
Cutinase ACut2 from Blastobotrysraffinosifermentans for the Selective Desymmetrization of the Symmetric Diester Diethyl Adipate to the Monoester Monoethyl Adipate

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Production of ACut2-6hp Enzyme Preparations
2.3. Assay for Determination of ACut2-6hp Activity
2.4. DEA Hydrolysis by ACut2-6hp
2.5. DEA, MEA and AA Extraction from the Solution and Analysis
2.6. Determination of the Kinetic Constants of ACut2-6hp Supernatant to DEA and MEA
2.7. Immobilization of ACut2-6hp on Purolite Carriers
2.8. Up-Scaling of DEA Hydrolysis
3. Results
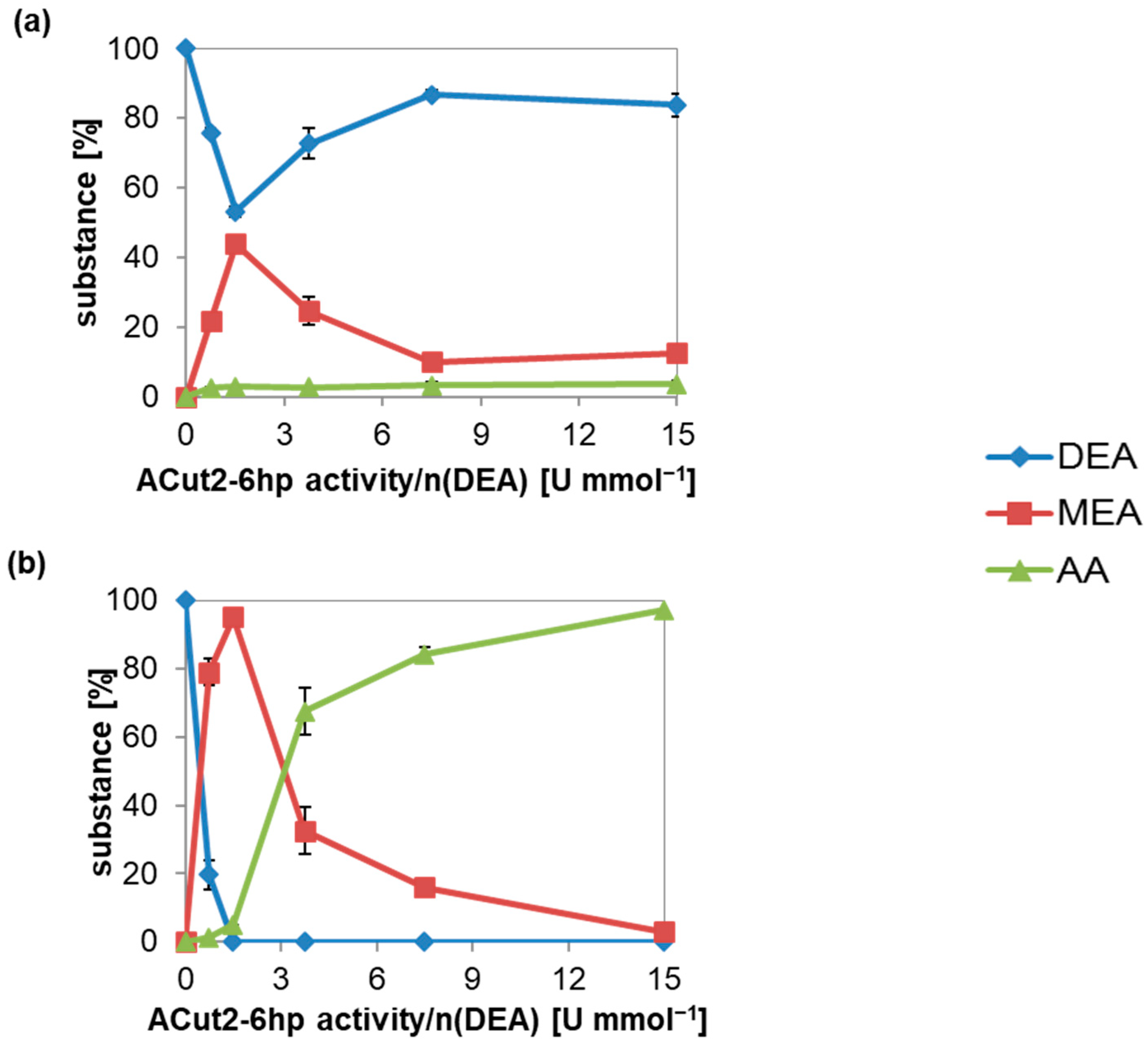
3.1. DEA Hydrolysis by Culture Supernatant of B. raffinosifermentans Strain G1212/YRC102-ACut2-6H
3.2. Diacid Reduction at DEA Hydrolysis by Culture Supernatant Containing Cutinase ACut2-6hp from B. raffinosifermentans
3.3. Kinetic Constant KM of ACut2-6hp Culture Supernatant for the Hydrolysis of DEA and MEA
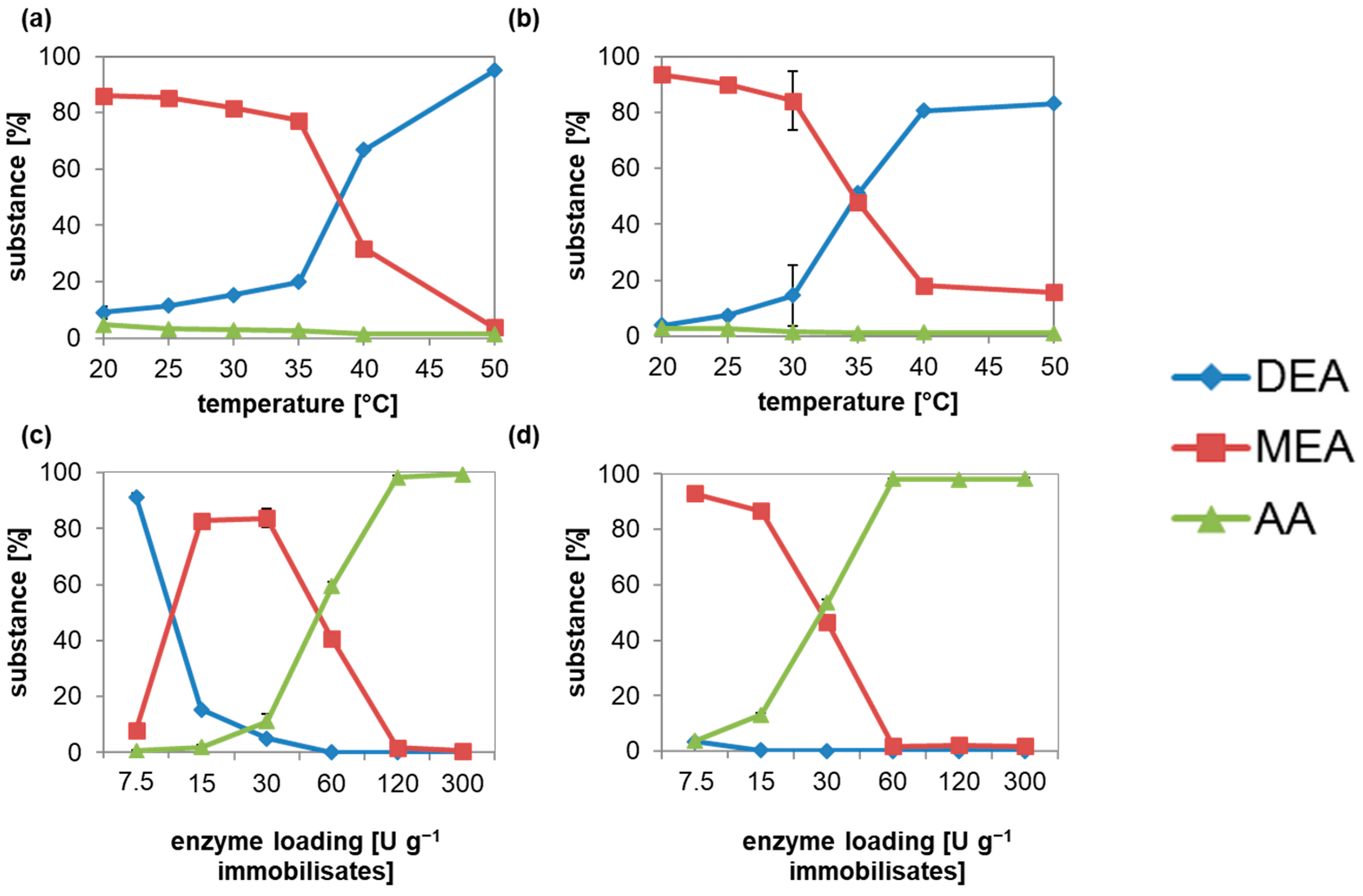
3.4. ACut2-6hp Immobilization by Adsorption and Covalent Bonding to Carriers
3.5. Decreased Diacid Formation at DEA Hydrolysis by Immobilized ACut2-6hp Culture Supernatant
3.6. Up-Scaling of DEA Hydrolysis by Culture Supernatant Containing Cutinase ACut2-6hp or Immobilized Cutinase ACut2-6hp from B. raffinosifermentans at Constant pH
3.7. Determination of Substrate Spectrum of ACut2-6hp Culture Supernatant
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jin, X.; Li, M.; Yin, L.; Zhou, J.; Zhang, Z.; Lv, H. Tyroservatide-TPGS-paclitaxel liposomes: Tyroservatide as a targeting ligand for improving breast cancer treatment. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hao, J.; Xiao, Y. Synthesis and in vitro antitumor activities of lupeol dicarboxylic acid monoester derivatives. Arch. Pharm. Res. 2013, 36, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Erythropel, H.C.; Brown, T.; Maric, M.; Nicell, J.A.; Cooper, D.G.; Leask, R.L. Designing greener plasticizers: Effects of alkyl chain length and branching on the biodegradation of maleate based plasticizers. Chemosphere 2015, 134, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Korsager, S.; Nielsen, D.U.; Taaning, R.H.; Skrydstrup, T. Access to β-Keto Esters by Palladium-Catalyzed Carbonylative Coupling of Aryl Halides with Monoester Potassium Malonates. Angew. Chem. Int. Ed. 2013, 52, 9763–9766. [Google Scholar] [CrossRef]
