Computational Modelling of Metabolic Burden and Substrate Toxicity in Escherichia coli Carrying a Synthetic Metabolic Pathway


Abstract
:1. Introduction
2. Materials and Methods
2.1. Laboratory Methods
2.1.1. Bacterial Strains and Plasmids
2.1.2. Preparation of Pre-Induced Cells
2.1.3. Short Term Toxicity Test
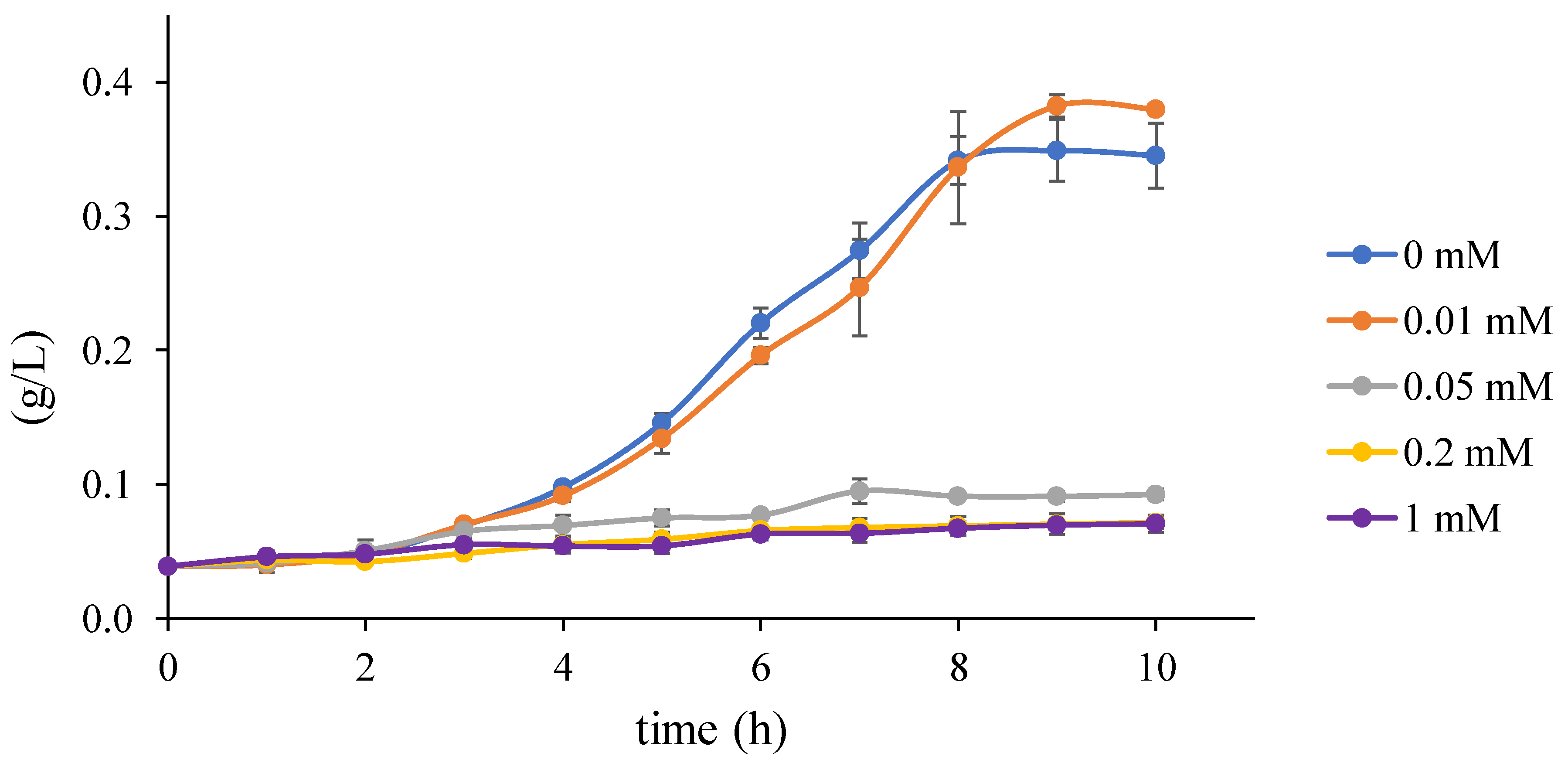
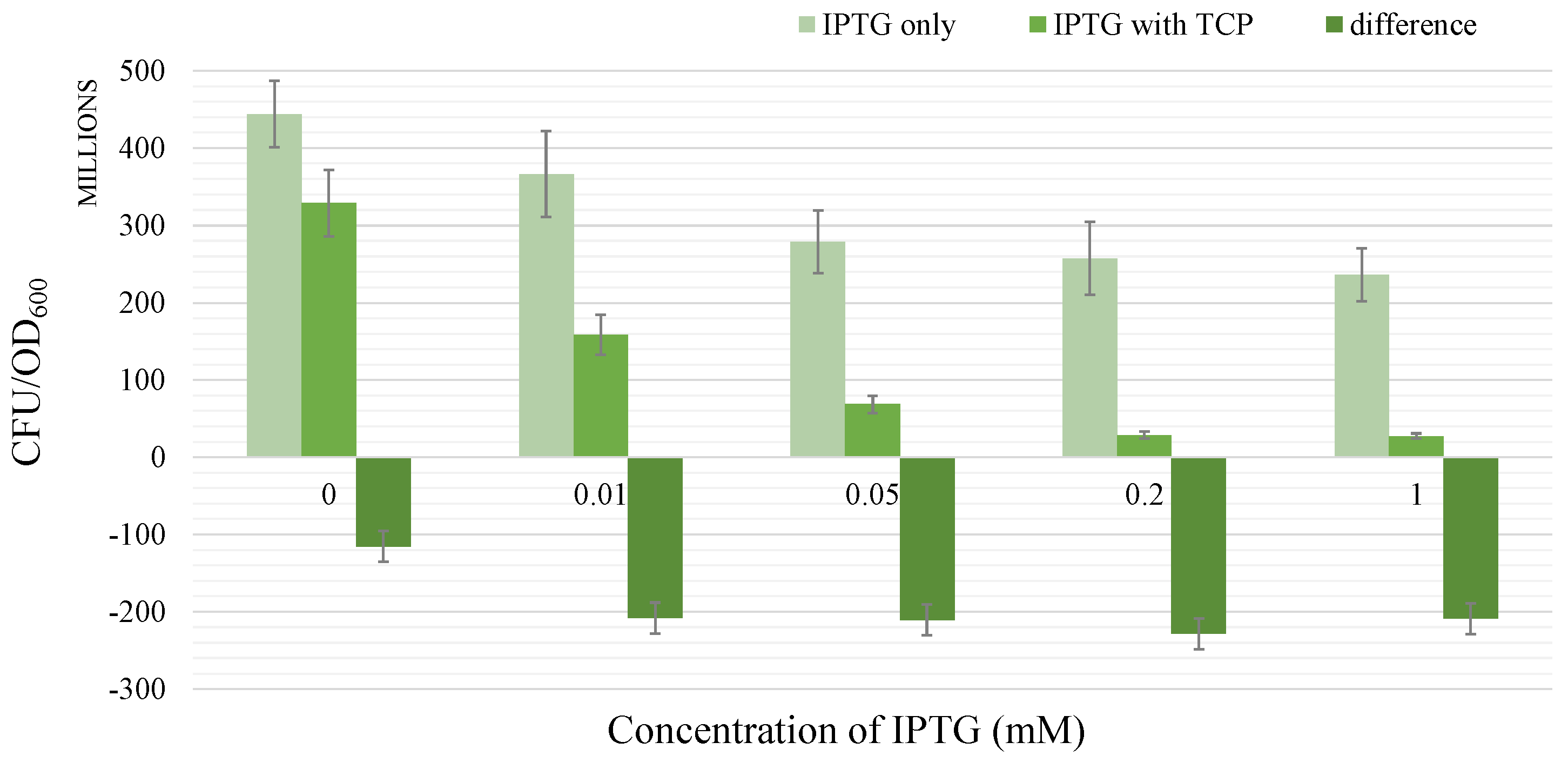
2.1.4. Growth Test
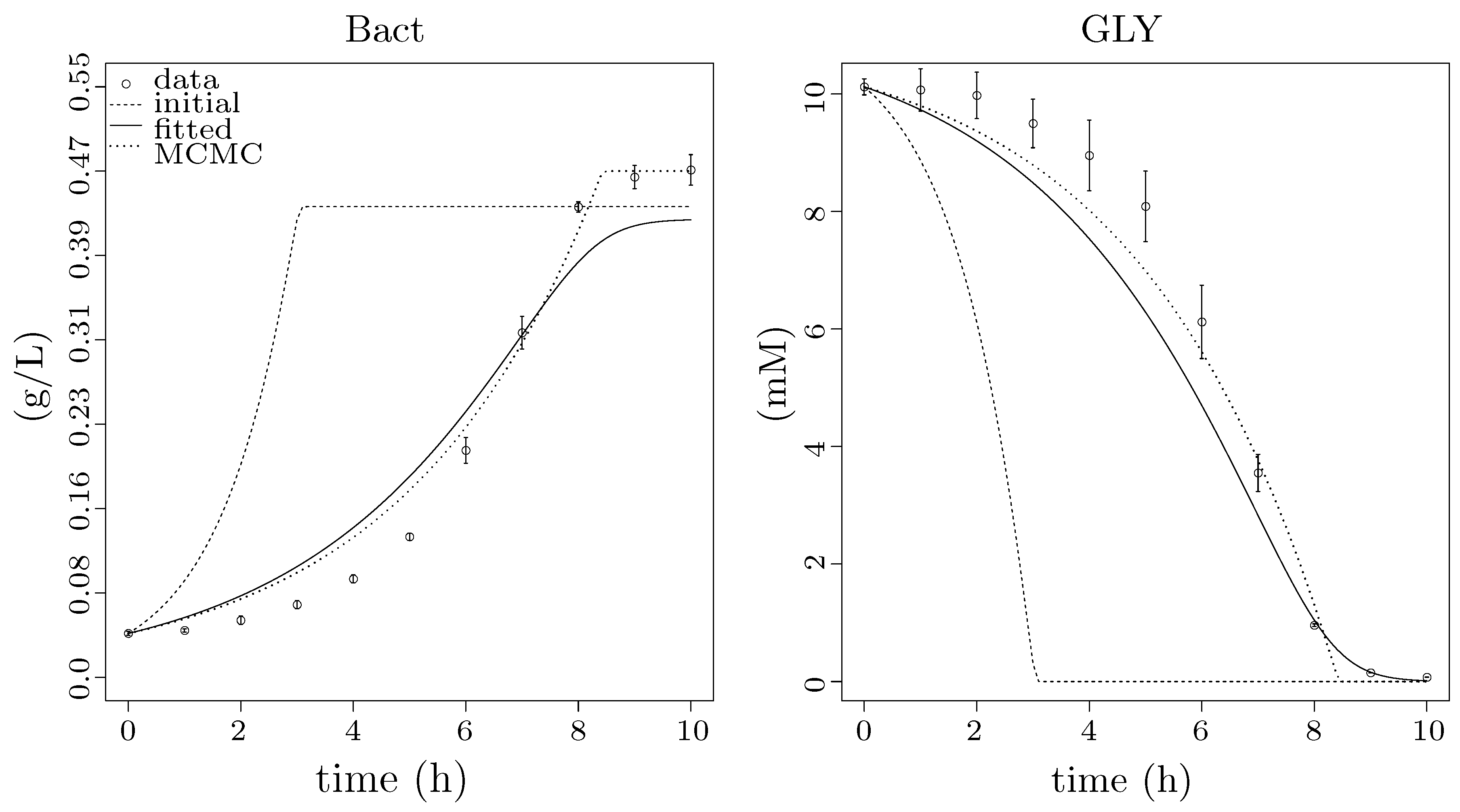
2.1.5. Glycerol Analysis
2.2. Parameter Constraints
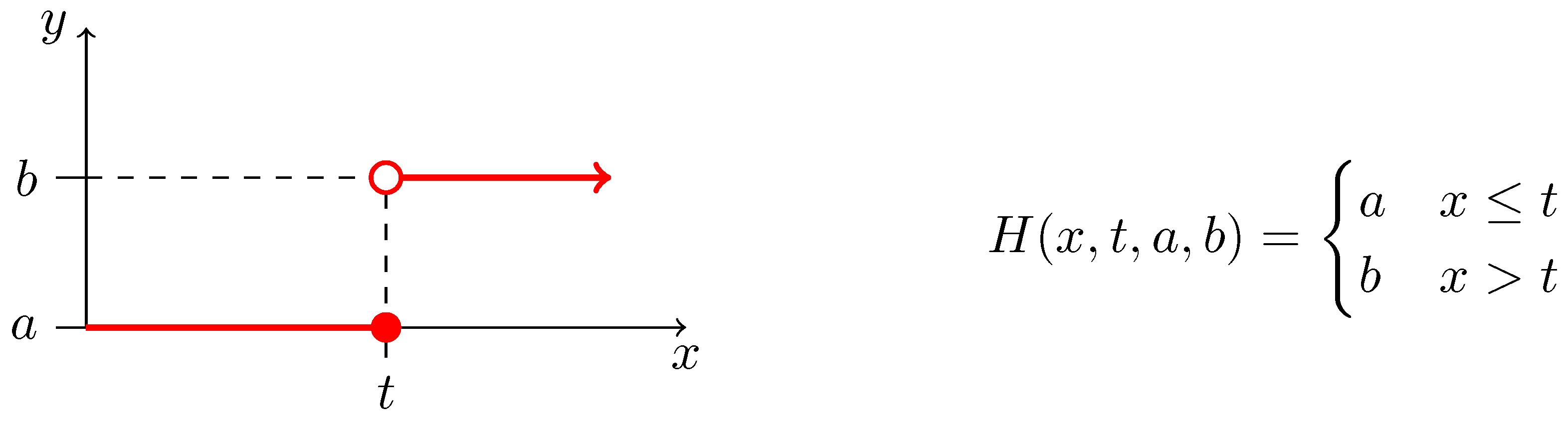
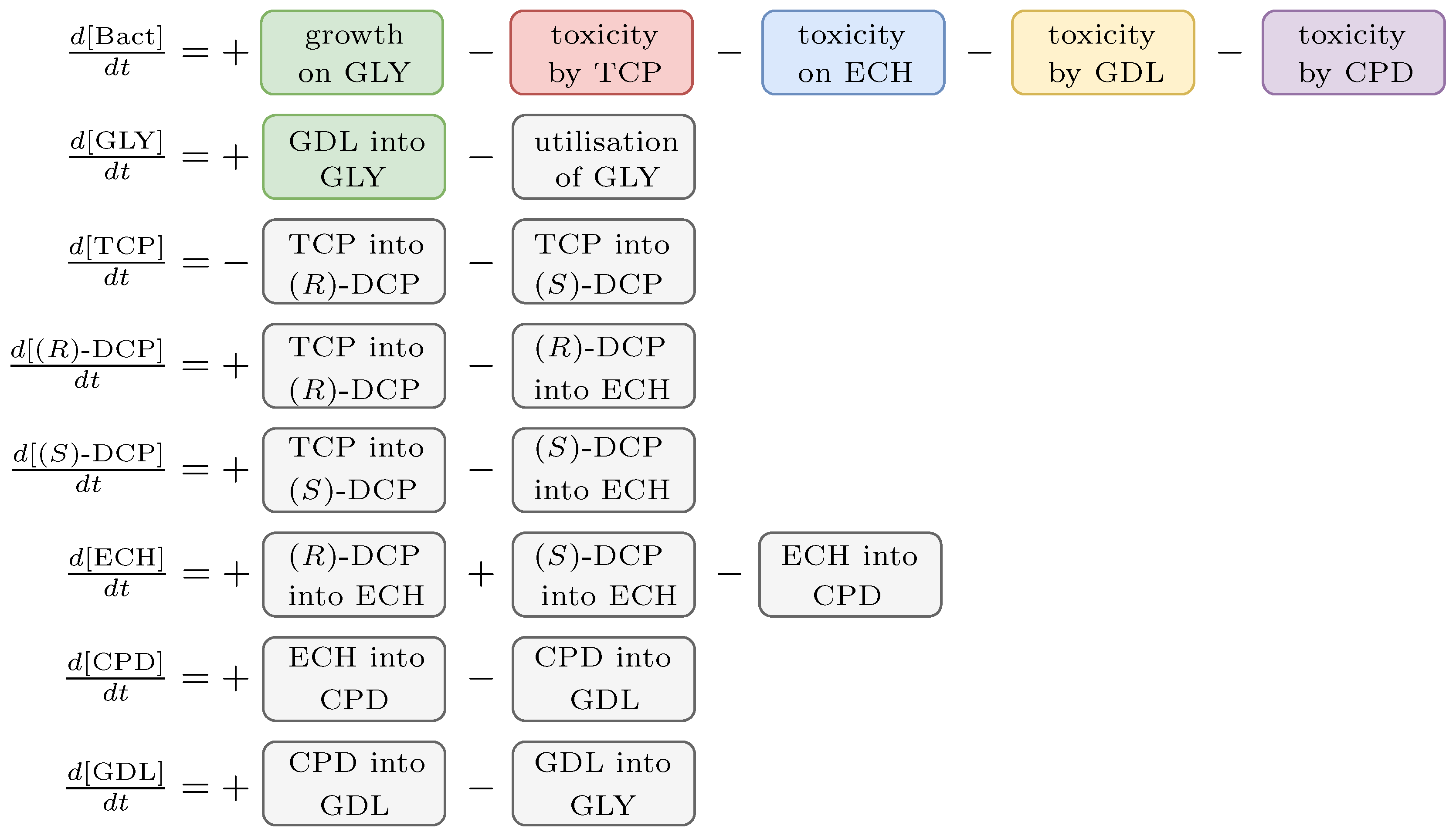
