Top-Down Proteomic Identification of Shiga Toxin 1 and 2 from Pathogenic Escherichia coli Using MALDI-TOF-TOF Tandem Mass Spectrometry

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culturing
2.2. Mass Spectrometry Analysis
2.3. Gene and Genomic Sequencing
2.4. Top-Down Proteomic Analysis
3. Results and Discussion
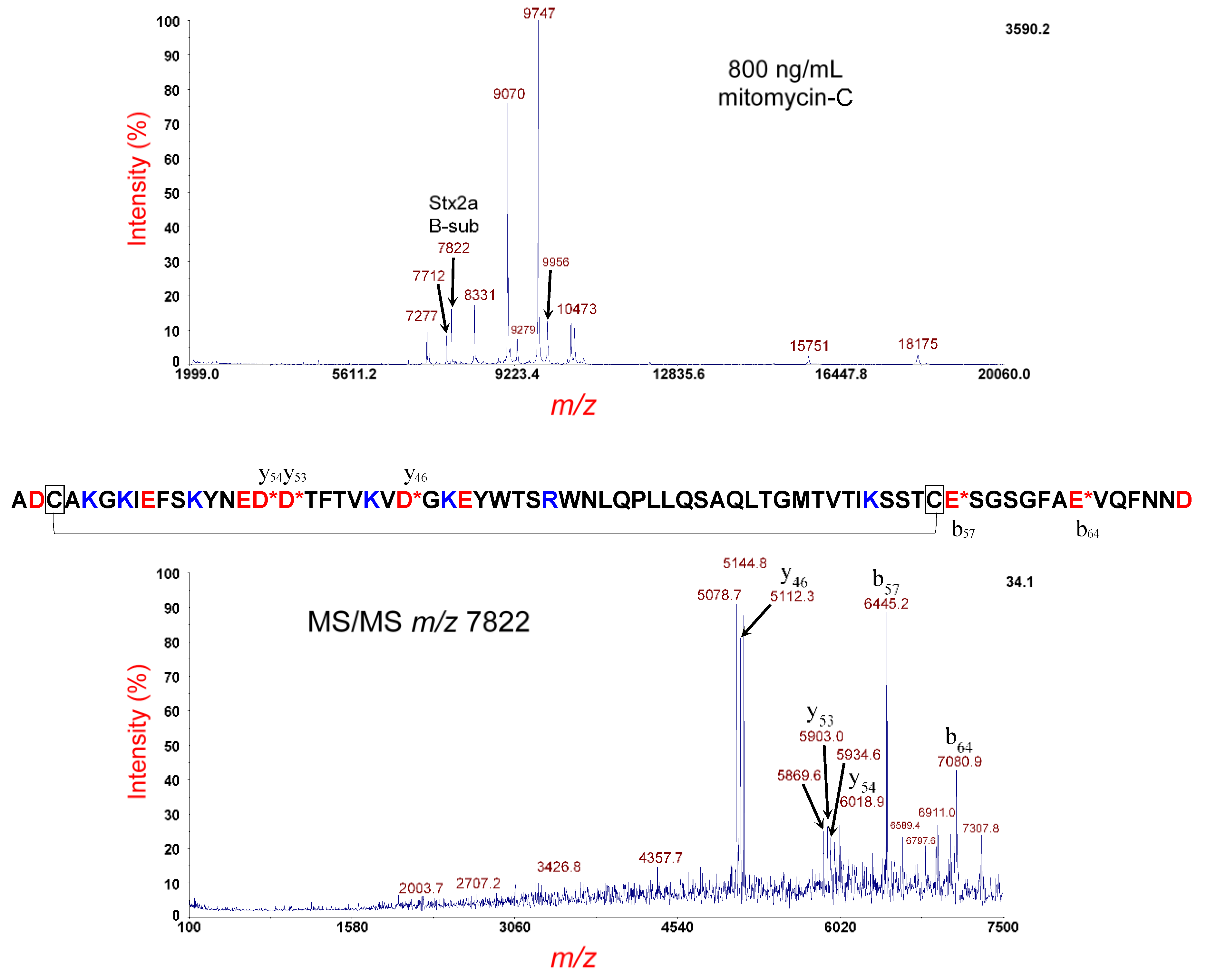
3.1. Analysis of E. coli O157:H- Strain 493/89
3.2. Analysis of E. coli O145:H28 Strain RM12367-C1
3.3. Analysis of E. coli O145:H28 Strain RM14496-C1
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dewey-Mattia, D.; Manikonda, K.; Hall, A.J.; Wise, M.E.; Crowe, S.J. Surveillance for Foodborne Disease Outbreaks—United States, 2009–2015. MMWR Surveill. Summ. 2018, 67, 1–11. [Google Scholar] [CrossRef] [PubMed]
- FAO/WHO STEC EXPERT GROUP. Hazard Identification and Characterization: Criteria for Categorizing Shiga Toxin-Producing Escherichia coli on a Risk Basis. J. Food Prot. 2018, 26, 7–21. [Google Scholar]
- Johannes, L.; Römer, W. Shiga toxins–from cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar] [CrossRef]
- Golshani, M.; Oloomi, M.; Bouzari, S. In silico analysis of Shiga toxins (Stxs) to identify new potential vaccine targets for Shiga toxin-producing Escherichia coli. In Silico. Pharmacol. 2017, 5, 2. [Google Scholar] [CrossRef]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef]
- O’Brien, A.D.; Melton, A.R.; Schmitt, C.K.; McKee, M.L.; Batts, M.L.; Griffin, D.E. Profile of Escherichia coli O157:H7 pathogen responsible for hamburger-borne outbreak of hemorrhagic colitis and hemolytic uremic syndrome in Washington. J. Clin. Microbiol. 1993, 31, 2799–2801. [Google Scholar]
- Kampmeier, S.; Berger, M.; Mellmann, A.; Karch, H.; Berger, P. The 2011 German Enterohemorrhagic Escherichia coli O104:H4 Outbreak-The Danger Is Still Out There. Curr. Top Microbiol. Immunol. 2018, 416, 117–148. [Google Scholar]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Osburne, M.S.; Brin-Jones, H.; Tai, A.K.; Leong, J.M. Prophage induction, but not production of phage particles, is required for lethal disease in a microbiome-replete murine model of enterohemorrhagic E. coli infection. PLoS Pathog. 2019, 15, e1007494. [Google Scholar] [CrossRef]
- Holmes, A.; Dallman, T.J.; Shabaan, S.; Hanson, M.; Allison, L. Validation of Whole-Genome Sequencing for Identification and Characterization of Shiga Toxin-Producing Escherichia coli To Produce Standardized Data To Enable Data Sharing. J. Clin. Microbiol. 2018, 56, e01388-17. [Google Scholar] [CrossRef]
- Yokoyama, E.; Hirai, S.; Ishige, T.; Murakami, S. Application of whole genome sequence data in analyzing the molecular epidemiology of Shiga toxin-producing Escherichia coli O157:H7/H. Int. J. Food Microbiol. 2018, 264, 39–45. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Patfield, S.; Rasooly, R.; Mavrici, D. Novel monoclonal antibodies against Stx1d and 1e and their use for improving immunoassays. J. Immunol. Methods 2017, 447, 52–56. [Google Scholar] [CrossRef] [PubMed]
