
A Pathogenic Role for FcγRI in the Immune Response against Chlamydial Respiratory Infection
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Mouse Model of C. trachomatis Respiratory Infection
2.3. IFUs Detection
2.4. Histology Analysis and Semi-Quantitative Pathological Scoring
2.5. Immunofluorescent Staining
2.6. Lung and Spleen Single Cells Preparation
2.7. Antibodies and Flow Cytometry
2.8. RNA Extraction and Quantitative Real-Time PCR (qPCR)
2.9. Statistical Analysis
3. Results
3.1. FcγRI Participates in and Regulates the Immune Response against Chlamydial Respiratory Infection
3.2. Chlamydial Respiratory Infection Induces Increased Expression Pattern of FcγRI in the Innate and Adaptive Immune System
3.3. Deficiency of FcγRI Differently Regulates the Recruitments of Innate and Adaptive Immune Cells Following C. Muridarum Infection
3.4. Deficiency of FcγRI Promotes Th1 Response against C. muridarum Infection
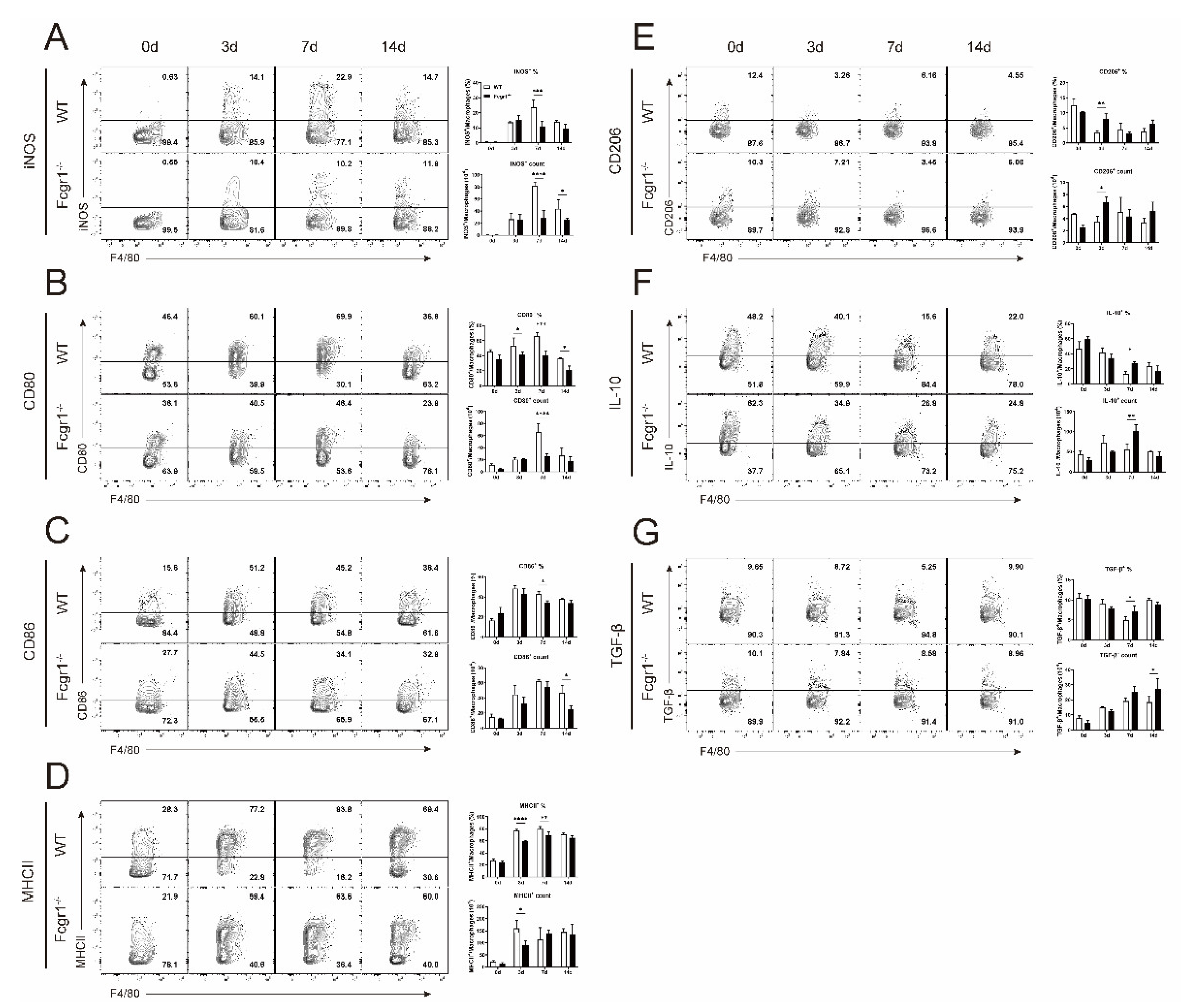
3.5. Deficiency of FcγRI Restricts the Macrophage Polarization toward M1 Phenotype during C. muridarum Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rother, M.; Teixeira da Costa, A.R.; Zietlow, R.; Meyer, T.F.; Rudel, T. Modulation of Host Cell Metabolism by Chlamydia trachomatis. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Elwell, C.; Mirrashidi, K.; Engel, J. Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol. 2016, 14, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Roan, N.R.; Starnbach, M.N. Immune-mediated control of Chlamydia infection. Cell Microbiol. 2008, 10, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.; Yu, N.; Lei, A.; Li, X.; Tan, S.; Huang, L.; Zhou, Z. Insights Into Host Cell Cytokines in Chlamydia Infection. Front. Immunol. 2021, 12, 639834. [Google Scholar] [CrossRef]
- Finethy, R.; Coers, J. Sensing the enemy, containing the threat: Cell-autonomous immunity to Chlamydia trachomatis. FEMS Microbiol. Rev. 2016, 40, 875–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, L.L.; Feilzer, K.; Caldwell, H.D. Immunity to Chlamydia trachomatis is mediated by T helper 1 cells through IFN-gamma-dependent and -independent pathways. J. Immunol. 1997, 158, 3344–3352. [Google Scholar] [PubMed]
- Bournazos, S.; Ravetch, J.V. Fcgamma Receptor Function and the Design of Vaccination Strategies. Immunity 2017, 47, 224–233. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. FcgammaRs in health and disease. Curr. Top. Microbiol. Immunol. 2011, 350, 105–125. [Google Scholar] [CrossRef]
- Bruhns, P.; Jonsson, F. Mouse and human FcR effector functions. Immunol. Rev. 2015, 268, 25–51. [Google Scholar] [CrossRef]
- Zeng, J.; Xu, Y.; Tan, L.; Zha, X.; Yang, S.; Zhang, H.; Tuo, Y.; Sun, R.; Niu, W.; Pang, G.; et al. IL-21/IL-21R Regulates the Neutrophil-Mediated Pathologic Immune Response during Chlamydial Respiratory Infection. Mediat. Inflamm. 2022, 2022, 4322092. [Google Scholar] [CrossRef]
