
Highly Variable Bacterial Communities Associated with the Octocoral Antillogorgia elisabethae

Abstract
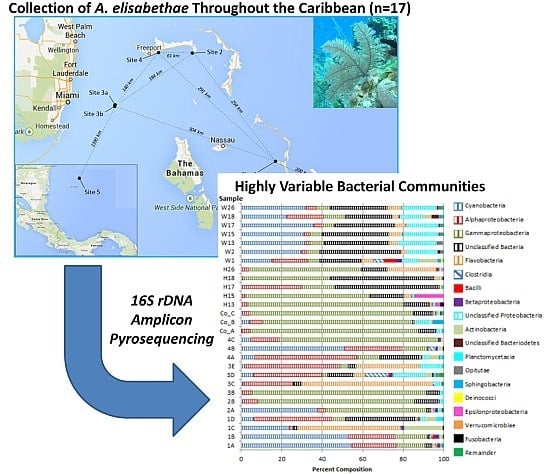
:
1. Introduction
2. Experimental Section
2.1. Sample Collection
2.2. DNA Isolation
2.3. Template Preparation for 16S rDNA Amplicon Pyrosequencing
2.4. 16S rDNA Amplicon Pyrosequencing 16S rDNA Amplicon Pyrosequencing
2.5. Pyrosequencing Data Analysis
2.6. Bacterial Community Analysis
2.7. Accession Numbers
3. Results
3.1. Bacterial Community Analysis
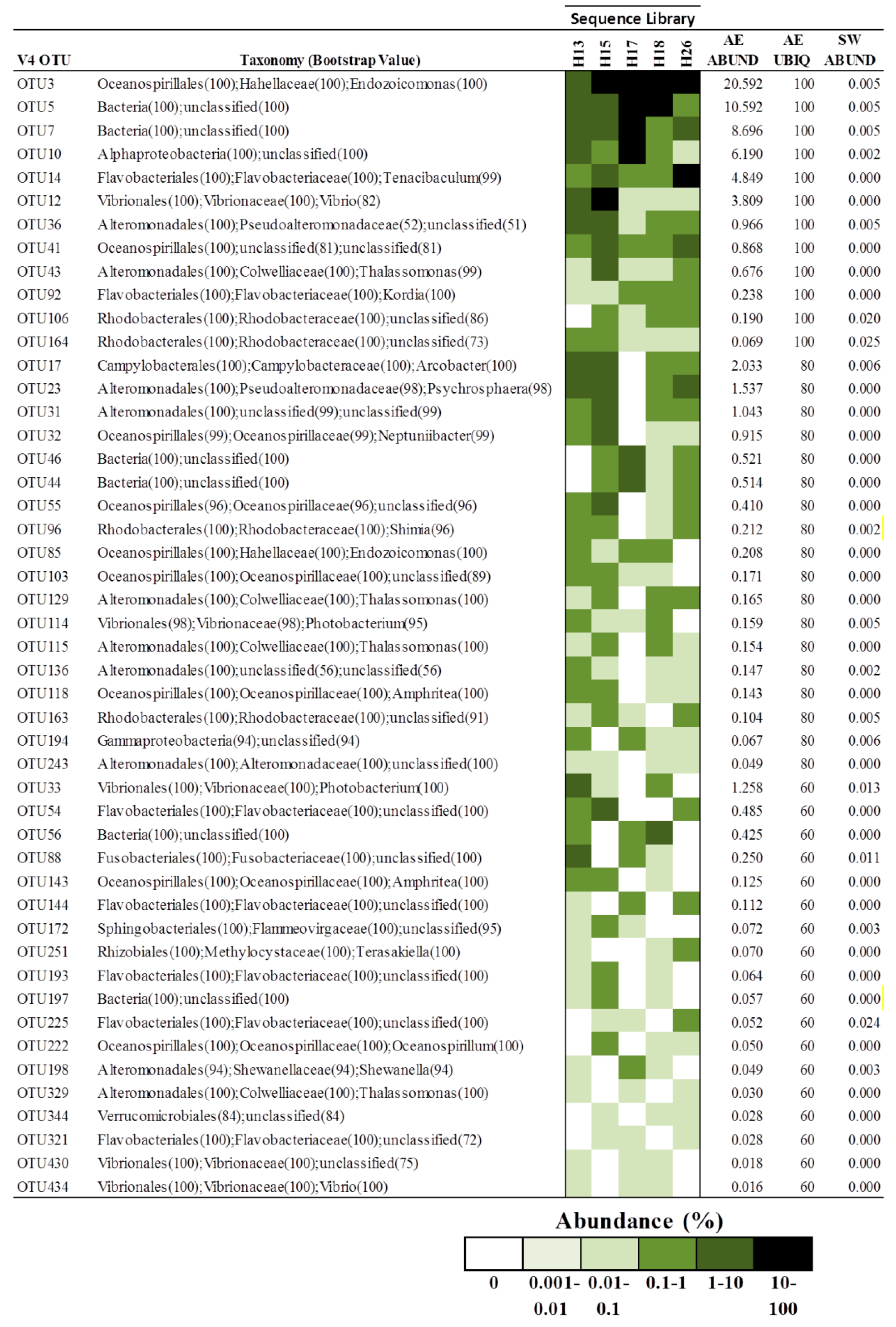
3.2. Core Microbiome of A. elisabethae
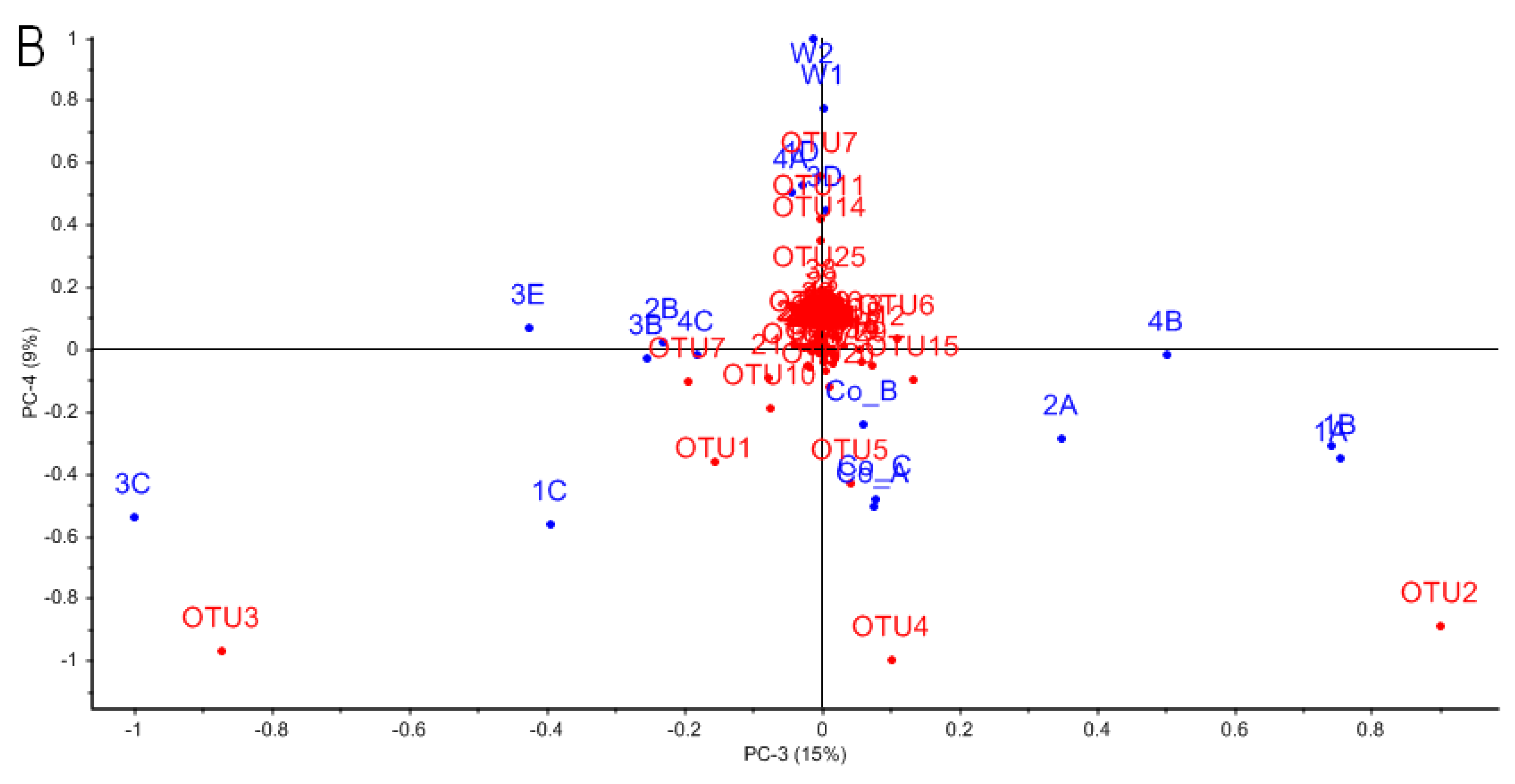
3.3. Characterizing Dominant Bacteria Driving the Separation of Coral-Associated Bacterial Communities by PCA
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McFadden, C.S.; Sánchez, J.A.; France, S.C. Molecular phylogenetic insights into the evolution of Octocorallia: A review. Integr. Comp. Biol. 2010, 50, 389–410. [Google Scholar] [CrossRef] [PubMed]
- Fenical, W. Marine soft corals of the genus Pseudopterogorgia—A resource for novel anti-inflammatory diterpenoids. J. Nat. Prod. 1987, 50, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Bayer, F. The Shallow Water Octocorallia of the West; Indian Region; Martinus Nijhoff: The Hague, The Netherlands, 1961; p. 373. [Google Scholar]
- Heckrodt, T.J.; Mulzer, J. Marine natural products from Pseudopterogorgia elisabethae: Structures, biosynthesis, pharmacology, and total synthesis. Top. Curr. Chem. 2005, 244, 1–41. [Google Scholar]
- Williams, G.C.; Chen, J.Y. Resurrection of the octocorallian genus Antillogorgia for Caribbean species previously assigned to Pseudopterogorgia, and a taxonomic assessment of the relationship of these genera with Leptogorgia (Cnidaria, Anthozoa, Gorgoniidae). Zootaxa 2012, 3505, 39–52. [Google Scholar]
- Guitiérrez-Rodriguez, C.; Barbeitos, M.S.; Sánchez, J.A.; Lasker, H.R. Phylogeography and morphological variation of the branching octocoral Pseudopterogorgia elisabethae. Mol. Phylogenet. Evol. 2009, 50, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Guitiérrez-Rodriguez, C.; Lasker, H.R. Microsatellite variation reveals high levels of genetic variability and population structure in the gorgonian coral Pseudopterogorgia elisabethae across The Bahamas. Mol. Ecol. 2004, 13, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Lasker, H.R.; Porto-Hannes, I. Population structure among octocoral adults and recruits identifies scale dependent patterns of population isolation in The Bahamas. PeerJ 2015, 3, e1019. [Google Scholar] [CrossRef] [PubMed]