2.3. Biochemical Model
2.4. Inverse Problem
2.4.1. Parameter Estimation and Regression
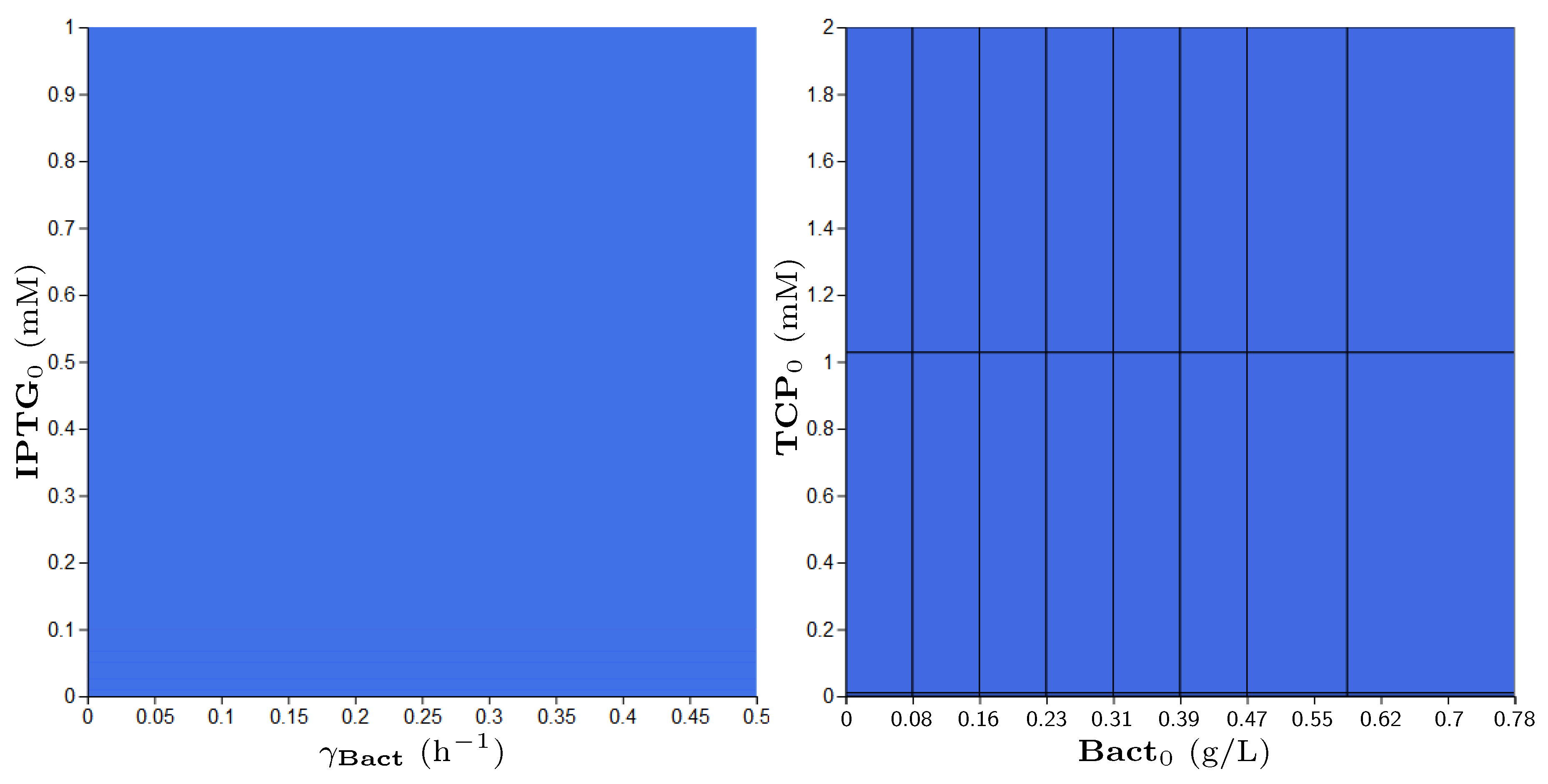
2.4.2. Parameter Synthesis and Robustness Monitoring
2.5. Analysis Workflow
2.5.1. General Assumptions
- We assume the total inducer concentration to be constant in the time frame of our interest. An inducer is supposed to have a function of an input parameter, and it would be an inadequate parameter should it be adjusted spontaneously over time. Otherwise, the inducer degradation rate would be needed either found in literature or extracted from experimental data.