- Schuster, L.; Rothmund, L.; He, X.; Van Landuyt, K.L.; Schweikl, H.; Hellwig, E.; Carell, T.; Hickel, R.; Reichl, F.X.; Högg, C. Effect of Opalescence® bleaching gels on the elution of dental composite components. Dent. Mater. 2015, 31, 745–757. [Google Scholar] [CrossRef]
- Yadav, A.; Mathur, R.; Samim, M.; Lomash, V.; Kushwaha, P.; Pathak, U.; Babbar, A.; Flora, S.; Mishra, A.; Parshad Kaushik, M. Nanoencapsulation of DMSA monoester for better therapeutic efficacy of the chelating agent against arsenic toxicity. Nanomedicine 2014, 9, 465–481. [Google Scholar] [CrossRef]
- Rajagopalan, A.; Kroutil, W. Biocatalytic reactions: Selected highlights. Mater. Today 2011, 14, 144–152. [Google Scholar] [CrossRef]
- Cabrera, Z.; Palomo, J.M.; Fernandez-Lorente, G.; Fernandez-Lafuente, R.; Guisan, J.M. Partial and enantioselective hydrolysis of diethyl phenylmalonate by immobilized preparations of lipase from Thermomyces lanuginose. Enzym. Microb. Technol. 2007, 40, 1280–1285. [Google Scholar] [CrossRef]
- Tan, J.N.; Dou, Y. Deep eutectic solvents for biocatalytic transformations: Focused lipase-catalyzed organic reactions. Appl. Microbiol. Biotechnol. 2020, 104, 1481–1496. [Google Scholar] [CrossRef]
- Aldabbagh, F. Acid halides. In Comprehensive Organic Functional Group Transformations II; Katritzky, A.R., Taylor, R.J.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 5, pp. 1–17. [Google Scholar] [CrossRef]
- Zhang, X.; Breslav, M.; Grimm, J.; Guan, K.; Huang, A.; Liu, F.; Maryanoff, C.A.; Palmer, D.; Patel, M.; Qian, Y.; et al. A new procedure for preparation of carboxylic acid hydrazides. J. Org. Chem. 2002, 67, 9471–9474. [Google Scholar] [CrossRef]
- Lanigan, R.M.; Starkov, P.; Sheppard, T.D. Direct Synthesis of Amides from Carboxylic Acids and Amines Using B(OCH2CF3)3. J. Org. Chem. 2013, 78, 4512–4523. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Yanagi, R.; Oe, Y. Recent advances in the synthesis of carboxylic acid esters. In Carboxylic Acid-Key Role in Life Sciences; Badea, G.-I., Radu, G.L., Eds.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Gopinath, P.; Watanabe, T.; Shibasaki, M. Studies on Catalytic Enantioselective Total Synthesis of Caprazamycin B: Construction of the Western Zone. J. Org. Chem. 2012, 77, 9260–9267. [Google Scholar] [CrossRef] [PubMed]