- Whalen, K.E.; Starczak, V.R.; Nelson, D.R.; Goldstone, J.V.; Hahn, M.E. Cytochrome P450 diversity and induction by gorgonian allelochemicals in the marine gastropod Cyphoma gibbosum. BMC Ecol. 2010, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Berrué, F.; Kerr, R.G. Diterpenes from gorgonian corals. Nat. Prod. Rep. 2009, 26, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Look, S.A.; Fenical, W.; Jacobs, R.S.; Clardy, J. The pseudopterosins: Anti-inflammatory and analgesic natural products from the sea whip Pseudopterogorgia elisabethae. Proc. Natl. Acad. Sci. USA 1986, 83, 6238–6240. [Google Scholar] [CrossRef] [PubMed]
- Lasker, H.R. Recruitment and resilience of a harvested Caribbean octocoral. PLoS ONE 2013, 8, e74587. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Breitbart, M.; Jara, J.; Azam, F.; Knowlton, N. Diversity of bacteria associated with the Caribbean coral Montastraea franksi. Coral Reefs 2001, 20, 86–91. [Google Scholar]
- Rohwer, F.; Knowlton, N. Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 2002, 243, 1–10. [Google Scholar] [CrossRef]
- Bourne, D.G.; Munn, C.B. Diversity of bacteria associated with the coral Pocillopora damicornis from the great barrier reef. Environ. Microbiol. 2005, 7, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Wegley, L.; Edwards, R.; Rodriguez-Brito, B.; Liu, H.; Rohwer, F. Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ. Microbiol. 2007, 9, 2707–2719. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.; Koren, O.; Reshef, L.; Efrony, R.; Zilber-Rosenberg, I. The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microbiol. 2007, 5, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Penn, K.; Wu, D.; Eisen, J.A.; Ward, N. Characterization of bacterial communities associated with deep-sea corals on Gulf of Alaska seamounts. Appl. Environ. Microbiol. 2006, 72, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.A.; Stone, R.P.; McLaughlin, M.R.; Kellogg, C.A. Microbial consortia of gorgonian corals from the Aleutian Islands. FEMS Microbiol. Ecol. 2011, 76, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Roder, C.; Bayer, T.; Aranda, M.; Kruse, M.; Voolstra, C.R. Microbiome structure of the fungid coral Ctenactis echinata aligns with environmental differences. Mol. Ecol. 2015, 24, 3501–3511. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, C.A.; Lisle, J.T.; Galkiewicz, J.P. Culture-independent characterization of bacterial communities associated with the cold-water coral Lophelia purtusa in the north eastern Gulf of Mexico. Appl. Environ. Microbiol. 2009, 75, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Kvennefors, E.C.; Sampayo, E.; Ridgway, T.; Barnes, A.C.; Hoegh-Guldberg, O. Bacterial communities of two ubiquitous Great Barrier Reef corals reveals both site- and species-specificity of common bacterial associates. PLoS ONE 2010, 5, e10401. [Google Scholar] [CrossRef] [PubMed]
- Neulinger, S.C.; Jarnegren, J.; Ludvigsen, M.; Lochte, K.; Dullo, W.C. Phenotype-specific bacterial communities in the cold-water coral Lophelia pertusa (Scleractinia) and their implications for the coral‘s nutrition, health, and distribution. Appl. Environ. Microbiol. 2008, 74, 7272–7285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littman, R.A.; Willis, B.L.; Bourne, D.G. Bacterial communities of juvenile corals infected with different Symbiodinium (dinoflagellate) clades. Mar. Ecol. Prog. Ser. 2009, 389, 45–59. [Google Scholar] [CrossRef]
- Sweet, M.J.; Croquer, A.; Bythell, J.C. Dynamics of bacterial community development in the reef coral Acropora muricata following experimental antibiotic treatment. Coral Reefs 2011, 30, 1121–1133. [Google Scholar] [CrossRef]
- Frias-Lopez, J.; Zerkle, A.L.; Bonheyo, G.T.; Fouke, B.W. Partitioning of bacterial communities between seawater and healthy, black band diseased, and dead coral surfaces. Appl. Environ. Microbiol. 2002, 68, 2214–2228. [Google Scholar] [CrossRef] [PubMed]
- Bourne, D.; Iida, Y.; Uthicke, S.; Smith-Keune, C. Changes in coral-associated microbial communities during a bleaching event. ISME J. 2008, 2, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Bourne, D.G.; Sato, Y. Changes in sulfate-reducing bacterial populations during the onset of black band disease. ISME J. 2011, 5, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Brück, T.B.; Brück, W.M.; Santiago-Vázquez, L.Z.; McCarthy, P.J.; Kerr, R.G. Diversity of the bacterial communities associated with the azooxanthellate deep water octocorals Leptogorgia minimata, Iciligorgia schrammi, and Swiftia exertia. Mar. Biotechnol. (N. Y.) 2007, 9, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.; Arif, C.; Ferrier-Pagès, C.; Zoccola, D.; Aranda, M.; Voolstra, C.R. Bacteria of the genus Endozoicomonas dominate the microbiome of the Mediterranean gorgonian coral Eunicella cavolini. Mar. Ecol. Prog. Ser. 2013, 479, 75–84. [Google Scholar] [CrossRef]