- The workflow is limited to protease-deficient bacterial strains (e.g., E. coli BL21). In particular, we assume the total concentration of every enzyme affecting the studied pathway is constant in the time frame of our interest. Moreover, no influx of the enzymes is permitted as a consequence of time-limited induction phase where the proteosynthesis takes place [50,51]. Additional synthetic processes are considered negligible in a microbial population stressed enough by the massive expression of (heterologous) genes during induction.
- There occurs a metabolic burden effect caused by the heterologous genes expression during the induction process and possibly by the presence of an inducer itself which in a combined way affects the bacterial growth rate.
- Finally, we assume the bacterial population is in the stationary phase after the induction process is finished.
2.5.2. Workflow Description
2.6. Software Tools
3. Results and Discussion
3.1. Extended Assumptions
- We define a new variable called Bact standing for CDW (g/L) of E. coli population taken as 0.39 g/L = 1 [24].
- We assume IPTG to be the only inducer for the synthetic pathway. Concentration of IPTG is considered to be constant in the given time frame.
- Reversible reactions in the TCP-degradation pathway are considered negligible.
- The initial concentration of substances (e.g., , , ) and the population (i.e., ) determines the input for the system.
- Dynamics of individual enzymes is approximated as a constant function of time in the given time frame. Moreover, enzymes dynamics is considered to be independent on the size of the bacterial population in the given time frame.
- Total conversion of TCP into GLY is assumed to occur in a sufficiently long time reflecting the known behaviour of the pathway.
- Viability of the bacterial population is given as the function of the pathway compounds toxicity, metabolic burden and the presence of nutrients (i.e., GLY).
- Toxic effects of the pathway compounds are considered to be mutually independent.
- Glycerol is the only assumed nutrient.
- We assume natural degradation (death rate) of the bacterial population.
3.2. Workflow Input: Synthetic TCP Degradation Pathway
3.3. Step 1: Enzymatic Space Settings and Reduction
3.4. Step 2: Integration with Population Growth
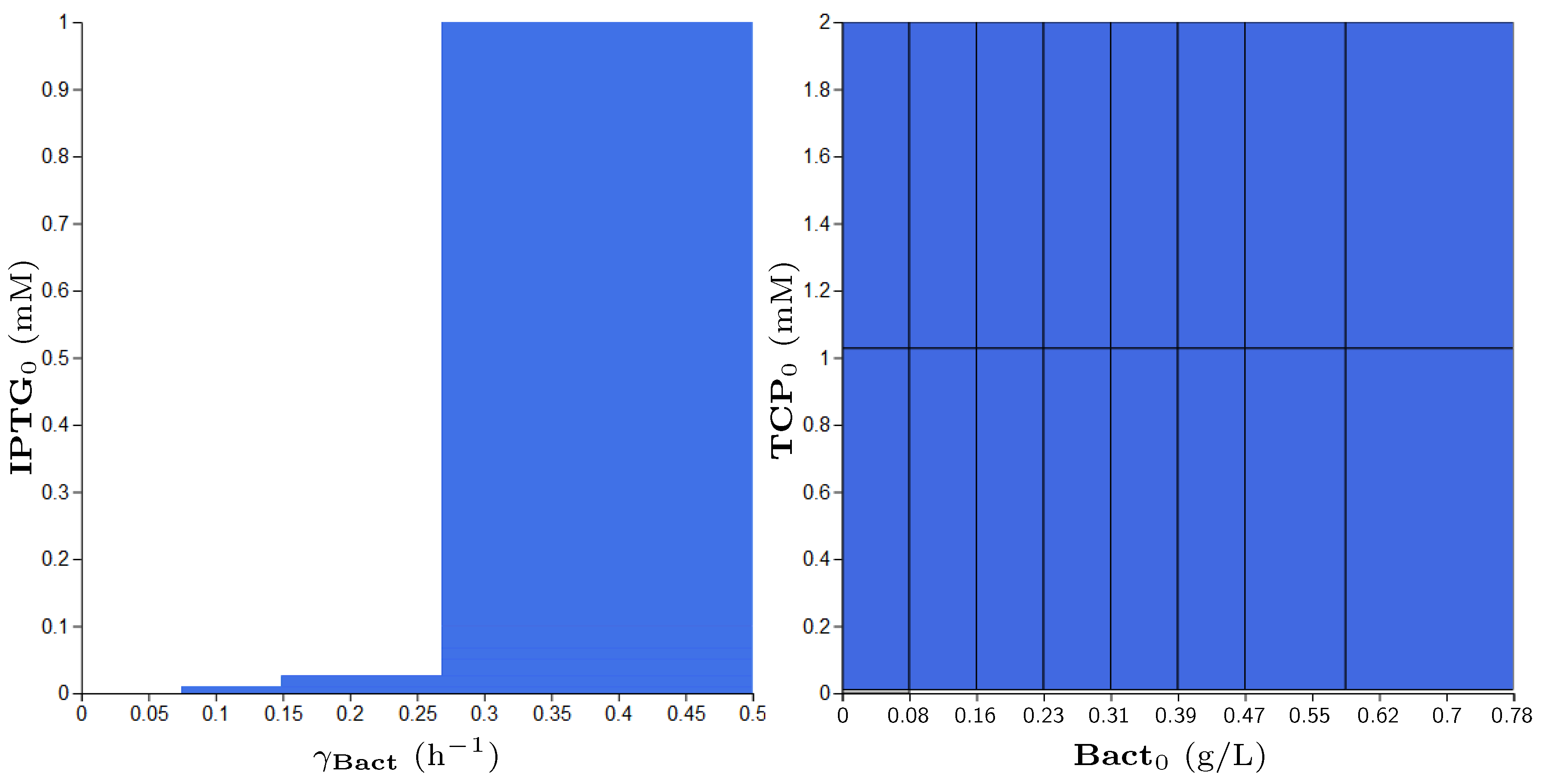
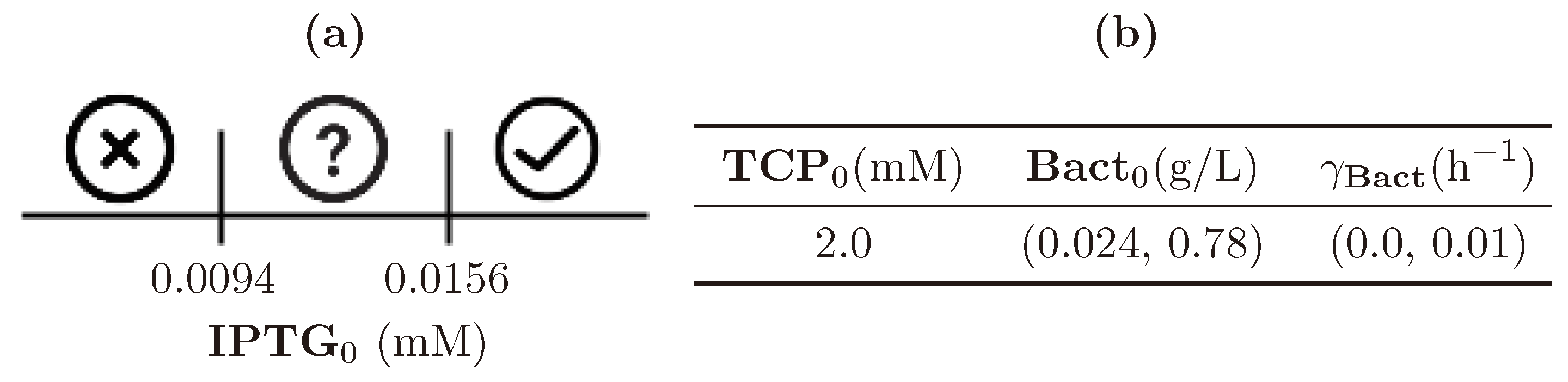
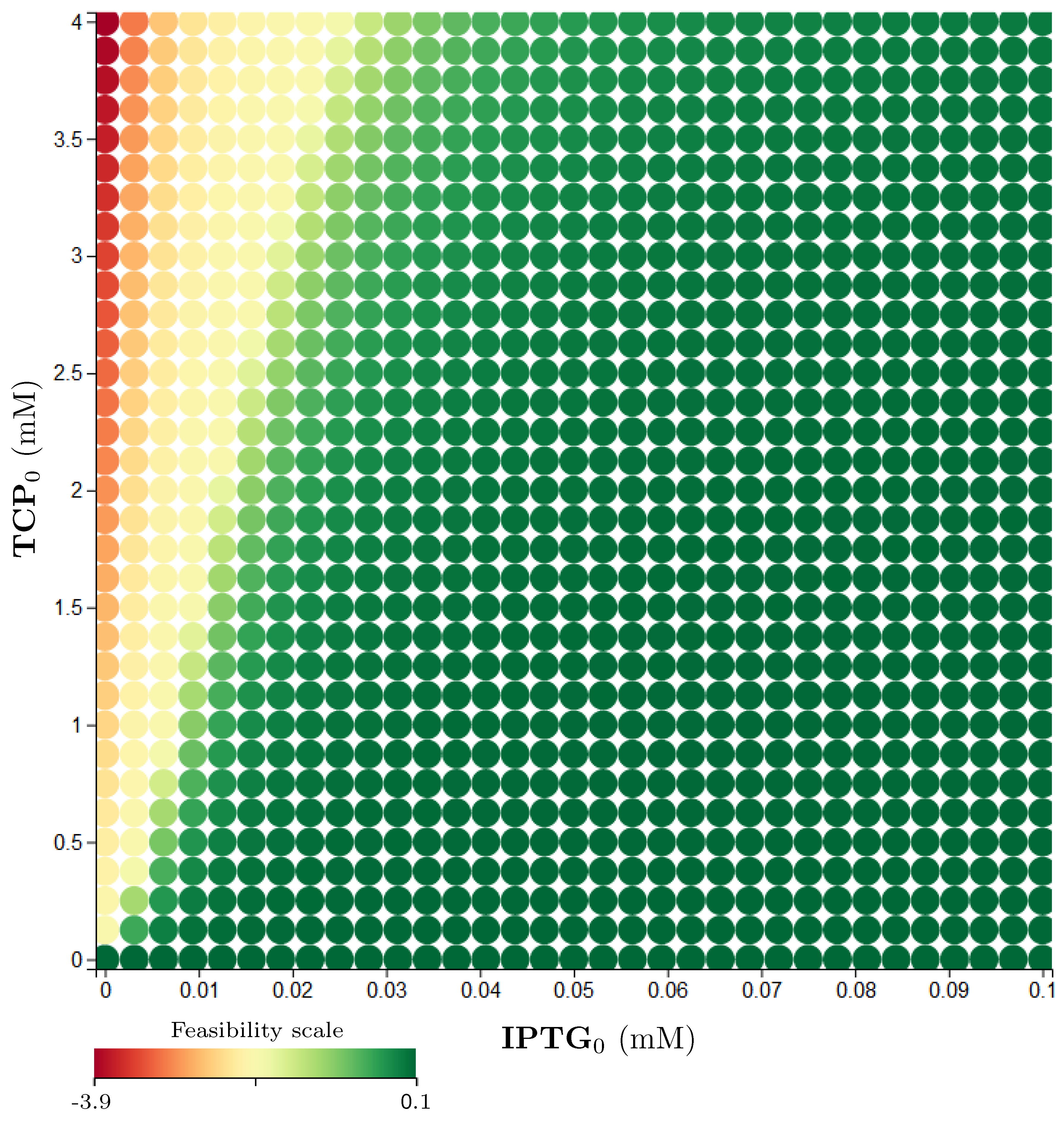
3.5. Step 3: Model Dynamics Exploration
- Concentration of IPTG in mM: IPTG is an obvious candidate for tuning because many aspects of the model depend on it and it can be controlled easily in the experimental environment (since its concentration is considered constant during the experiment, it can be referred by its initial concentration, denoted ).
- Size of bacterial population (Bact) in g/L: The initial population size, denoted , makes the crucial input of the model and it affects the model output—the final population size (reached in a given time). In general, the initial population size can be controlled in experiments.
- Initial concentration of TCP () in mM: The key input of the model that must be set in order to make the modelled metabolic pathway work; it can be easily set to any arbitrary value during the experiments.
- Death rate of the population () in: The death rate is considered as a parameter because we are interested in the dynamics of the microbial culture and the effects affecting the growth.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IPTG | isopropyl-beta-D-thiogalactopyranoside |
| TCP | 1,2,3-trichloropropane |
| DCP | 2,3-dichloropropane-1-ol |
| ECH | epichlorohydrin |
| CPD | 3-chloropropane-1,2-diol |
| GDL | glycidol |
| GLY | glycerol |
| DhaA | haloalkane dehalogenase |
| EchA | epoxide hydrolase |
| HheC | haloalcohol dehalogenase |
| MM | Michaelis-Menten |
| CDW | cell dry weight |
| ODE | ordinary differential equations |