- Acker, D.S.; Park, B. Process of Preparing Alpha-Lipoic Acid Using Dichlorooctanoate and Metal Disulfide. U.S. Patent US2792406A, 14 May 1957. [Google Scholar] [CrossRef] [Green Version]
- Ye, W.; Zhao, Y.; Na, R.; Li, F.; Mei, Q.; Zhao, M.; Zhou, S. Actively Targeted Delivery of Doxorubicin to Bone Metastases by a pH-Sensitive Conjugation. J. Pharm. Sci. 2015, 104, 2293–2303. [Google Scholar] [CrossRef]
- Stieber, F.; Waldmann, H. Development of new acid-functionalized resins for combinatorial synthesis on solid supports. Chem. Commun. 2002, 2002, 1748–1749. [Google Scholar] [CrossRef]
- Pon, R.T.; Yu, S. Tandem oligonucleotide synthesis using linker (First BaseTM) phosphoramidites. Nucleic Acids Res. 2005, 33, 1940–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogl, O. Polymers With Functional Groups. Pure Appl. Chem. 1979, 51, 2409–2419. [Google Scholar] [CrossRef]
- Escamilla, G.H. Dendritic bolaamphiphiles and related molecules. In Advances in Dendritic Macromolecules; Newkome, G.R., Ed.; JAI Press: Stamford, CT, USA, 1995; Volume 2, pp. 157–190. [Google Scholar] [CrossRef]
- Tejero, R.; Gutiérrez, B.; López, D.; López-Fabal, F.; Gómez-Garcés, J.L.; Muñoz-Bonilla, A.; Fernández-García, M. Tailoring Macromolecular Structure of Cationic Polymers towards Efficient Contact Active Antimicrobial Surfaces. Polymers 2018, 10, 241. [Google Scholar] [CrossRef] [Green Version]
- Janović, Z.; Jukić, A.; Vogl, O. Spacer groups in macromolecular structures. Polimeri 2010, 31, 14–21. [Google Scholar]
- Anyanwu, U.K.; Venkataraman, D. Effect of spacers on the activity of soluble polymer supported catalysts for the asymmetric addition of diethylzinc to aldehydes. Tetrahedron Lett. 2003, 44, 6445–6448. [Google Scholar] [CrossRef]
- Guo, Z.; Wong, M.K.Y.; Hickey, M.R.; Patel, B.P.; Qian, X.; Goswami, A. Enzyme-Catalyzed Hydrolysis of Bicycloheptane and Cyclobutene Diesters to Monoesters. Org. Process Res. Dev. 2014, 18, 774–780. [Google Scholar] [CrossRef]
- Süss, P.; Illner, S.; von Langermann, J.; Borchert, S.; Bornscheuer, U.T.; Wardenga, R.; Kragl, U. Scale-Up of a Recombinant Pig Liver Esterase-Catalyzed Desymmetrization of Dimethyl Cyclohex-4-ene-cis-1,2-dicarboxylate. Org. Process Res. Dev. 2014, 18, 897–903. [Google Scholar] [CrossRef]