- La Rivière, M.; Roumagnac, M.; Garrabou, J.; Bally, M. Transient shifts in bacterial communities associated with the temperate gorgonian Paramuricea clavata in the northwestern Mediterranean Sea. PLoS ONE 2013, 8, e57385. [Google Scholar] [CrossRef] [PubMed]
- Vezzulli, L.; Pezzati, E.; Huete-Stauffer, C.; Pruzzo, C.; Cerrano, C. 16S rDNA pyrosequencing of the Mediterranean gorgonian Paramuricea clavata reveals a link among alterations in bacterial holobiont members, anthropogenic influence and disease outbreaks. PLoS ONE 2013, 8, e67745. [Google Scholar] [CrossRef] [PubMed]
- Ransome, E.; Rowley, S.J.; Thomas, S.; Tait, K.; Munn, C.B. Disturbance to conserved bacterial communities in the cold-water gorgonian coral Eunicella verrucosa. FEMS Microbiol. Ecol. 2014, 90, 404–416. [Google Scholar] [PubMed]
- Correa, H.; Haltli, B.; Duque, C.; Kerr, R. Bacterial communities of the gorgonian octocoral Pseudopterogorgia elisabethae. Microb. Ecol. 2013, 66, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.D.; Shi, Y. Structurally diverse terpenoids from the sea whip Pseudopterogorgia elisabethae (Bayer). Tetrahedron 2000, 56, 9015–9023. [Google Scholar] [CrossRef]
- Edwards, U.; Rogall, T.; Blocker, H.; Emde, M.; Bottger, E.C. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 1989, 17, 7843–7853. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, L.M.; Blackwood, C.B. Assessment of bias associated with incomplete extraction of microbial DNA from soil. Appl. Environ. Microbiol. 2009, 75, 5428–5433. [Google Scholar] [CrossRef] [PubMed]
- Callaway, T.R.; Dowd, S.E.; Wolcott, R.D.; Sun, Y.; McReynolds, J.L.; Edrington, T.S.; Byrd, J.A.; Anderson, R.C.; Krueger, N.; Nisbet, D.J. Evaluation of the bacterial diversity in cecal contents of laying hens fed various molting diets by using bacterial tag-encoded FLX amplicon pyrosequencing. Poult. Sci. 2009, 88, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Wolcott, R.D.; Gontcharova, V.; Sun, Y.; Dowd, S.E. Evaluation of the bacterial diversity among and within individual venous leg ulcers using bacterial tag-encoded FLX and titanium amplicon pyrosequencing and metagenomic approaches. BMC Microbiol. 2009, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D. 454 SOP. Available online: http://www.mothur.org/wiki/454_SOP (accessed on 27 February 2015).