| MCMC | Markov chain Monte Carlo |
| GUI | graphical user interface |
| CLI | command-line interface |
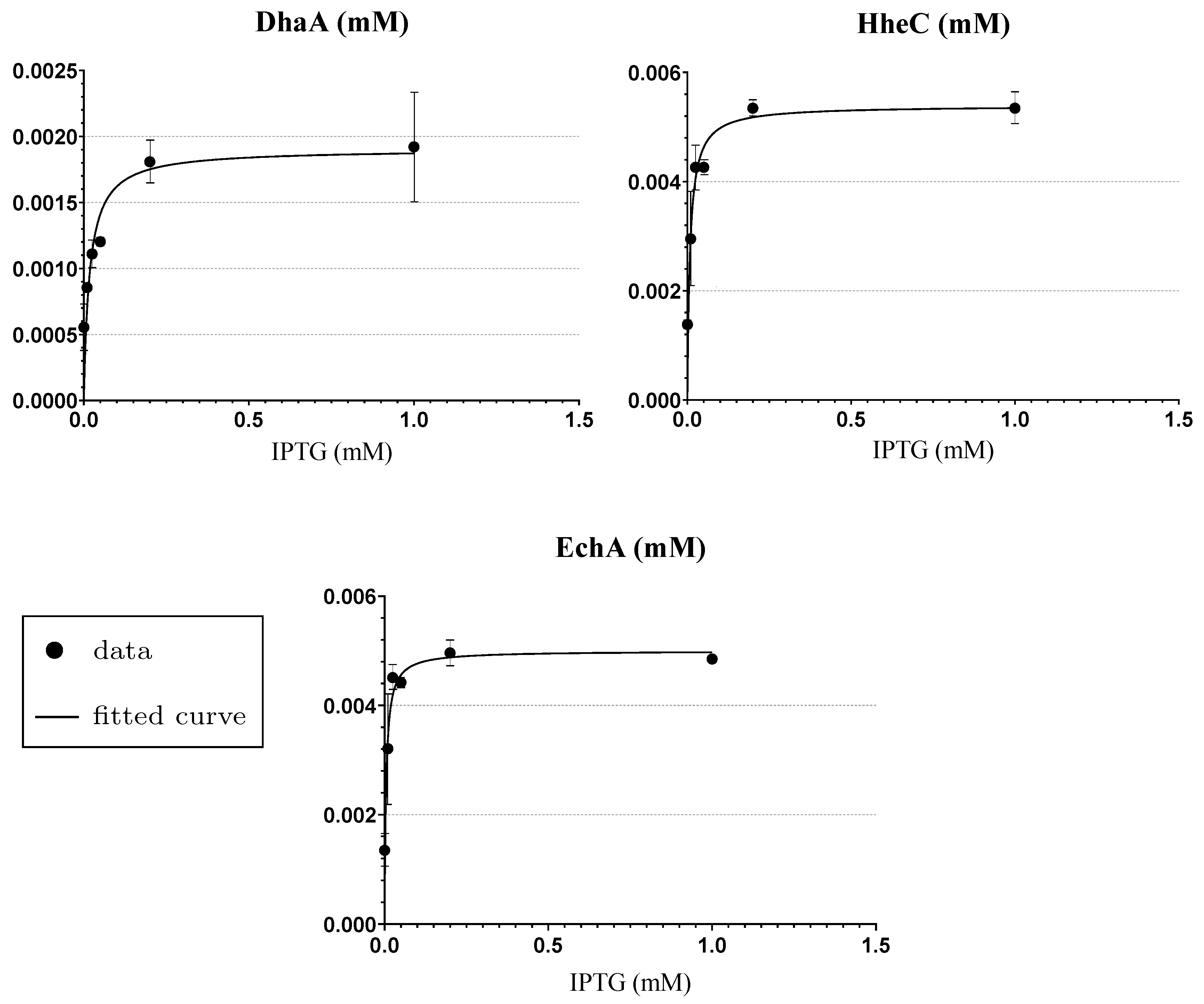
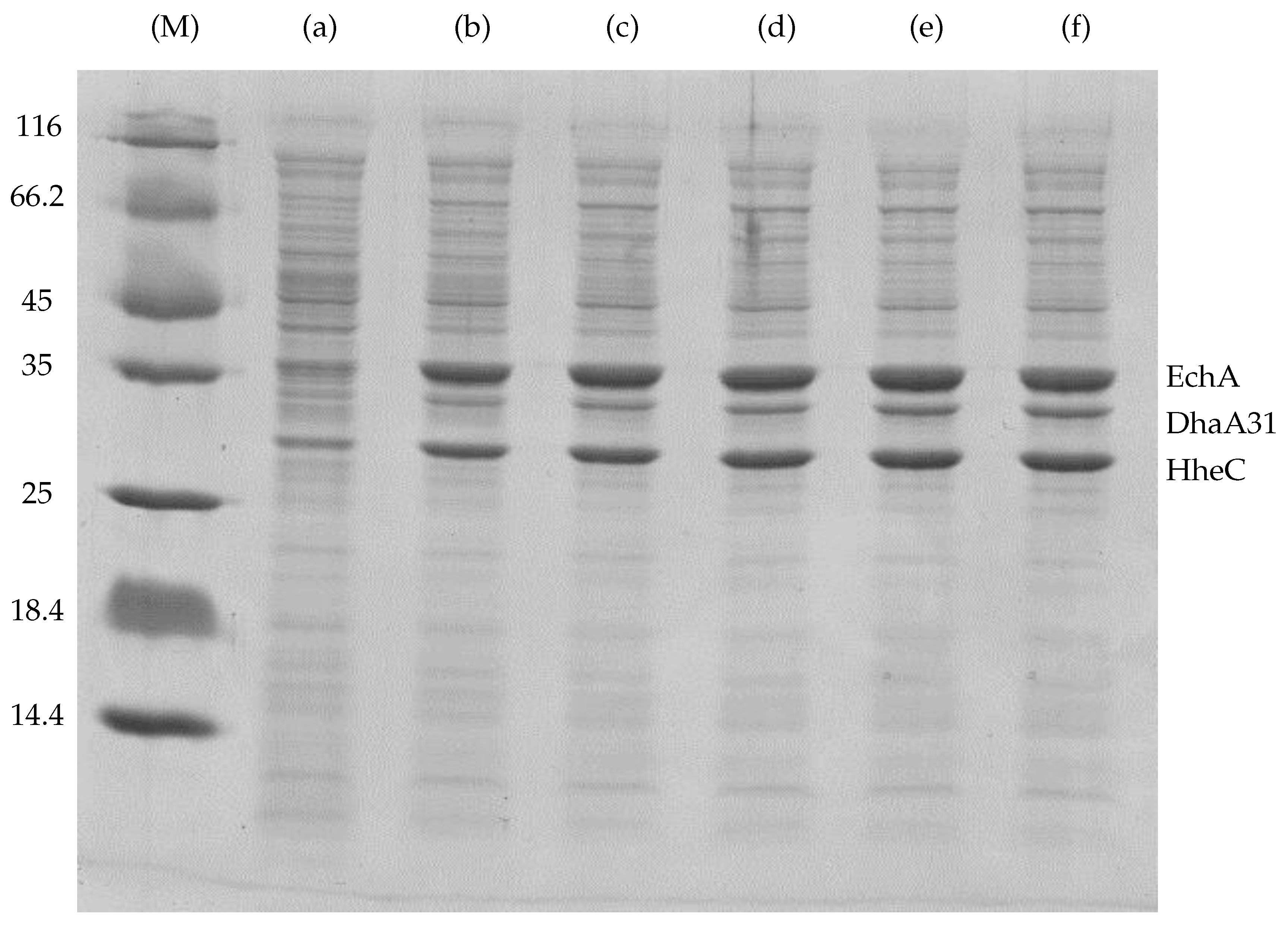
Appendix A. The Experimental Data Used for the Fitting of Functions Explaining the Enzymes Concentration Determined at Respective IPTG Concentrations

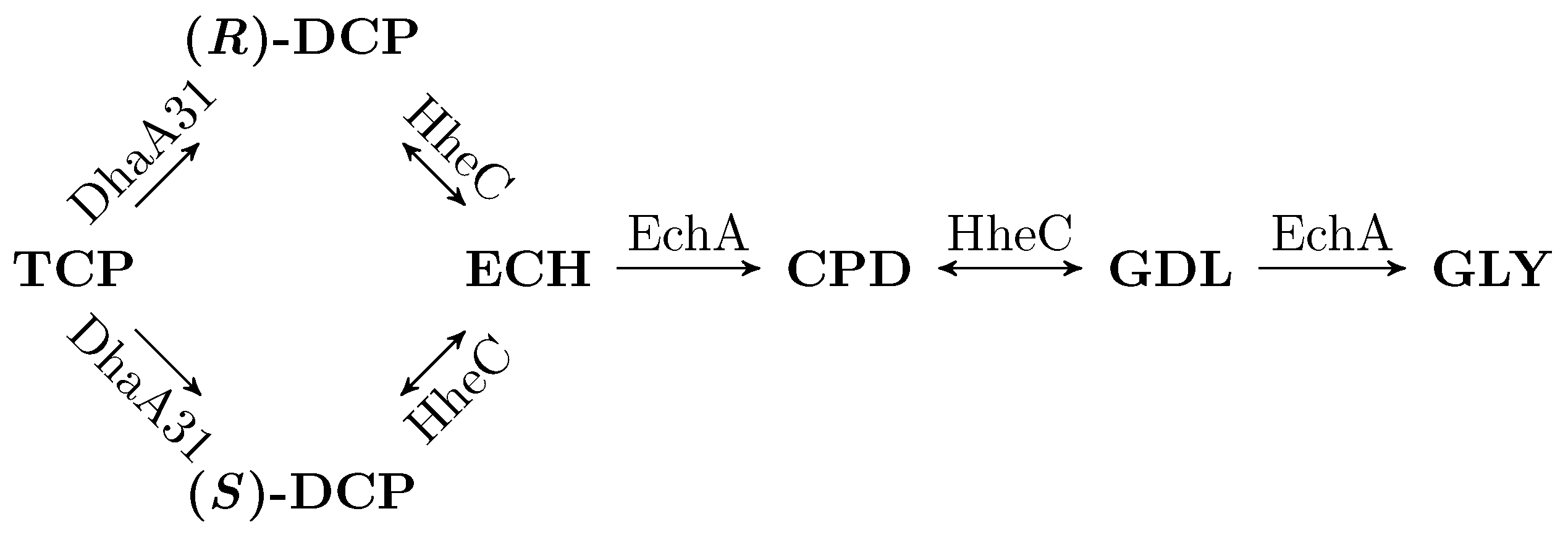{kind=link}
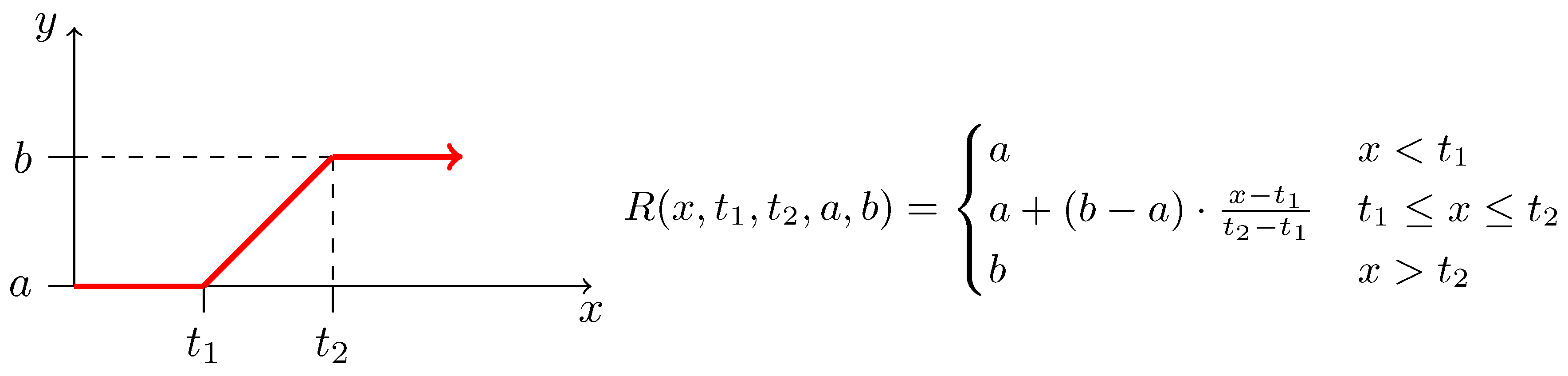{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
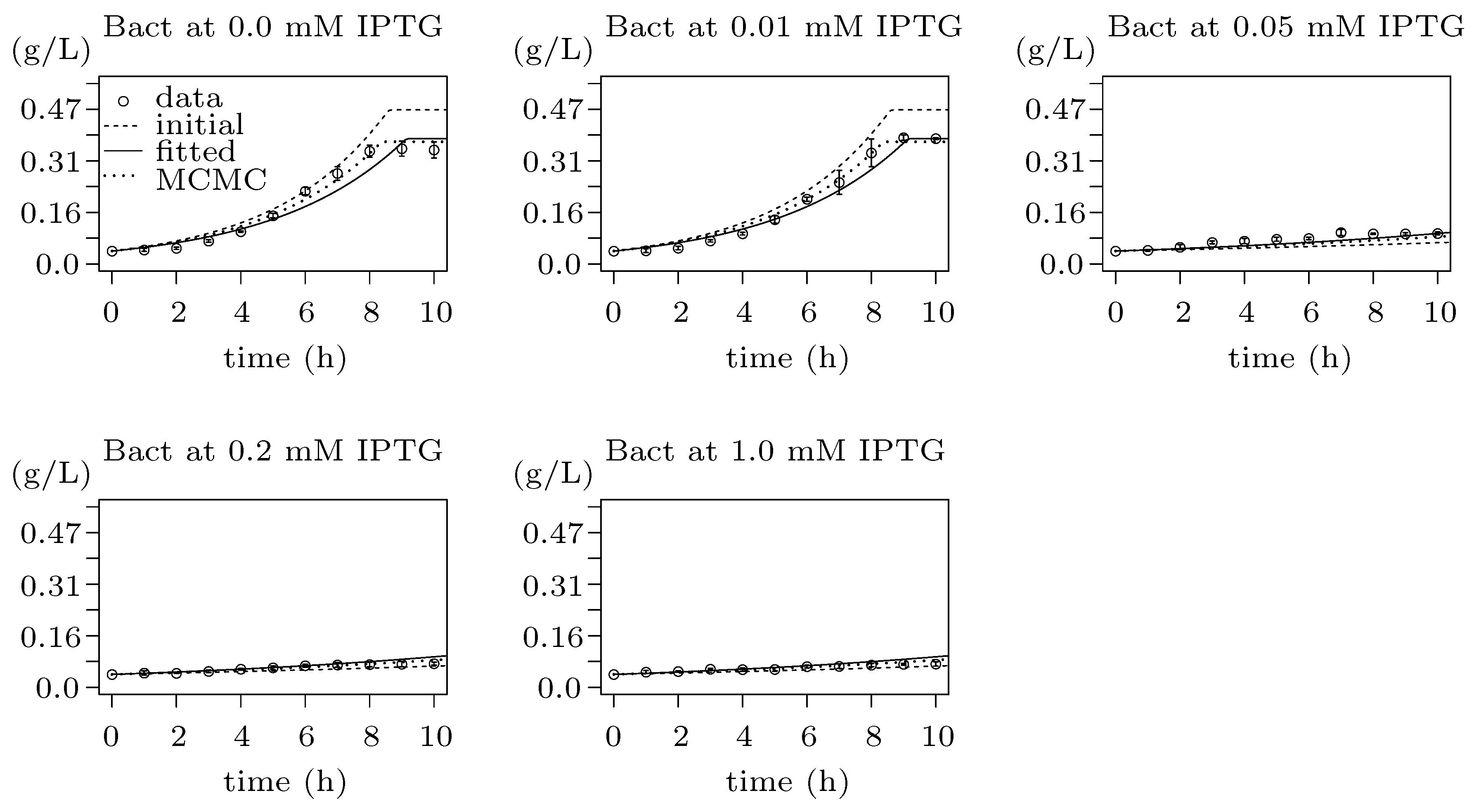{kind=link}
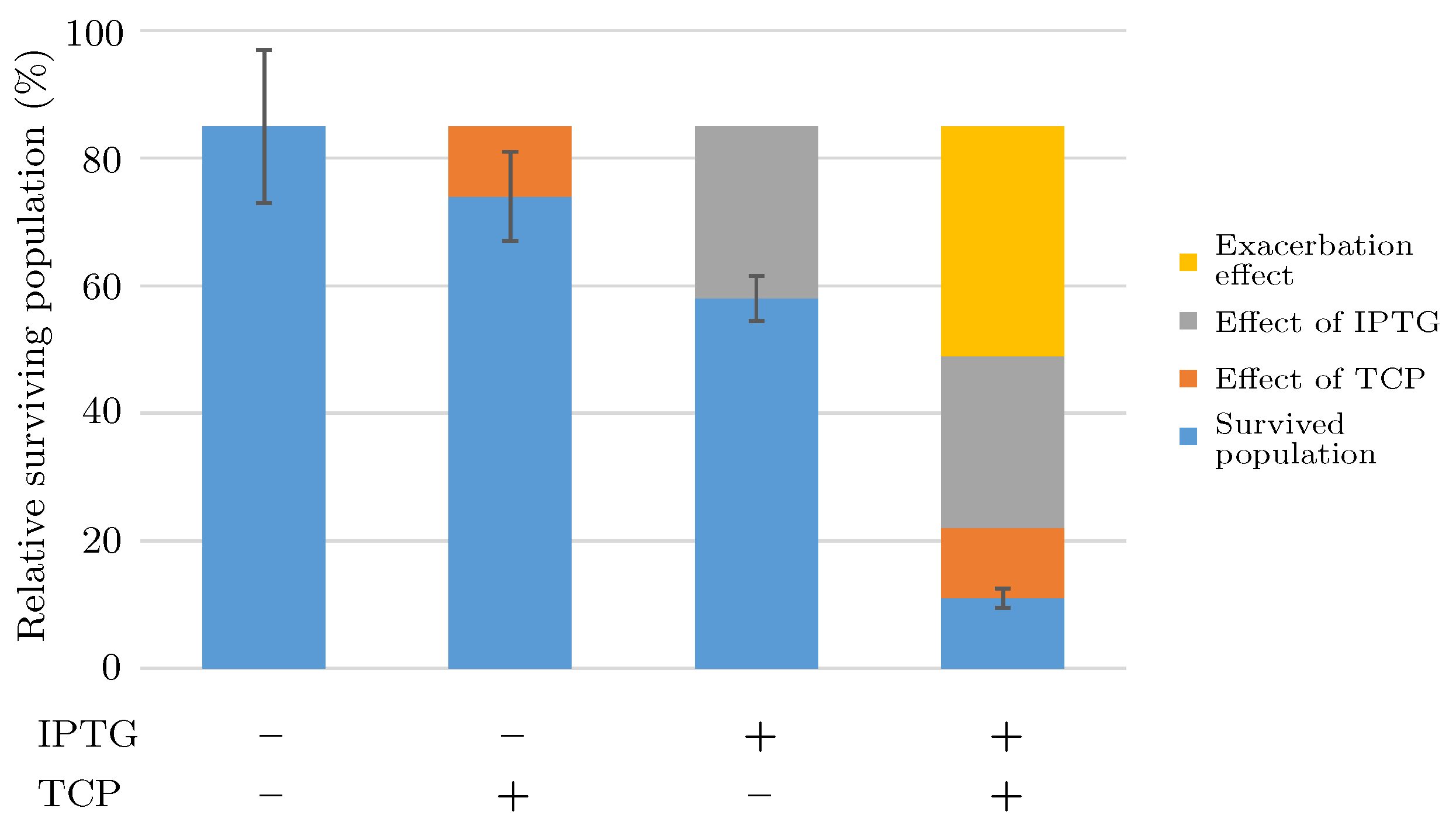{kind=link}
{kind=link}
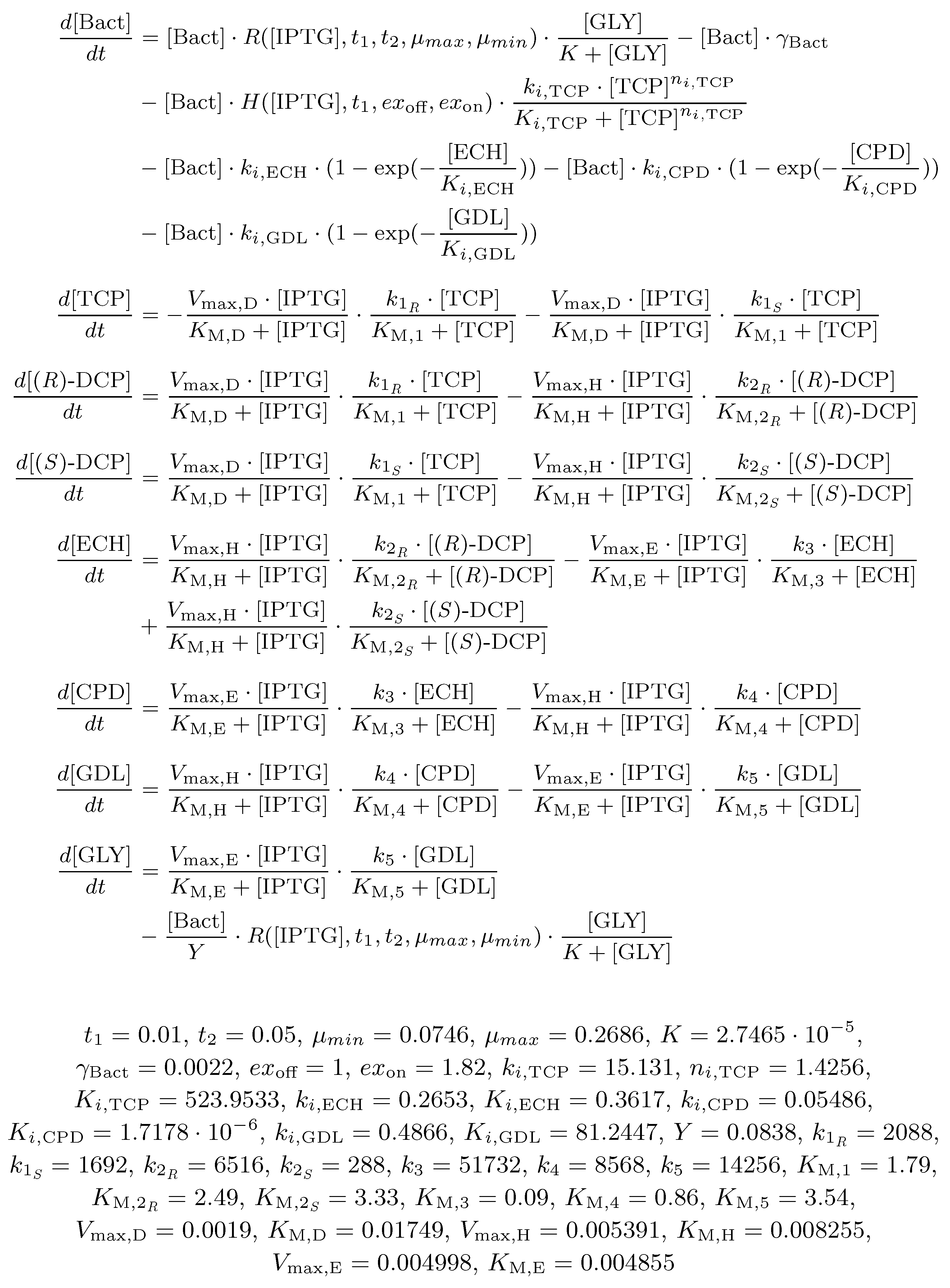{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IPTG (mM) | Content in Cell-Free Extract (%) * | DhaA31 (Relative Portion) | HheC (Relative Portion) | EchA (Relative Portion) | Total Mass () |
|---|---|---|---|---|---|
| 1.0 | 50 | 0.19 | 0.39 | 0.42 | 4.188 |
| 0.2 | 48 | 0.17 | 0.40 | 0.43 | 4.02 |
| 0.05 | 40 | 0.13 | 0.38 | 0.49 | 3.350 |
| 0.025 | 40 | 0.12 | 0.41 | 0.47 | 3.350 |
| 0.01 | 22 | 0.16 | 0.36 | 0.48 | 1.843 |
| 0.0 | 15 | 0.20 | 0.33 | 0.47 | 1.256 |
| IPTG (mM) | Content in Cell-Free Extract (%) * | DhaA31 (Relative Portion) | HheC (Relative Portion) | EchA (Relative Portion) | Total Mass () |
|---|---|---|---|---|---|
| 1.0 | 60 | 0.15 | 0.39 | 0.46 | 3.890 |
| 0.2 | 62 | 0.15 | 0.39 | 0.46 | 4.020 |
| 0.05 | 50 | 0.13 | 0.38 | 0.49 | 3.242 |
| 0.025 | 50 | 0.11 | 0.37 | 0.52 | 3.242 |
| 0.01 | 42 | 0.11 | 0.37 | 0.52 | 2.723 |
| 0.0 | 15 | 0.17 | 0.40 | 0.44 | 0.973 |

| IPTG | Total Mass | DhaA31 | HheC | EchA | DhaA31 | HheC | EchA |
|---|---|---|---|---|---|---|---|
| (mM) | * | * | * | (mM) # | (mM) # | (mM) # | |
| 1.0 | 4.188 | 79.6 | 163.3 | 175.9 | 2.24 × 10−3 | 5.57 × 10−3 | 4.82 × 10−3 |
| 0.2 | 4.02 | 68.3 | 160.8 | 172.9 | 1.92 × 10−3 | 5.48 × 10−3 | 4.74 × 10−3 |
| 0.05 | 3.350 | 43.6 | 127.3 | 164.2 | 1.22 × 10−3 | 4.34 × 10−3 | 4.50 × 10−3 |
| 0.025 | 3.350 | 40.2 | 137.4 | 157.5 | 1.13 × 10−3 | 4.68 × 10−3 | 4.32 × 10−3 |
| 0.01 | 1.843 | 29.5 | 66.3 | 88.4 | 8.29 × 10−4 | 2.26 × 10−3 | 2.43 × 10−3 |
| 0.0 | 1.256 | 25.1 | 41.5 | 59.0 | 7.06 × 10−4 | 1.41 × 10−3 | 1.62 × 10−3 |
| IPTG | Total Mass | DhaA31 | HheC | EchA | DhaA31 | HheC | EchA |
|---|---|---|---|---|---|---|---|
| (mM) | * | * | * | (mM) # | (mM) # | (mM) # | |
| 1.0 | 3.89 | 58.4 | 151.7 | 179.0 | 1.64 × 10−3 | 5.17 × 10−3 | 4.91 × 10−3 |
| 0.2 | 4.02 | 60.3 | 156.8 | 184.9 | 1.69 × 10−3 | 5.34 × 10−3 | 5.07 × 10−3 |
| 0.05 | 3.242 | 42.1 | 123.2 | 158.9 | 1.18 × 10−3 | 4.20 × 10−3 | 4.36 × 10−3 |
| 0.025 | 3.242 | 35.7 | 120.0 | 168.6 | 1.00 × 10−3 | 4.09 × 10−3 | 4.62 × 10−3 |
| 0.01 | 2.723 | 30.0 | 100.8 | 141.6 | 8.42 × 10−4 | 3.44 × 10−3 | 3.88 × 10−3 |
| 0.0 | 0.973 × 10−1 | 16.5 | 38.9 | 42.8 | 4.65 × 10−4 | 1.33 × 10−3 | 1.17 × 10−3 |
| DhaA31 (Da) | HheC (Da) | EchA (Da) |
|---|---|---|
| 35,576.37 | 29,333.07 | 36,465.11 |
| IPTG | DhaA31 (mM) | HheC (mM) | EchA (mM) | |||
|---|---|---|---|---|---|---|
| (mM) | Median | stdev | Median | stdev | Median | stdev |
| 1.0 | 1.94 × 10−3 | 4.22 × 10−4 | 5.37 × 10−3 | 2.79 × 10−4 | 4.87 × 10−3 | 5.97 × 10−5 |
| 0.2 | 1.81 × 10−3 | 1.60 × 10−4 | 5.41 × 10−3 | 9.69 × 10−5 | 4.91 × 10−3 | 2.34 × 10−4 |
| 0.05 | 1.20 × 10−3 | 2.78 × 10−5 | 4.27 × 10−3 | 9.90 × 10−5 | 4.43 × 10−3 | 1.03 × 10−4 |
| 0.025 | 1.07 × 10−3 | 9.02 × 10−5 | 4.39 × 10−3 | 4.19 × 10−4 | 4.47 × 10−3 | 2.16 × 10−4 |
| 0.01 | 8.35 × 10−4 | 9.45 × 10−6 | 2.85 × 10−3 | 8.30 × 10−4 | 3.15 × 10−3 | 1.03 × 10−3 |
| 0.0 | 5.85 × 10−4 | 1.71 × 10−4 | 1.37 × 10−3 | 6.15 × 10−5 | 1.40 × 10−3 | 3.15 × 10−4 |
Appendix B. The Origin of the Death Rate Coefficient
| g () | (h) | () as 0.5% p.g. | () as 1% p.g. |
|---|---|---|---|
| 0.26 | 2.58 | ||
| 0.07 | 9.29 |
Appendix C. The List of Considered Inhibition Models
| Monod + Hill [27]: | (A3) | |
| Monod + Aiba-Edward [65]: | (A4) | |