- De Rauw, K.; Breynaert, J.; Piérard, D. Evaluation of the Alere SHIGA TOXIN QUIK CHEK™ in comparison to multiplex Shiga toxin PCR. Diagn. Microbiol. Infect. Dis. 2016, 86, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Cherubin, P.; Quiñones, B.; Elkahoui, S.; Yokoyama, W.; Teter, K. A cell-based fluorescent assay to detect the activity of AB toxins that inhibit protein synthesis. Methods Mol. Biol. 2017, 1600, 25–36. [Google Scholar]
- Massey, S.; Quiñones, B.; Teter, K. A cell-based fluorescent assay to detect the activity of Shiga toxin and other toxins that inhibit protein synthesis. Methods Mol. Biol. 2011, 739, 49–59. [Google Scholar]
- Fagerquist, C.K.; Zaragoza, W.J.; Sultan, O.; Woo, N.; Quiñones, B.; Cooley, M.B.; Mandrell, R.E. Top-down proteomic identification of Shiga toxin 2 subtypes from Shiga toxin-producing Escherichia coli by matrix-assisted laser desorption ionization-tandem time of flight mass spectrometry. Appl. Envrion. Microbiol. 2014, 80, 2928–2940. [Google Scholar] [CrossRef]
- Fagerquist, C.K.; Zaragoza, W.J.; Lee, B.G.; Yambao, J.C.; Quiñones, B. Clinically-relevant Shiga toxin 2 subtypes from environmental Shiga toxin-producing Escherichia coli identified by top-down/middle-down proteomics and DNA sequencing. Clin. Mass Spectrom. 2019, 11, 27–36. [Google Scholar] [CrossRef]
- Silva, C.J.; Erickson-Beltran, M.L.; Skinner, C.B.; Dynin, I.; Hui, C.; Patfield, S.A.; Carter, J.M.; He, X. Safe and effective means of detecting and quantitating Shiga-like toxins in attomole amounts. Anal. Chem. 2014, 86, 4698–4706. [Google Scholar] [CrossRef]
- Janka, A.; Bielaszewska, M.; Dobrindt, U.; Karch, H. Identification and distribution of the enterohemorrhagic Escherichia coli factor for adherence (efa1) gene in sorbitol-fermenting Escherichia coli O157: H-. Int. J. Med. Microbiol. 2002, 292, 207–214. [Google Scholar] [CrossRef]
- Karch, H.; Wiss, R.; Gloning, H.; Emmrich, P.; Aleksić, S.; Bockemühl, J. Hemolytic-uremic syndrome in infants due to verotoxin-producing Escherichia coli. Dtsch. Med. Wochenschr. 1990, 115, 489–495. [Google Scholar] [CrossRef]
- Carter, M.Q.; Quinones, B.; He, X.; Zhong, W.; Louie, J.W.; Lee, B.G.; Yambao, J.C.; Mandrell, R.E.; Cooley, M.B. An Environmental Shiga Toxin-Producing Escherichia coli O145 Clonal Population Exhibits High-Level Phenotypic Variation That Includes Virulence Traits. Appl. Environ. Microbiol. 2015, 82, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Fagerquist, C.K.; Zaragoza, W.J. Bacteriophage cell lysis of Shiga toxin-producing Escherichia coli for top-down proteomic identification of Shiga toxins 1 & 2 using matrix-assisted laser desorption/ionization tandem time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2016, 30, 671–680. [Google Scholar] [PubMed]