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 15 March 2015).
- Clark, K. Non-parmetric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Ainsworth, T.D.; Krause, L.; Bridge, T.; Torda, G.; Raina, J.B.; Zakrzewski, M.; Gates, R.D.; Padilla-Gamino, J.L.; Spalding, H.L.; Smith, C.; et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 2015, 9, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Engelbrektson, A.; Kunin, V.; Wrighton, K.C.; Zvenigorodsky, N.; Chen, F.; Ochman, H.; Hugenholtz, P. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J. 2010, 4, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Woodley, C.M.; Medina, M. Threatened corals provide underexplored microbial habitats. PLoS ONE 2010, 5, e9554. [Google Scholar] [CrossRef] [PubMed]
- Pike, R.E.; Haltli, B.; Kerr, R.G. Description of Endozoicomonas euniceicola sp. nov. and Endozoicomonas gorgoniicola sp. nov., bacteria isolated from the octocorals Eunicea fusca and Plexaura sp., and an emended description of the genus Endozoicomonas. Int. J. Syst. Evol. Microbiol. 2013, 63, 4294–4302. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Huang, H.; Lai, Q.; Shao, Z. Aquimarina penaei sp. nov., isolated from intestinal tract contents of pacific white shrimp, Penaeus vannamei. Antonie Van Leeuwenhoek 2014, 106, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Friedline, C.J.; Franklin, R.B.; McCallister, S.L.; Rivera, M.C. Microbial community diversity of the eastern Atlantic Ocean reveals geographic differences. Biogeosci. Discuss. 2012, 9, 109–150. [Google Scholar] [CrossRef]
- Webster, N.S.; Taylor, M.W.; Behnam, F.; Lucker, S.; Rattei, T.; Whalan, S.; Horn, M.; Wagner, M. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 2010, 12, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- White, J.R.; Patel, J.; Ottesen, A.; Arce, G.; Blackwelder, P.; Lopez, J.V. Pyrosequencing of bacterial symbionts within Axinella corrugata sponges: Diversity and seasonal variability. PLoS ONE 2012, 7, e38204. [Google Scholar] [CrossRef] [PubMed]
- Flombaum, P.; Gallegos, J.L.; Gordillo, R.A.; Rincon, J.; Zabala, L.L.; Jiao, N.; Karl, D.M.; Li, W.K.W.; Lomas, M.W.; Veneziano, D.; et al. Present and future global distributions of the marine cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. USA 2013, 110, 9824–9829. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.M.; Rappe, M.S.; Connon, S.A.; Vergin, K.L.; Siebold, W.A.; Carlson, C.A.; Giovannoni, S.J. Sar11 clade dominates ocean surface bacterioplankton communities. Nature 2002, 420, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Ribes, M.; Coma, R.; Atkinson, M.J.; Kinzie, R.A. Particle removal by coral reef communities: Picoplankton is a major source of nitrogen. Mar. Ecol. Prog. Ser. 2003, 257, 13–23. [Google Scholar] [CrossRef]
- Lesser, M.P.; Falcón, L.I.; Rodríguez-Román, A.; Enríquez, S.; Hoegh-Guldberg, O.; Iglesias-Prieto, R. Nitrogen fixation by symbiotic Cyanobacteria provides a source of nitrogen for the scleractinian coral Montastraea cavernosa. Mar. Ecol. Prog. Ser. 2007, 346, 143–152. [Google Scholar] [CrossRef]
- Carella, F.; Aceto, S.; Saggioma, M.; Mangioni, O.; de Vico, G. Gorgonian disease outbreak in the Gulf of Naples: Pathology reveals cyanobacterial infection linked to eleveated sea temperatures. Dis. Aquat. Organ. 2014, 111, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Morrow, K.M.; Moss, A.G.; Chadwick, N.E.; Liles, M.R. Bacterial associates of two Caribbean coral species reveal species-specific distribution and geographic variability. Appl. Environ. Microbiol. 2012, 78, 6438–6449. [Google Scholar] [CrossRef] [PubMed]