- Wang, Y.; Jonsson, F. Expression, Role, and Regulation of Neutrophil Fcgamma Receptors. Front. Immunol. 2019, 10, 1958. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.; Gavin, A.L.; Tan, P.S.; Mottram, P.; Koentgen, F.; Hogarth, P.M. FcgammaRI-deficient mice show multiple alterations to inflammatory and immune responses. Immunity 2002, 16, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioan-Facsinay, A.; de Kimpe, S.J.; Hellwig, S.M.; van Lent, P.L.; Hofhuis, F.M.; van Ojik, H.H.; Sedlik, C.; da Silveira, S.A.; Gerber, J.; de Jong, Y.F.; et al. FcgammaRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity 2002, 16, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Su, Y.; Li, Y.; Zhang, L.; Lu, D.; Li, R.; Zhang, L.; Huang, J. The alternatively spliced porcine FcgammaRI regulated PRRSV-ADE infection and proinflammatory cytokine production. Dev. Comp. Immunol. 2019, 90, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, S.; Peng, Y.; Wang, S.; Yang, X. CD103+ lung dendritic cells (LDCs) induce stronger Th1/Th17 immunity to a bacterial lung infection than CD11b(hi) LDCs. Cell Mol. Immunol. 2018, 15, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Zha, X.; Yang, S.; Niu, W.; Tan, L.; Xu, Y.; Zeng, J.; Tang, Y.; Sun, L.; Pang, G.; Qiao, S.; et al. IL-27/IL-27R Mediates Protective Immunity against Chlamydial Infection by Suppressing Excessive Th17 Responses and Reducing Neutrophil Inflammation. J. Immunol. 2021, 206, 2160–2169. [Google Scholar] [CrossRef]
- Bai, H.; Cheng, J.; Gao, X.; Joyee, A.G.; Fan, Y.; Wang, S.; Jiao, L.; Yao, Z.; Yang, X. IL-17/Th17 promotes type 1 T cell immunity against pulmonary intracellular bacterial infection through modulating dendritic cell function. J. Immunol. 2009, 183, 5886–5895. [Google Scholar] [CrossRef] [Green Version]
- Rottenberg, M.E.; Gigliotti-Rothfuchs, A.; Wigzell, H. The role of IFN-gamma in the outcome of chlamydial infection. Curr. Opin. Immunol. 2002, 14, 444–451. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.; Ananaba, G.A.; Bolier, J.; Bowers, S.; Belay, T.; Eko, F.O.; Igietseme, J.U. Fc receptor regulation of protective immunity against Chlamydia trachomatis. Immunology 2002, 105, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Ochoa, M.C.; Cordeiro, L.; Berraondo, P.; Lopez-Diaz de Cerio, A.; Cabo, M.; Lopez-Botet, M.; Melero, I. Targeting NK-cell checkpoints for cancer immunotherapy. Curr. Opin. Immunol. 2017, 45, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yin, C.; Lum, L.G.; Simons, A.; Weiner, G.J. Bispecific antibody-activated T cells enhance NK cell-mediated antibody-dependent cellular cytotoxicity. J. Hematol. Oncol. 2021, 14, 204. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Sitnicka, E.; Bryceson, Y.T. NK cell development and function--plasticity and redundancy unleashed. Semin. Immunol. 2014, 26, 114–126. [Google Scholar] [CrossRef]
