Physiology and genomic characteristics of Geotoga petraea, a bacterium isolated from a low-temperature oil reservoir (Russia). Microbiology 2019, 88, 662–670. [Google Scholar] [CrossRef]
- Bidzhieva, S.K.; Sokolova, D.S.; Grouzdev, D.S.; Kostrikina, N.A.; Poltaraus, A.B.; Tourova, T.P.; Shcherbakova, V.A.; Troshina, O.Y.; Nazina, T.N. Sphaerochaeta halotolerans sp. nov.; a novel spherical halotolerant spirochete from a Russian heavy oil reservoir, emended description of the genus Sphaerochaeta, reclassification of Sphaerochaeta coccoides to a new genus Parasphaerochaeta gen. nov. as Parasphaerochaeta coccoides comb. nov. and proposal of Sphaerochaetaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 4748–4759. [Google Scholar] [CrossRef]
- Collins, M.D.; Jones, D. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol. Rev. 1981, 45, 316–354. [Google Scholar] [CrossRef]
- Ianutsevich, E.A.; Danilova, O.A.; Groza, N.V.; Kotlova, E.R.; Tereshina, V.M. Heat shock response of thermophilic fungi: Membrane lipids and soluble carbohydrates under elevated temperatures. Microbiology 2016, 162, 989–999. [Google Scholar] [CrossRef]
- Nichols, B.W. Separation of the lipids of photosynthetic tissues: Improvements in analysis by thin-layer chromatography. Biochim. Biophys. Acta 1963, 70, 417–422. [Google Scholar] [CrossRef]
- Benning, C.; Huang, Z.H.; Gage, D.A. Accumulation of a novel glycolipid and a betaine lipid in cells of Rhodobacter sphaeroides grown under phosphate limitation. Arch. Biochem. Biophys. 1995, 317, 103–111. [Google Scholar] [CrossRef]
- Vaskovsky, V.E.; Kostetsky, E.X.; Vasendin, J.M. A universal reagent for phospholipids analysis. J. Chromatogr. 1975, 111, 129–141. [Google Scholar] [CrossRef]
- Kates, M. Techniques of lipidology: Isolation, analysis and identification of lipids. In Laboratory Techniques in Biochemistry and Molecular Biology; Work, T.S., Work, E., Eds.; North-Holland Publishing: Amsterdam, The Netherlands, 1972; Volume 3, pp. 267–610. [Google Scholar] [CrossRef]
- Potekhina, N.V.; Streshinskaya, G.M.; Tul’skaya, E.M.; Shashkov, A.S. Cell wall teichoic acids in the taxonomy and characterization of Gram-positive bacteria. In Methods in Microbiology; Rainey, F.A., Oren, A., Eds.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2011; Volume 38, Chapter 6; pp. 132–164. [Google Scholar] [CrossRef]
- Trofimova, L.; Ksenofontov, A.L.; Mkrtchyan, G.; Graf, A.; Baratova, L.; Bunik, V. Quantification of rat brain amino acids: Analysis of the data consistency. Curr. Anal. Chem. 2016, 12, 349–356. [Google Scholar] [CrossRef]
- Borzenkov, I.A.; Milekhina, E.I.; Gotoeva, M.T.; Rozanova, E.P.; Belyaev, S.S. The properties of hydrocarbon-oxidizing bacteria isolated from the oilfields of Tatarstan, Western Siberia, and Vietnam. Microbiology 2006, 75, 66–72. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 005056. [Google Scholar] [CrossRef]
- Breitenstein, A.; Wiegel, J.; Haertig, C.; Weiss, N.; Andreesen, J.R.; Lechner, U. Reclassification of Clostridium hydroxybenzoicum as Sedimentibacter hydroxybenzoicus gen. nov.; comb. nov.; and description of Sedimentibacter saalensis sp. nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Kleinsteuber, S.; Schleinitz, K.; Vogt, C. Key players and team play: Anaerobic microbial communities in hydrocarbon-contaminated aquifers. Appl. Microbiol. Biotechnol. 2012, 94, 851–873. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, J.; Lin, J.; Zhao, J.; Xu, T.; Yang, Q.; Zhao, J.; Zhao, Z.; Song, X. Changes in the microbial community diversity of oil exploitation. Genes 2019, 10, 556. [Google Scholar] [CrossRef]