- Cabrera, Z.; Palomo, J.M. Enantioselective desymmetrization of prochiral diesters catalyzed by immobilized Rhizopus oryzae lipase. Tetrahedron Asymm. 2011, 22, 2080–2084. [Google Scholar] [CrossRef]
- Nietz, D.; Bode, R.; Kunze, G.; Rauter, M. A new lipase (Alip2) with high potential for enzymatic hydrolysis of the diester diethyladipate to the monoester monoethyladipate. Enzym. Microb. Technol. 2022, 153, 109898. [Google Scholar] [CrossRef]
- Ehlert, J.; Kronemann, J.; Zumbrägel, N.; Preller, M. Lipase-catalyzed chemoselective ester hydrolysis of biomimetically coupled aryls for the synthesis of unsymmetric biphenyl Esters. Molecules 2019, 24, 4272. [Google Scholar] [CrossRef] [Green Version]
- Chênevert, R.; Ngatcha, B.T.; Rose, Y.S.; Goupil, D. Regio- and enantioselectivity of the enzyme catalysed hydrolysis of citric acid derivatives. Tetrahedron Asymm. 1998, 9, 2827–2831. [Google Scholar] [CrossRef]
- Torres, C.; Otero, C. Part I. Enzymatic synthesis of lactate and glycolate esters of fatty alcohols. Enzym. Microb. Technol. 1999, 25, 745–752. [Google Scholar] [CrossRef]
- Bianchi, D.; Bortolo, R.; Bernardi, A.; Gagliardi, I.; Ingrassia, A.G.M. Selective enzymatic hydrolysis of aromatic diesters using esterase from Brevibacterium imperiale B222. Biotechnol. Lett. 1995, 17, 711–716. [Google Scholar] [CrossRef]
- Kashima, Y.; Liu, J.; Takenami, S.; Niwayama, S. Asymmetric desymmetrization of dialkyl bicyclo[2.2.1]hept-2,5-diene-2,3-dicarboxylates by a thermophilic esterase/lipase. Tetrahedron Asymm. 2002, 13, 953–956. [Google Scholar] [CrossRef]
- Goswami, A.; Kissick, T.P. Enzymatic Desymmetrization of Dimethyl Cylcohex-4-ene-cis-1,2-dicarboxylate to (1S,2R)-2-(Methoxycarbonyl)cyclohex-4-ene-1-carboxylic Acid. Org. Process Res. Dev. 2009, 13, 483–488. [Google Scholar] [CrossRef]
- Flors, V.; Miralles, C.; Cerezo, M.; González-Bosch, C.; García-Agustín, P. Effect of a Novel Chemical Mixture on Senescence Processes and Plant−Fungus Interaction in Solanaceae Plants. J. Agric. Food Chem. 2001, 49, 2569–2575. [Google Scholar] [CrossRef]
- Flors, V.; Miralles, C.; González-Bosch, C.; Carda, M.; García-Agustín, P. Three novel synthetic amides of adipic acid protect Capsicum anuum plants against the necrotrophic pathogen Alternaria solani. Physiol. Mol. Plant Pathol. 2003, 63, 151–158. [Google Scholar] [CrossRef]