- Punyana, M.; Marvaez, G.; Paz, A.; Osorno, O.; Duque, C. Pseudopterosin content variability of the purple sea whip Pseudopterogorgia elisabethae at the islands of San Andreas and Providencia (SW Caribbean). J. Chem. Ecol. 2004, 30, 1183–1201. [Google Scholar] [CrossRef]
- McCulloch, M.W.; Haltli, B.; Marchbank, D.H.; Kerr, R.G. Evaluation of pseudopteroxazole and pseudopterosin derivatives against Mycobacterium tuberculosis and other pathogens. Mar. Drugs 2012, 10, 1711–1728. [Google Scholar] [CrossRef] [PubMed]
- Correa, H.; Aristizabal, F.; Duque, C.; Kerr, R. Cytotoxic and antimicrobial activity of pseudopterosins and seco-pseudopterosins isolated from the octocoral Pseudopterogorgia elisabethae of San Andres and Providencia Islands (southwest Caribbean Sea). Mar. Drugs 2011, 9, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, L.; Deng, Y.; Zhi, X.; Jiang, Y.H.; Tu, Q.; Xie, J.; Van Nostrand, J.D.; He, Z.; Yang, Y. Reproducibility and quantitation of amplicon sequencing-based detection. ISME J. 2011, 5, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Cruaud, P.; Vigneron, A.; Lucchetti-Miganeh, C.; Ciron, P.; Godfroy, A.; Cambon-Bonavita, M. Influence of DNA extraction method, 16S rRNA targeted hypervariable regions, and sample origin on microbial diversity detected by 454 pyrosequencing in marine chemosynthetic ecosystems. Appl. Environ. Microbiol. 2014, 80, 4426–4639. [Google Scholar] [CrossRef] [PubMed]
- Webster, N.S.; Bourne, D. Bacterial community structure associated with the antarctic soft coral, Alcyonium antarcticum. FEMS Microbiol. Ecol. 2007, 59, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Hester, E.R.; Barott, K.L.; Nulton, J.; Vermeij, M.J.; Rohwer, F.L. Stable and sporadic symbiotic communities of coral and algal holobionts. ISME J. 2016, 10, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.; Leininger, S.; Schmaljohann, R.; Brummer, F.; Imhoff, J.F. Sponge-specific bacterial associations of the Mediterranean sponge Chondrilla nucula (Demospongiae, Tetractinomorpha). Microb. Ecol. 2007, 54, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, M.; Adachi, K.; Katsuta, A.; Shizuri, Y.; Yamasato, K. Endozoicomonas numazuensis sp. nov., a gammaproteobacterium isolated from marine sponges, and emended description of the genus Endozoicomonas Kurahashi and Yokota 2007. Int. J. Syst. Evol. Microbiol. 2013, 63, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, M.; Yokota, A. Endozoicomonas elysicola gen. nov., sp. nov., a gamma-proteobacterium isolated from the sea slug Elysia ornata. Syst. Appl. Microbiol. 2007, 30, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, M.; Diaz-Valdez, M.; Wanner, G.; Ramos-Espla, A.; Anton, J. Microbial community associated with the colonial Ascidian Cystodytes dellechiajei. Environ. Microbiol. 2007, 9, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Goffredi, S.K.; Orphan, V.J.; Rouse, G.W.; Jahnke, L.; Embaye, T.; Turk, K.; Lee, R.; Vrijenhoek, R.C. Evolutionary innovation: A bone-eating marine symbiosis. Environ. Microbiol. 2005, 7, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Kwon, H.C.; Sohn, Y.C.; Yang, H.O. Kistimonas asteriae gen. nov., sp. nov., a gammaproteobacterium isolated from Asterias amurensis. Int. J. Syst. Evol. Microbiol. 2010, 60, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhang, W.; Xia, H.; Lu, G.; Chen, G. Isolation and diversity analysis of heterotrophic bacteria associated with sea anemones. Acta Oceanol. Sin. 2010, 29, 62–69. [Google Scholar] [CrossRef]