- Imachi, H.; Sakai, S.; Kubota, T.; Miyazaki, M.; Saito, Y.; Takai, K. Sedimentibacter acidaminivorans sp. nov.; an anaerobic, amino-acid-utilizing bacterium isolated from marine subsurface sediment. Int. J. Syst. Evol. Microbiol. 2016, 66, 1293–1300. [Google Scholar] [CrossRef]
- Yi, H.; Schumann, P.; Chun, J. Demequina aestuarii gen. nov.; sp. nov.; a novel actinomycete of the suborder Micrococcineae, and reclassification of Cellulomonas fermentans Bagnara et al. 1985 as Actinotalea fermentans gen. nov.; comb. nov. Int. J. Syst. Evol. Microbiol. 2007, 57, 151–156. [Google Scholar] [CrossRef]
- Li, Y.; Chen, F.; Dong, K.; Wang, G. Actinotalea ferrariae sp. nov.; isolated from iron mine, and emended description of the genus Actinotalea. Int. J. Syst. Evol. Microbiol. 2013, 63, 3398–3403. [Google Scholar] [CrossRef]
- Jin, L.; Ko, S.-R.; Lee, C.S.; Ahn, C.-Y.; Lee, J.-S.; Lee, K.C.; Oh, H.-M.; Lee, H.-G. Actinotalea caeni sp. nov.; isolated from a sludge sample of a biofilm reactor. Int. J. Syst. Evol. Microbiol. 2017, 67, 1595–1599. [Google Scholar] [CrossRef]
- Yan, Z.-F.; Lin, P.; Li, C.-T.; Kook, M.; Yi, T.-H. Actinotalea solisilvae sp. nov.; isolated from forest soil and emended description of the genus Actinotalea. Int. J. Syst. Evol. Microbiol. 2018, 68, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Salam, N.; Jiao, J.Y.; Zhang, X.T.; Li, W.J. Update on the classification of higher ranks in the phylum Actinobacteria. Int. J. Syst. Evol. Microbiol. 2020, 70, 1331–1355, Erratum in Int. J. Syst. Evol. Microbiol. 2020, 70, 2958. [Google Scholar] [CrossRef] [PubMed]
- Shlimon, A.G.; Friedrich, M.W.; Niemann, H.; Ramsing, N.B.; Finster, K. Methanobacterium aarhusense sp. nov.; a novel methanogen isolated from a marine sediment (Aarhus Bay, Denmark). Int. J. Syst. Evol. Microbiol. 2004, 54, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Oh, H.-S.; Park, S.-C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef]
- Shi, Z.; Luo, G.; Wang, G. Cellulomonas carbonis sp. nov.; isolated from coal mine soil. Int. J. Syst. Evol. Microbiol. 2012, 62, 2004–2010. [Google Scholar] [CrossRef]
- Jones, B.E.; Grant, W.D.; Duckworth, A.W.; Schumann, P.; Weiss, N.; Stackebrandt, E. Cellulomonas bogoriensis sp. nov.; an alkaliphilic cellulomonad. Int. J. Syst. Evol. Microbiol. 2005, 55, 1711–1714. [Google Scholar] [CrossRef]
- Felsenstein, J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Zool. 1978, 27, 401–410. [Google Scholar] [CrossRef]
- Bagnara, C.; Toci, R.; Gaudin, C.; Belaich, J.P. Isolation and characterization of a cellulolytic microorganism, Cellulomonas fermentans sp. nov. Int. J. Syst. Bacteriol. 1985, 35, 502–507. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Cho, H.; Hamada, M.; Ahn, J.-H.; Weon, H.-Y.; Joa, J.-H.; Suzuki, K.-I.; Kwon, S.-W.; Kim, S.-J. Pseudactinotalea terrae gen. nov., sp. nov., isolated from greenhouse soil, and reclassification of Actinotalea suaedae as Pseudactinotalea suaedae comb. nov. Int. J. Syst. Evol. Microbiol. 2017, 67, 704–709. [Google Scholar] [CrossRef]