- Flors, V.; Paradís, M.; García-Andrade, J.; Cerezo, M.; González-Bosch, C.; García-Agustín, P. Tolerant Behavior in Salt-Sensitive Tomato Plants can be Mimicked by Chemical Stimuli. Plant Signal Behav. 2007, 2, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicedo, B.; de la, O.; Leyva, M.; Flors, V.; Finiti, I.; del Amo, G.; Walters, D.; Real, M.D.; García-Agustín, P.; González-Bosch, C. Control of the phytopathogen Botrytis cinerea using adipic acid monoethyl ester. Arch. Microbiol. 2006, 184, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Barros, D.; Azevedo, A.M.; Cabral, J.; Fonseca, L.P. Optimization of Flavor Esters Synthesis by Fusarium solani pisi Cutinase. J. Food Biochem. 2012, 36, 275–284. [Google Scholar] [CrossRef]
- Baker, P.J.; Poultney, C.; Liu, Z.; Gross, R.; Montclar, J.K. Identification and comparison of cutinases for synthetic polyester degradation. Appl. Microbiol. Biotechnol. 2012, 93, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, F.; Litwińska, K.; Cordes, A.; Baronian, K.; Bode, R.; Schauer, F.; Kunze, G. Three New Cutinases from the Yeast Arxula adeninivorans that Are Suitable for Biotechnological Applications. Appl. Environ. Microbiol. 2015, 81, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Sanya, D.R.A.; Fouchard, F.; Nguyen, H.-V.; Kunze, G.; Neuvéglise, C.; Crutz-Le Coq, A.-M. Blastobotrys adeninivorans and B. raffinosifermentans, two sibling yeast species which accumulate lipids at elevated temperatures and from diverse sugars. Biotechnol. Biofuels 2019, 12, 154–165. [Google Scholar] [CrossRef]
- Pohanka, M. Biosensors and Bioassays Based on Lipases, Principles and Applications, a Review. Molecules 2019, 24, 616. [Google Scholar] [CrossRef] [Green Version]
- Kazlauskas, R.J. Quantitative assay of hydrolases for activity and selectivity using color changes. In Enzyme Assays; Reymond, J.-L., Ed.; Wiley-VCH: Hoboken, NJ, USA, 2005; pp. 15–39. [Google Scholar] [CrossRef]
- Sheldon, R.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef]
- Zisis, T.; Freddolino, P.L.; Turunen, P.; van Teeseling, M.C.F.; Rowan, A.E.; Blank, K.G. Interfacial Activation of Candida antarctica Lipase B: Combined Evidence from Experiment and Simulation. Biochemistry 2015, 54, 5969–5979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Galan, C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R.; Rodrigues, R.C. Potential of Different Enzyme Immobilization Strategies to Improve Enzyme Performance. Adv. Synth. Catal. 2011, 353, 2885–2904. [Google Scholar] [CrossRef]