- Raina, J.B.; Tapiolas, D.; Willis, B.L.; Bourne, D.G. Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl. Environ. Microbiol. 2009, 75, 3492–3501. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.R.; Gutiérrez-Rodríguez, C.; Lasker, H.R.; Coffroth, M.A. Symbiodinium sp. associations in the gorgonian Pseudopterogorgia elisabethae in The Bahamas: High levels of genetic variability and population structure in symbiotic dinoflagellates. Mar. Biol. 2003, 143, 111–120. [Google Scholar] [CrossRef]
- Jeong, H.; Yim, J.H.; Lee, C.; Choi, S.H.; Park, Y.K.; Yoon, S.H.; Hur, C.G.; Kang, H.Y.; Kim, D.; Lee, H.H.; et al. Genomic blueprint of Hahella chejuensis, a marine microbe producing an algicidal agent. Nucleic Acids Res. 2005, 33, 7066–7073. [Google Scholar] [CrossRef] [PubMed]
- Klaus, J.S.; Janse, I.; Heikoop, J.M.; Sanford, R.A.; Fouke, B.W. Coral microbial communities, zooxanthellae and mucus along gradients of seawater depth and coastal pollution. Environ. Microbiol. 2007, 9, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Lema, K.A.; Bourne, D.G.; Willis, B.L. Onset and establishment of diazotrophs and other bacterial associates in the early life history stages of the coral Acropora millepora. Mol. Ecol. 2014, 23, 4682–4695. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.C.; de Marco, P. Methanesulfonate (MSA) catabolic genes from marine and estuarine bacteria. PLoS ONE 2015, 10, e0125735. [Google Scholar] [CrossRef] [PubMed]
- Galkiewicz, J.P.; Pratte, Z.A.; Gray, M.A.; Kellogg, C.A. Characterization of culturable bacteria isolated from the cold-water coral Lophelia pertusa. FEMS Microbiol. Ecol. 2011, 77, 333–346. [Google Scholar] [CrossRef] [PubMed]
- La Rivière, M.; Garrabou, J.; Bally, M. Evidence for host specificity among dominant bacterial symbionts in temperate gorgonian corals. Coral Reefs 2015, 34, 1087–1098. [Google Scholar] [CrossRef]
- Brinkhoff, T.; Giebel, H.A.; Simon, M. Diversity, ecology, and genomics of the Roseobacter clade: A short overview. Arch. Microbiol. 2008, 189, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Gignoux-Wolfsohn, S.A.; Vollmer, S.V. Identification of candidate coral pathogens on white band disease-infected staghorn coral. PLoS ONE 2015, 10, e0134416. [Google Scholar]
- Xu, T.; Yu, M.; Lin, H.; Zhang, Z.; Liu, J.; Zhang, X.H. Genomic insight into Aquimarina longa SW024 T: Its ultra-oligotrophic adapting mechanisms and biogeochemical functions. BMC Genom. 2015, 16, 772. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.M.; Sheu, F.S.; Sheu, S.Y. Aquimarina salinaria sp. nov., a novel algicidal bacterium isolated from a saltpan. Arch. Microbiol. 2012, 194, 103–112. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Library | Site | Sample | Year | DNA Isol. Meth. | Pyroseq. Template | 16S rDNA Region | No. Reads | Avg. Length (bp) | Sobs a | Sest a | H′ a | E a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1A b | 1 | AE1 | 2006 | PC | PCR | V1/V2 | 4685 | 222 | 253 | 514 | 2.47 | 0.44 |
| 162 | 385 | 2.43 | 0.48 | |||||||||
| 1B b | 1 | AE1 | 2006 | PC | PCR | V1/V2 | 4969 | 222 | 260 | 444 | 2.25 | 0.41 |
| 162 | 366 | 2.21 | 0.43 | |||||||||
| 1C | 1 | AE2 | 2006 | PC | PCR | V1/V2 | 6962 | 240 | 162 | 216 | 2.01 | 0.40 |
| 90 | 179 | 1.98 | 0.44 | |||||||||
| 1D | 1 | AE3 | 2006 | PC | PCR | V1/V2 | 5221 | 228 | 886 | 1978 | 5.13 | 0.79 |
| 524 | 1295 | 5.05 | 0.81 | |||||||||
| 2A | 2 | AE4 | 2006 | PC | PCR | V1/V2 | 7200 | 233 | 310 | 533 | 1.90 | 0.37 |
| 151 | 379 | 1.85 | 0.41 | |||||||||