| Monod + Andrews [66]: | (A5) | |
| Monod + Haldane-Andrews [66]: | (A6) | |
| Monod + Monod: | (A7) | |
| Monod + Moser [64]: | (A8) | |
| Monod + Tessier [63]: | (A9) | |
| Monod + Tessier-type [65]: | (A10) | |
| competitive inhibition: | (A11) | |
| non-competitive inhibition: | (A12) | |
| uncompetitive inhibition: | (A13) | |
| non-competitive inhibition using negative Hill | (A14) | |
| non-competitive inhibition using negative Moser | (A15) | |
| non-competitive exponential inhibition | (A16) |
References
- Marisch, K.; Bayer, K.; Cserjan-Puschmann, M.; Luchner, M.; Striedner, G. Evaluation of three industrial Escherichia coli strains in fed-batch cultivations during high-level SOD protein production. Microb. Cell Factories 2013, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kuipers, G.; Niemiec, Ł.; Baumgarten, T.; Slotboom, D.J.; de Gier, J.W.; Hjelm, A. High-level production of membrane proteins in E. coli BL21 (DE3) by omitting the inducer IPTG. Microb. Cell Factories 2015, 14, 142. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Keum, K.C.; Lee, S.Y. Production of recombinant proteins by high cell density culture of Escherichia coli. Chem. Eng. Sci. 2006, 61, 876–885. [Google Scholar] [CrossRef]
- Balzer, S.; Kucharova, V.; Megerle, J.; Lale, R.; Brautaset, T.; Valla, S. A comparative analysis of the properties of regulated promoter systems commonly used for recombinant gene expression in Escherichia coli. Microb. Cell Factories 2013, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Tolia, N.H.; Joshua-Tor, L. Strategies for protein coexpression in Escherichia coli. Nat. Methods 2006, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Vansiri, A.; Bhan, N.; Koffas, M.A. ePathBrick: A synthetic biology platform for engineering metabolic pathways in E. coli. ACS Synth. Biol. 2012, 1, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Du, G.; Zhou, J.; Chen, J. Metabolic engineering of Escherichia coli for (2S)-pinocembrin production from glucose by a modular metabolic strategy. Metab. Eng. 2013, 16, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Gu, Q.; Wang, W.; Wong, L.; Bower, A.G.; Collins, C.H.; Koffas, M.A. Modular optimization of multi-gene pathways for fatty acids production in E. coli. Nat. Commun. 2013, 4, 1409. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Y.; Zhang, C.; Yang, S.; Cui, J.Y.; Jiang, P.X.; Lou, K.; Wachi, M.; Xing, X.H. High crude violacein production from glucose by Escherichia coli engineered with interactive control of tryptophan pathway and violacein biosynthetic pathway. Microb. Cell Factories 2015, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Men, X.; Chen, H.; Li, M.; Ding, Z.; Chen, G.; Wang, F.; Liu, H.; Wang, Q.; Zhu, Y.; et al. A systematic optimization of styrene biosynthesis in Escherichia coli BL21 (DE3). Biotechnol. Biofuels 2018, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Fordjour, E.; Adipah, F.K.; Zhou, S.; Du, G.; Zhou, J. Metabolic engineering of Escherichia coli BL21 (DE3) for de novo production of l-DOPA from d-glucose. Microb. Cell Factories 2019, 18, 74. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Database; CID=656894. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/656894 (accessed on 18 December 2018).
- Glick, B.R. Metabolic load and heterologous gene expression. Biotechnol. Adv. 1995, 13, 247–261. [Google Scholar] [CrossRef]
- Wu, G.; Yan, Q.; Jones, J.A.; Tang, Y.J.; Fong, S.S.; Koffas, M.A. Metabolic burden: Cornerstones in synthetic biology and metabolic engineering applications. Trends Biotechnol. 2016, 34, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Kim, S.W.; Keasling, J. Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metab. Eng. 2000, 2, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.; Queiroz, J.A.; Domingues, F.C. Evaluating metabolic stress and plasmid stability in plasmid DNA production by Escherichia coli. Biotechnol. Adv. 2012, 30, 691–708. [Google Scholar] [CrossRef] [PubMed]