- Fagerquist, C.K.; Sultan, O. A new calibrant for matrix-assisted laser desorption/ionization time-of-flight-time-of-flight post-source decay tandem mass spectrometry of non-digested proteins for top-down proteomic analysis. Rapid Commun. Mass Spectrom. 2012, 26, 1241–1248. [Google Scholar] [CrossRef]
- Yu, W.; Vath, J.E.; Huberty, M.C.; Martin, S.A. Identification of the facile gas-phase cleavage of the Asp-Pro and Asp-Xxx peptide bonds in matrix-assisted laser desorption time-of-flight mass spectrometry. Anal. Chem. 1993, 65, 3015–3023. [Google Scholar] [CrossRef]
- Carter, M.Q.; Pham, A.; Huynh, S.; He, X. Complete Genome Sequence of a Shiga Toxin-Producing Enterobacter cloacae Clinical Isolate. Genome Announc. 2017, 5, e00883-17. [Google Scholar] [CrossRef] [Green Version]
- Fagerquist, C.K.; Garbus, B.R.; Williams, K.E.; Bates, A.H.; Boyle, S.; Harden, L.A. Web-based software for rapid top-down proteomic identification of protein biomarkers with implications for bacterial identification. Appl. Environ. Microbiol. 2009, 75, 4341–4353. [Google Scholar] [CrossRef]
- Demirev, P.A.; Feldman, A.B.; Kowalski, P.; Lin, J.S. Top-down proteomics for rapid identification of intact microorganisms. Anal. Chem. 2005, 77, 7455–7461. [Google Scholar] [CrossRef]
- Rozman, M. Aspartic acid side chain effect-experimental and theoretical insight. J. Am. Soc. Mass Spectrom. 2007, 18, 121–127. [Google Scholar] [CrossRef]
- Fagerquist, C.K.; Sultan, O. Induction and identification of disulfide-intact and disulfide-reduced β-subunit of Shiga toxin 2 from Escherichia coli O157:H7 using MALDI-TOF-TOF-MS/MS and top-down proteomics. Analyst 2011, 136, 1739–1746. [Google Scholar] [CrossRef]
- Harrison, A.G. The gas-phase basicities and proton affinities of amino acids and peptides. Mass Spectrom. 1997, 16, 201–217. [Google Scholar] [CrossRef]
- Fagerquist, C.K.; Lee, B.G.; Zaragoza, W.J.; Yambao, J.C.; Quiñones, B. Software for top-down proteomic identification of a plasmid-borne factor (and other proteins) from genomically sequenced pathogenic bacteria using MALDI-TOF-TOF-MS/MS and post-source decay. Int. J. Mass Spectrom. 2019, 438, 1–12. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fagerquist, C.K.; Zaragoza, W.J.; Carter, M.Q. Top-Down Proteomic Identification of Shiga Toxin 1 and 2 from Pathogenic Escherichia coli Using MALDI-TOF-TOF Tandem Mass Spectrometry. Microorganisms 2019, 7, 488. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7110488
Fagerquist CK, Zaragoza WJ, Carter MQ. Top-Down Proteomic Identification of Shiga Toxin 1 and 2 from Pathogenic Escherichia coli Using MALDI-TOF-TOF Tandem Mass Spectrometry. Microorganisms. 2019; 7(11):488. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7110488
Chicago/Turabian StyleFagerquist, Clifton K., William J. Zaragoza, and Michelle Q. Carter. 2019. "Top-Down Proteomic Identification of Shiga Toxin 1 and 2 from Pathogenic Escherichia coli Using MALDI-TOF-TOF Tandem Mass Spectrometry" Microorganisms 7, no. 11: 488. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7110488