- Poznanski, S.M.; Ashkar, A.A. What Defines NK Cell Functional Fate: Phenotype or Metabolism? Front. Immunol. 2019, 10, 1414. [Google Scholar] [CrossRef]
- Shekhar, S.; Peng, Y.; Gao, X.; Joyee, A.G.; Wang, S.; Bai, H.; Zhao, L.; Yang, J.; Yang, X. NK cells modulate the lung dendritic cell-mediated Th1/Th17 immunity during intracellular bacterial infection. Eur. J. Immunol. 2015, 45, 2810–2820. [Google Scholar] [CrossRef]
- Jiao, L.; Gao, X.; Joyee, A.G.; Zhao, L.; Qiu, H.; Yang, M.; Fan, Y.; Wang, S.; Yang, X. NK cells promote type 1 T cell immunity through modulating the function of dendritic cells during intracellular bacterial infection. J. Immunol. 2011, 187, 401–411. [Google Scholar] [CrossRef]
- Zhao, L.; Li, J.; Zhou, X.; Pan, Q.; Zhao, W.; Yang, X.; Wang, H. Natural Killer Cells Regulate Pulmonary Macrophages Polarization in Host Defense Against Chlamydial Respiratory Infection. Front. Cell Infect. Microbiol. 2021, 11, 775663. [Google Scholar] [CrossRef]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef]
- Liu, Y.C.; Zou, X.B.; Chai, Y.F.; Yao, Y.M. Macrophage polarization in inflammatory diseases. Int. J. Biol. Sci. 2014, 10, 520–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Schultze, J.L.; Freeman, T.; Hume, D.A.; Latz, E. A transcriptional perspective on human macrophage biology. Semin. Immunol. 2015, 27, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. T Helper Cell Differentiation, Heterogeneity, and Plasticity. Cold Spring Harb. Perspect. Biol. 2018, 10, a030338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruterbusch, M.; Pruner, K.B.; Shehata, L.; Pepper, M. In Vivo CD4(+) T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu. Rev. Immunol. 2020, 38, 705–725. [Google Scholar] [CrossRef]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, J.; Yang, S.; Sun, R.; Tuo, Y.; Tan, L.; Zhang, H.; Zhang, Y.; Che, X.; Lu, T.; Zhang, X.; et al. A Pathogenic Role for FcγRI in the Immune Response against Chlamydial Respiratory Infection. Microorganisms 2023, 11, 39. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms11010039
Zeng J, Yang S, Sun R, Tuo Y, Tan L, Zhang H, Zhang Y, Che X, Lu T, Zhang X, et al. A Pathogenic Role for FcγRI in the Immune Response against Chlamydial Respiratory Infection. Microorganisms. 2023; 11(1):39. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms11010039
Chicago/Turabian StyleZeng, Jiajia, Shuaini Yang, Ruoyuan Sun, Yuqing Tuo, Lu Tan, Hong Zhang, Yongci Zhang, Xuchun Che, Tingsha Lu, Xuejun Zhang, and et al. 2023. "A Pathogenic Role for FcγRI in the Immune Response against Chlamydial Respiratory Infection" Microorganisms 11, no. 1: 39. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms11010039