- Mairhofer, J.; Scharl, T.; Marisch, K.; Cserjan-Puschmann, M.; Striedner, G. Comparative transcription profiling and in-depth characterization of plasmid-based and plasmid-free Escherichia coli expression systems under production conditions. Appl. Environ. Microbiol. 2013, 79, 3802–3812. [Google Scholar] [CrossRef] [PubMed]
- Kurumbang, N.P.; Dvorak, P.; Bendl, J.; Brezovsky, J.; Prokop, Z.; Damborsky, J. Computer-assisted engineering of the synthetic pathway for biodegradation of a toxic persistent pollutant. ACS Synth. Biol. 2013, 3, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Dvořák, P.; Chrást, L.; Nikel, P.I.; Fedr, R.; Souček, K.; Sedláčková, M.; Chaloupková, R.; Lorenzo de, V.; Prokop, Z.; Damborský, J. Exacerbation of substrate toxicity by IPTG in Escherichia coli BL21 (DE3) carrying a synthetic metabolic pathway. Microb. Cell Factories 2015, 14, 201. [Google Scholar] [CrossRef] [PubMed]
- de Jong, H.; Casagranda, S.; Giordano, N.; Cinquemani, E.; Ropers, D.; Geiselmann, J.; Gouzé, J.L. Mathematical modelling of microbes: Metabolism, gene expression and growth. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Weiße, A.Y.; Oyarzún, D.A.; Danos, V.; Swain, P.S. Mechanistic links between cellular trade-offs, gene expression, and growth. Proc. Natl. Acad. Sci. USA 2015, 112, E1038–E1047. [Google Scholar] [CrossRef] [PubMed]
- Kitano, H. Systems biology: Towards system-level understanding of biological systems. Found. Syst. Biol. 2001, 1. [Google Scholar]
- Dvořák, P. Engineering of the Synthetic Metabolic Pathway for Biodegradation of an Environmental Pollutant. Ph.D. Thesis, Masaryk University, Faculty of Science, Brno, Czech Republic, 2014. [Google Scholar]
- Glazyrina, J.; Materne, E.M.; Dreher, T.; Storm, D.; Junne, S.; Adams, T.; Greller, G.; Neubauer, P. High cell density cultivation and recombinant protein production with Escherichia coli in a rocking-motion-type bioreactor. Microb. Cell Factories 2010, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Keener, J.P.; Sneyd, J. Mathematical Physiology; Springer: New York, NY, USA, 1998; Volume 1. [Google Scholar]
- Michaelis, L.; Menten, M.L. Die kinetik der invertinwirkung. Biochem. Ztg. 1913, 49, 333–369. [Google Scholar]
- Hill, A.V. The possible effects of the aggregation of the molecules of hæmoglobin on its dissociation curves. J. Physiol. 1910, 40, 4–7. [Google Scholar]
- Monod, J. Recherches sur la Croissance des Cultures Bactériennes; Hermann & Cie: Paris, France, 1942. [Google Scholar]
- Yordanov, B.; Belta, C. Parameter synthesis for piecewise affine systems from temporal logic specifications. In Hybrid Systems: Computation and Control; Springer: New York, NY, USA, 2008; pp. 542–555. [Google Scholar]
- Donzé, A.; Clermont, G.; Langmead, C.J. Parameter synthesis in nonlinear dynamical systems: Application to systems biology. J. Comput. Biol. 2010, 17, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Donzé, A. Breach, a toolbox for verification and parameter synthesis of hybrid systems. In Proceedings of the Computer Aided Verification, Edinburgh, UK, 15–19 July 2010; Springer: New York, NY, USA, 2010; pp. 167–170. [Google Scholar]
- Barnat, J.; Brim, L.; Krejčí, A.; Streck, A.; Šafránek, D.; Vejnár, M.; Vejpustek, T. On parameter synthesis by parallel model checking. IEEE/ACM Trans. Comput. Biol. Bioinform. (TCBB) 2012, 9, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Češka, M.; Dannenberg, F.; Kwiatkowska, M.; Paoletti, N. Precise parameter synthesis for stochastic biochemical systems. In Computational Methods in Systems Biology; Springer: Cham, Switzerland, 2014; pp. 86–98. [Google Scholar]
- Brim, L.; Češka, M.; Demko, M.; Pastva, S.; Šafránek, D. Parameter Synthesis by Parallel Coloured CTL Model Checking. In Computational Methods in Systems Biology; Springer: Cham, Switzerland, 2015; Volume 9308, pp. 251–263. [Google Scholar]