| 2B | 2 | AE5 | 2006 | PC | PCR | V1/V2 | 8884 | 245 | 404 | 785 | 1.71 | 0.28 |
| 181 | 414 | 1.64 | 0.32 | |||||||||
| 3B | 3a | AE6 | 2006 | PC | PCR | V1/V2 | 2424 | 245 | 182 | 471 | 1.20 | 0.23 |
| 170 | 442 | 1.19 | 0.23 | |||||||||
| 3C | 3a | AE7 | 2006 | PC | PCR | V1/V2 | 5906 | 243 | 366 | 1032 | 1.99 | 0.34 |
| 184 | 563 | 1.93 | 0.37 | |||||||||
| 3D | 3a | AE8 | 2006 | PC | PCR | V1/V2 | 2754 | 235 | 565 | 1245 | 4.77 | 0.75 |
| 497 | 1054 | 4.73 | 0.76 | |||||||||
| 3E | 3b | AE9 | 2009 | PC | PCR | V1/V2 | 7038 | 228 | 530 | 1434 | 3.52 | 0.56 |
| 248 | 726 | 3.44 | 0.62 | |||||||||
| 4A | 4 | AE10 | 2006 | PC | PCR | V1/V2 | 5560 | 231 | 598 | 1290 | 4.48 | 0.70 |
| 356 | 768 | 4.38 | 0.75 | |||||||||
| 4B | 4 | AE11 | 2006 | PC | PCR | V1/V2 | 6450 | 221 | 233 | 556 | 2.74 | 0.50 |
| 128 | 280 | 2.71 | 0.56 | |||||||||
| 4C | 4 | AE12 | 2006 | PC | PCR | V1/V2 | 7269 | 241 | 144 | 338 | 1.30 | 0.26 |
| 72 | 165 | 1.27 | 0.30 | |||||||||
| Co_A c | 5 | AE13 | 2010 | PC | gDNA | V1/V2 | 5831 | 247 | 152 | 333 | 2.03 | 0.40 |
| 85 | 210 | 2.00 | 0.45 | |||||||||
| Co_B c | 5 | AE14 | 2010 | PC | gDNA | V1/V2 | 2258 | 248 | 91 | 197 | 2.41 | 0.54 |
| 90 | 195 | 2.41 | 0.54 | |||||||||
| Co_C c | 5 | AE15 | 2010 | PC | gDNA | V1/V2 | 4846 | 244 | 411 | 842 | 2.58 | 0.43 |
| 247 | 615 | 2.52 | 0.46 | |||||||||
| W1 | 3a | W1 | 2009 | UW | gDNA | V1/V2 | 3149 | 232 | 155 | 206 | 3.86 | 0.76 |
| 140 | 184 | 3.85 | 0.78 | |||||||||
| W2 | 3b | W2 | 2009 | UW | gDNA | V1/V2 | 6294 | 223 | 325 | 563 | 3.25 | 0.56 |
| 195 | 366 | 3.19 | 0.61 | |||||||||
| H13 | 6a | AE16 | 2013 | PS | gDNA | V4 | 3759 | 201 | 219 | 457 | 3.10 | 0.58 |
| 178 | 395 | 3.08 | 0.59 | |||||||||
| H15 | 6b | AE17 | 2013 | PS | gDNA | V4 | 5511 | 201 | 164 | 314 | 3.10 | 0.61 |
| 116 | 219 | 3.10 | 0.65 | |||||||||
| H17 | 6b | AE18 | 2011 | PS | gDNA | V4 | 6247 | 201 | 147 | 351 | 2.12 | 0.42 |
| 94 | 180 | 2.10 | 0.46 | |||||||||
| H18 | 6b | AE19 | 2011 | PS | gDNA | V4 | 4775 | 201 | 249 | 514 | 2.57 | 0.47 |
| 172 | 422 | 2.54 | 0.49 | |||||||||
| H26 | 6a | AE20 | 2011 | PS | gDNA | V4 | 2659 | 201 | 102 | 192 | 2.25 | 0.49 |
| 101 | 190 | 2.25 | 0.49 | |||||||||
| W13 | 6a | W3 | 2011 | UW | gDNA | V4 | 3341 | 201 | 216 | 300 | 3.35 | 0.62 |
| 197 | 276 | 3.33 | 0.63 | |||||||||
| W15 | 6b | W4 | 2011 | UW | gDNA | V4 | 4278 | 201 | 235 | 358 | 3.34 | 0.61 |
| 194 | 296 | 3.32 | 0.60 | |||||||||
| W17 | 6b | W5 | 2011 | UW | gDNA | V4 | 7706 | 201 | 352 | 518 | 3.27 | 0.57 |
| 213 | 331 | 3.22 | 0.60 | |||||||||
| W18 | 6b | W6 | 2011 | UW | gDNA | V4 | 7979 | 201 | 409 | 695 | 3.80 | 0.63 |
| 254 | 414 | 3.74 | 0.68 | |||||||||
| W26 | 6a | W7 | 2011 | UW | gDNA | V4 | 8491 | 201 | 317 | 526 | 3.36 | 0.58 |
| 195 | 324 | 3.31 | 0.63 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robertson, V.; Haltli, B.; McCauley, E.P.; Overy, D.P.; Kerr, R.G. Highly Variable Bacterial Communities Associated with the Octocoral Antillogorgia elisabethae. Microorganisms 2016, 4, 23. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms4030023
Robertson V, Haltli B, McCauley EP, Overy DP, Kerr RG. Highly Variable Bacterial Communities Associated with the Octocoral Antillogorgia elisabethae. Microorganisms. 2016; 4(3):23. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms4030023
Chicago/Turabian StyleRobertson, Veronica, Brad Haltli, Erin P. McCauley, David P. Overy, and Russell G. Kerr. 2016. "Highly Variable Bacterial Communities Associated with the Octocoral Antillogorgia elisabethae" Microorganisms 4, no. 3: 23. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms4030023