- Tang, S.-K.; Zhi, X.-Y.; Wang, Y.; Wu, J.-Y.; Lee, J.-C.; Kim, C.-J.; Lou, K.; Xu, L.-H.; Li, W.-J. Haloactinobacterium album gen. nov., sp. nov., a halophilic actinobacterium, and proposal of Ruaniaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2010, 60, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Nazina, T.N.; Shestakova, N.M.; Ivoilov, V.S.; Kostrukova, N.K.; Belyaev, S.S.; Ivanov, M.V. Radiotracer assay of microbial processes in petroleum reservoirs. Adv. Biotech. Microbiol. 2017, 2, 555591. [Google Scholar] [CrossRef]
- Cheng, L.; Shi, S.B.; Yang, L.; Zhang, Y.; Dolfing, J.; Sun, Y.-G.; Liu, L.-Y.; Li, Q.; Tu, B.; Dai, L.-R.; et al. Preferential degradation of long-chain alkyl substituted hydrocarbons in heavy oil under methanogenic conditions. Org. Geochem. 2019, 138, 103927. [Google Scholar] [CrossRef]
- Martirani-Von Abercron, S.-M.; Marin, P.; Solsona-Ferraz, M.; Castaňeda-Cataňa, M.A.; Marques, S. Naphthalene biodegradation under oxygen-limiting conditions: Community dynamics and the relevance of biofilm-forming capacity. Microb. Biotechnol. 2017, 10, 1781–1796. [Google Scholar] [CrossRef]
- Radwan, S.S.; Al-Mailem, D.M.; Kansour, M.K. Bioaugmentation failed to enhance oil bioremediation in three soil samples from three different continents. Sci. Rep. 2019, 9, 19508. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Zhang, X.; Zhao, X.; Sun, Y.; Weng, L.; Li, Y. Long-term effect of biochar amendment on the biodegradation of petroleum hydrocarbons in soil microbial fuel cells. Sci. Total. Environ. 2019, 651, 796–806. [Google Scholar] [CrossRef]
- Vick, S.H.W.; Greenfield, P.; Tetu, S.G.; Midgley, D.J.; Paulsen, I.T. Genomic and phenotypic insights point to diverse ecological strategies by facultative anaerobes obtained from subsurface coal seams. Sci. Rep. 2019, 9, 16186. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | HO-Ch2T | A. ferrariae CF5-4T | A. fermentans MT [72,82] | A. solisilvae THG-T121T [75] | A. carbonis T26T [79] | A. bogoriensis 69B4T [80] |
|---|---|---|---|---|---|---|
| Motility | + | – | – | – | + | + |
| Temperature range (optimum), °C | 10–40 (28) | 4–40 (28) | 10–40 (30–37) | 10–40 (28–30) | 4–45 (28) | 20–37 (30–37) |
| pH range (optimum) | 6–9 (7.5–8.5) | 6–8 (7) | 6–11 a | 6–8 (7) | 6.0–10 (7) | 6.0–10.5 (9–10) |
| NaCl range (optimum), %, w/v | 0–4.5 (1) | 0–7 (3) | 0–7 (2–4) | 0–4 (1) | 0–7 | 0–8 |
| Catalase | + | + | – | + | + | + |
| Oxidase | – | – | + | + | – | |
| Hydrolysis of: | ||||||
| Chitin | + | – | – a | – a | ND | ND |
| Gelatin | – | + | + | + | + | + |
| Nitrate reduction to NO2− | + | + | + | – | + | – |
| Activity and assimilation of (API 20E or API 50CH) | ||||||
| Acetate | + | + | + | + | – | – |
| N-Acetylglucosamine | + | – | – a | + | + | – |
| Gluconate | – | – | – | + | + | – |
| Lactate | – | – | – | + | – | – |
| Propionate | – | – | – | + | + | ND |
| D-Galactose | + | + | + | ND | + | – |
| D-Glucose | + | + | + | – | + | + |
| Lactose | – | – | + | ND | + | – |
| Melibiose | – | – | – | + | + | – |
| Raffinose | W | – | + | ND | W | – |
| D-Ribose | – | + | – | + | – | – |
| L-Rhamnose | – | – | – | + | – | + |
| Inositol | – | – | + a | + | – | – |
| Glycerol | W | + | – | ND | ND | – |
| D-Mannitol | + | + | + | – | – | – |
| D-Sorbitol | – | – | + a | + | + | – |
| Enzyme activity: (API ZYM) | ||||||
| Acid phosphatase | – | + | – a | + | + | ND |
| Alkaline phosphatase | – | + | – a | + | – | – |
| Esterase lipase (C8) | + | + | – a | + | – | + |