- Bortolussi, L.; Milios, D.; Sanguinetti, G. U-Check: Model Checking and Parameter Synthesis Under Uncertainty. In Proceedings of the QEST’15, Madrid, Spain, 1–3 September 2015; Lecture Notes in Computer Science. Springer: New York, NY, USA, 2015; Volume 9259, pp. 89–104. [Google Scholar]
- Bogomolov, S.; Schilling, C.; Bartocci, E.; Batt, G.; Kong, H.; Grosu, R. Abstraction-Based Parameter Synthesis for Multiaffine Systems. In Hardware and Software: Verification and Testing; Springer: New York, NY, USA, 2015; pp. 19–35. [Google Scholar]
- Demko, M.; Beneš, N.; Brim, L.; Pastva, S.; Šafránek, D. High-performance symbolic parameter synthesis of biological models: A case study. In Computational Methods in Systems Biology; Springer: Cham, Switzerland, 2016; pp. 82–97. [Google Scholar]
- Beneš, N.; Brim, L.; Demko, M.; Pastva, S.; Šafránek, D. Parallel SMT-based parameter synthesis with application to piecewise multi-affine systems. In Proceedings of the International Symposium on Automated Technology for Verification and Analysis, Chiba, Japan, 17–20 October 2016; Springer: New York, NY, USA, 2016; Volume 9936, pp. 192–208. [Google Scholar]
- Pastva, S. Parallel Parameter Synthesis from Hybrid Logic HUCTL Formulas. Master’s Thesis, Masaryk University, Faculty of Informatics, Brno, Czech Republic, 2017. [Google Scholar]
- Beneš, N.; Brim, L.; Demko, M.; Pastva, S.; Šafránek, D. Pithya: A Parallel Tool for Parameter Synthesis of Piecewise Multi-affine Dynamical Systems. In Proceedings of the International Conference on Computer Aided Verification (CAV), Heidelberg, Germany, 24–28 July 2017; Springer: New York, NY, USA, 2017; pp. 591–598. [Google Scholar]
- Aster, R.C.; Borchers, B.; Thurber, C.H. Parameter Estimation and Inverse Problems; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Hughes, G.E.; Cresswell, M.J. A New Introduction to Modal Logic; Psychology Press: London, UK, 1996. [Google Scholar]
- Rizk, A.; Batt, G.; Fages, F.; Soliman, S. A general computational method for robustness analysis with applications to synthetic gene networks. Bioinformatics 2009, 25, i169–i178. [Google Scholar] [CrossRef] [PubMed]
- Rizk, A.; Batt, G.; Fages, F.; Soliman, S. Continuous valuations of temporal logic specifications with applications to parameter optimization and robustness measures. Theor. Comput. Sci. 2011, 412, 2827–2839. [Google Scholar] [CrossRef]
- Donzé, A.; Fanchon, E.; Gattepaille, L.M.; Maler, O.; Tracqui, P. Robustness analysis and behavior discrimination in enzymatic reaction networks. PLoS ONE 2011, 6, e24246. [Google Scholar] [CrossRef] [PubMed]
- Brim, L.; Dluhoš, P.; Šafránek, D.; Vejpustek, T. STL*: Extending signal temporal logic with signal-value freezing operator. Inf. Comput. 2014, 236, 52–67. [Google Scholar] [CrossRef]
- Vejpustek, T. Robustness Analysis of Extended Signal Temporal Logic STL*. Master’s Thesis, Masaryk University, Faculty of Informatics, Brno, Czech Republic, 2013. [Google Scholar]
- Brim, L.; Vejpustek, T.; Šafránek, D.; Fabriková, J. Robustness Analysis for Value-Freezing Signal Temporal Logic. Electron. Proc. Theor. Comput. Sci. 2013, 125, 20–36. [Google Scholar] [CrossRef]
- Nelson, D.L.; Lehninger, A.L.; Cox, M.M. Lehninger Principles of Biochemistry; Macmillan: New York, NY, USA, 2008. [Google Scholar]
- Studier, F.W.; Moffatt, B.A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986, 189, 113–130. [Google Scholar] [CrossRef]
- Ferrer-Miralles, N.; Saccardo, P.; Corchero, J.L.; Xu, Z.; García-Fruitós, E. General introduction: Recombinant protein production and purification of insoluble proteins. In Insoluble Proteins; Springer: New York, NY, USA, 2015; pp. 1–24. [Google Scholar]