- Kasche, V. Mechanism and yields in enzyme catalysed equilibrium and kinetically controlled synthesis of β-lactam antibiotics, peptides and other condensation products. Enzym. Microb. Technol. 1986, 8, 4–16. [Google Scholar] [CrossRef]
- Sousa, H.A.; Afonso, C.A.M.; Crespo, J.G. Kinetic study of the enantioselective hydrolysis of a meso-diester using pig liver esterase. J. Chem. Technol. Biotechnol. 2000, 75, 707–714. [Google Scholar] [CrossRef]
- Almilly, R.F.K.; Alobaidy, A.A.; Alhassani, M.H. Saponification of Diethyl Adipate with Sodium Hydroxide Using Reactive Distillation. J. Eng. 2014, 20, 20–34. [Google Scholar]
- Babler, J.H. Method of Preparing Monoesters. U.S. Patent US4314071A, 2 February 1982. [Google Scholar]
- Niwayama, S.; Cho, H. Practical large Scale Synthesis of Half-Esters of Malonic Acid. Chem. Pharm. Bull. 2009, 57, 508–510. [Google Scholar] [CrossRef] [Green Version]
- van Nuland, Y.; de Vogel, F.A.; Eggink, G.; Weusthuis, R. Expansion of the ω-oxidation system AlkBGTL of Pseudomonas putida GPo1 with AlkJ and AlkH results in exclusive mono-esterified dicarboxylic acid production in E. coli. Microb. Biotechnol. 2017, 10, 594–603. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Name and Function | Literature | |
|---|---|---|---|
| 1 |  | Building block for the synthesis of lipo-nucleoside antibiotic caprazamycin | [14] |
| 2 |  | Lipoic acid: natural antioxidant and cofactor of several enzymes | [15] |
| 3 |  | Alendronate-monoethyl adipate-(hydrazone)-doxorubicin: anti-tumor agent for active delivery of doxorubicin to bone metastasis | [16] |
| 4 |  | Acid functionalized polystyrene resin for use in solid phase chemistry | [17] |
| Substrate | pH | Indicator | Tested (Substrate) Range | KM [mM] | R2 for Regression Graph of the v-[S]-Plot |
|---|---|---|---|---|---|
| DEA | 7.2 | para-nitrophenol (pNP) | 0–15 mM | n.d. | 0.99 |
| DEA | 4.9 | bromcresol green | 0–15 mM | 4.98 | 0.95 |
| MEA | 7.2 | pNP | 0–150 mM | 67.69 | 0.99 |
| MEA | 4.9 | bromcresol green | 0–13 mM | 0.61 | 0.96 |
| Parameter | Free Enzyme * | Adsorbed Enzyme | Covalently Bound Enzyme |
|---|---|---|---|
| Yield | - | 99.2 | 97.5 |
| pH optimum | 5.0 (4.0–6.0) | 6.5 (6.0–6.5) | 6.0 (5.5–6.0) |