| Valine arylamidase | + | + | – a | – | – | ND |
| Cystine arylamidase | + | + | – a | + | – | ND |
| N-acetyl-β-glucosaminidase | + | + | – a | + | + | ND |
| α-Galactosidase | + | + | – a | – | + | ND |
| β-Galactosidase | + | – | + | + | + | ND |
| Lipase (C14) | + | – | –a | – | – | ND |
| Major fatty acids | ai-C15:0, C14: 0, C16:0, C15:0 | ai-C15:0, C16:0, C15:0 | C14:0, ai-C15:0, C16:0 | ai-C15:0, ai-C15:1 A, C16:0 | ai -C15:0, ai -C15:1 A, C16:0 | ai -C15:0, C16:0, C14: 0 |
| Peptidoglycan 1 | L-Orn (Lys)-D-Ser-D-Glu | L-Orn–D-Ser–D-Asp | L-Orn-D-Ser-L-Aspb | L–Orn–D–Ser–L–Asp | L-Orn– D-Glu | L-Orn–D-Asp |
| Cell–wall sugars 2 | Rha, Gal, Man, Glc | Rha, Fuc, Man, Galb | Rha, Rib, Glc | Rha, Rib, Man, Glc | Rha, Gal, Xyl, Ino | ND |
| Major polar lipids 3 | DPG, GL, PGL, PC, PG, PL | DPG, PC, PGL, PG, GL, PL | DPG, PGc | DPG, PG, PI, PIM, PL, GL, L | DPG, PG, PIM, PI, PL, PGL | DPG, PG, PIM, PI, PL, PGLb |
| Quinones | MK-9(H4), MK-9(H6), MK-8(H4) | MK–10(H4) | MK–10(H4), MK–9(H4), MK–8(H4) | MK–10(H4) | MK–9(H4) | MK-9(H4) |
| Isolation source | Methanogenic enrichment from the oil field | Iron mining powder | Methanogenic enrichment from dumping ground | Forest soil | Coal mine soil | Sediment of the littoral zone of the lake |
| Attribute | HO-Ch2T | A. ferrariae CF5-4 T | A. solisilvae KACC 19191T | A. fermentans DSM 3133T | A. carbonis T26T | A. bogoriensis 69B4T |
|---|---|---|---|---|---|---|
| Genome size (bp) | 4,027,363 | 3,987,077 | 4,296,322 | 3,737,518 | 3,904,075 | 3,183,361 |
| G+C content (%) | 73.4 | 73.8 | 75.2 | 74.1 | 74.2 | 72.2 |
| DNA scaffolds | 28 | 626 | 41 | 22 | 73 | 524 |
| Total genes | 3678 | 3907 | 3957 | 3450 | 3527 | 3241 |
| Protein coding genes | 3589 | 3697 | 3851 | 3369 | 3433 | 3015 |
| Number of tRNA | 45 | 45 | 46 | 45 | 45 | 45 |
| Number of rRNA | 3 | 8 | 5 | 3 | 3 | 4 |
| Pseudo genes | 38 | 154 | 51 | 30 | 43 | 174 |
| ANI (%) | 100 | 80.2 | 82.0 | 80.1 | 81.0 | 79.8 |
| dDDH (%) | 100 | 21.4 | 22.2 | 20.5 | 21.5 | 20.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semenova, E.M.; Grouzdev, D.S.; Sokolova, D.S.; Tourova, T.P.; Poltaraus, A.B.; Potekhina, N.V.; Shishina, P.N.; Bolshakova, M.A.; Avtukh, A.N.; Ianutsevich, E.A.; et al. Physiological and Genomic Characterization of Actinotalea subterranea sp. nov. from Oil-Degrading Methanogenic Enrichment and Reclassification of the Family Actinotaleaceae. Microorganisms 2022, 10, 378. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10020378
Semenova EM, Grouzdev DS, Sokolova DS, Tourova TP, Poltaraus AB, Potekhina NV, Shishina PN, Bolshakova MA, Avtukh AN, Ianutsevich EA, et al. Physiological and Genomic Characterization of Actinotalea subterranea sp. nov. from Oil-Degrading Methanogenic Enrichment and Reclassification of the Family Actinotaleaceae. Microorganisms. 2022; 10(2):378. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10020378
Chicago/Turabian StyleSemenova, Ekaterina M., Denis S. Grouzdev, Diyana S. Sokolova, Tatiyana P. Tourova, Andrey B. Poltaraus, Natalia V. Potekhina, Polina N. Shishina, Maria A. Bolshakova, Alexander N. Avtukh, Elena A. Ianutsevich, and et al. 2022. "Physiological and Genomic Characterization of Actinotalea subterranea sp. nov. from Oil-Degrading Methanogenic Enrichment and Reclassification of the Family Actinotaleaceae" Microorganisms 10, no. 2: 378. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10020378