| Temperature optimum | 30–45 °C | 55 °C 50–55 °C | 50 °C 45–55 °C |
| Temperature stability | Not stable | 30 °C | 30 °C |
| Cycles for re-use | - | >50 | >20 |
| Free Enzyme (40 mL Buffer + 4 mL DEA) | Adsorbed Enzyme (40 mL Buffer + 4 mL DEA) | Adsorbed Enzyme (40 mL Buffer + 20 mL DEA) | Covalently Bound Enzyme (40 mL Buffer + 4 mL DEA) | |
|---|---|---|---|---|
| Product (g) | 3.4 | 3.1 | 15.6 | 2.8 |
| Product analysis | 7.3% DEA, 86.5% MEA, 6.2% AA | 98% MEA, 2% AA | 5.7% DEA, 92.3% MEA, 2% AA | 98% MEA, 2% AA |
| Isolated yield MEA (%) | 84 | 87.8 | 89.7 | 77.3 |
| Incubation time (h) | 24 | 22 | 120 | 48 |
| Space-time yield (STY) (g L−1 h−1) | 2.45 | 2.6 | 1.5 | 1.12 |
| Substrate | Monoester in % after 2 h Incubation | Monoester/Diacid in % after 24 h Incubation |
|---|---|---|
| Dicarboxylic acid ester | ||
| Diethyl malonate (3C + 2C) | 0 | 0/0 |
| Diethyl succinate (4C + 2C) | 4 | 28.5/0 |
| Dimethyl adipate (6C + 1C) | 3 | 9.6/0 |
| Diethyl adipate (6C + 2C) | 42 | 99.1/0.9 |
| Diisopropyl adipate (6C + 3C) | 11 | 96.0/0 |
| Dimethyl suberate (8C + 1C) | 29 | 93.3/1.8 |
| Dimethyl sebacate (10C + 1C) | 15 | 74.8/3.7 |
| Diethyl sebacate (10C + 2C) | 16 | 42.5/10.3 |
| Diol diester | ||
| 1,4-Diacetoxy butane (4C + 2C) | 0 | 0/0 |
| 1,6-Diacetoxyhexane (6C + 2C) | 0 | 0/0 |
| 1,10-Diacetoxy decane (10C + 2C) | 0 | 0/0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rauter, M.; Nietz, D.; Kunze, G. Cutinase ACut2 from Blastobotrysraffinosifermentans for the Selective Desymmetrization of the Symmetric Diester Diethyl Adipate to the Monoester Monoethyl Adipate. Microorganisms 2022, 10, 1316. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10071316
Rauter M, Nietz D, Kunze G. Cutinase ACut2 from Blastobotrysraffinosifermentans for the Selective Desymmetrization of the Symmetric Diester Diethyl Adipate to the Monoester Monoethyl Adipate. Microorganisms. 2022; 10(7):1316. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10071316
Chicago/Turabian StyleRauter, Marion, Daniela Nietz, and Gotthard Kunze. 2022. "Cutinase ACut2 from Blastobotrysraffinosifermentans for the Selective Desymmetrization of the Symmetric Diester Diethyl Adipate to the Monoester Monoethyl Adipate" Microorganisms 10, no. 7: 1316. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10071316