- Sadhukhan, S.; Villa, R.; Sarkar, U. Microbial production of succinic acid using crude and purified glycerol from a Crotalaria juncea based biorefinery. Biotechnol. Rep. 2016, 10, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Gadd, G.M. Bacterial Physiology and Metabolism; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Soetaert, K.; Petzoldt, T. Inverse Modelling, Sensitivity and Monte Carlo Analysis in R Using Package FME. J. Stat. Softw. Artic. 2010, 33, 1–28. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Beneš, N.; Brim, L.; Demko, M.; Pastva, S.; Šafránek, D. A model checking approach to discrete bifurcation analysis. In FM 2016: Formal Methods: 21st International Symposium, Limassol, Cyprus, 9–11 November 2016; Springer: New York, NY, USA, 2016; pp. 85–101. [Google Scholar]
- Brim, L.; Demko, M.; Pastva, S.; Šafránek, D. High-Performance Discrete Bifurcation Analysis for Piecewise-Affine Dynamical Systems. In Hybrid Systems Biology; Springer: New York, NY, USA, 2015; pp. 58–74. [Google Scholar]
- Hajnal, M.; Šafránek, D.; Demko, M.; Pastva, S.; Krejčí, P.; Brim, L. Toward Modelling and Analysis of Transient and Sustained Behaviour of Signalling Pathways. In Proceedings of the HSB 2016, Grenoble, France, 20–21 October 2016; Springer: New York, NY, USA, 2016; pp. 57–66. [Google Scholar]
- Barnat, J.; Beneš, N.; Brim, L.; Demko, M.; Hajnal, M.; Pastva, S.; Šafránek, D. Detecting Attractors in Biological Models with Uncertain Parameters. In Proceedings of the International Conference on Computational Methods in Systems Biology (CMSB), Darmstadt, Germany, 27–29 September 2017; Springer: New York, NY, USA, 2017; pp. 40–56. [Google Scholar]
- Beneš, N.; Brim, L.; Pastva, S.; Šafránek, D.; Troják, M.; Červenỳ, J.; Šalagovič, J. Fully Automated Attractor Analysis of Cyanobacteria Models. In Proceedings of the 2018 22nd IEEE International Conference on System Theory, Control and Computing (ICSTCC), Sinaia, Romania, 10–12 October 2018; pp. 354–359. [Google Scholar]
- Jia, B.; Jeon, C.O. High-throughput recombinant protein expression in Escherichia coli: Current status and future perspectives. Open Biol. 2016, 6, 160196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar, J.M.; Guet, C.C.; Leibler, S. Modeling network dynamics: The lac operon, a case study. J. Cell Biol. 2003, 161, 471–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santillán, M.; Mackey, M.; Zeron, E. Origin of bistability in the lac operon. Biophys. J. 2007, 92, 3830–3842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessier, G. Les lois quantitatives de la croissance. Ann. Physiol. Physiochim. Biol. 1936, 12, 527–573. [Google Scholar]
- Moser, H. The Dynamics of Bacterial Populations Maintained in the Chemostat; Carnegie Institution of Washington: Washington, DC, USA, 1958. [Google Scholar]
- Edwards, V.H. The influence of high substrate concentrations on microbial kinetics. Biotechnol. Bioeng. 1970, 12, 679–712. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.F. A mathematical model for the continuous culture of microorganisms utilizing inhibitory substrates. Biotechnol. Bioeng. 1968, 10, 707–723. [Google Scholar] [CrossRef]
- Maier, R.M.; Pepper, I.L.; Gerba, C.P. Environmental Microbiology; Academic Press: New York, NY, USA, 2009; Volume 397. [Google Scholar]
- Wang, P.; Robert, L.; Pelletier, J.; Dang, W.L.; Taddei, F.; Wright, A.; Jun, S. Robust growth of Escherichia coli. Curr. Biol. 2010, 20, 1099–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]















| (0.0, 1.0) | (0.0, 4.0) | (0.024, 0.78) | (0.0, 0.01) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demko, M.; Chrást, L.; Dvořák, P.; Damborský, J.; Šafránek, D. Computational Modelling of Metabolic Burden and Substrate Toxicity in Escherichia coli Carrying a Synthetic Metabolic Pathway. Microorganisms 2019, 7, 553. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7110553
Demko M, Chrást L, Dvořák P, Damborský J, Šafránek D. Computational Modelling of Metabolic Burden and Substrate Toxicity in Escherichia coli Carrying a Synthetic Metabolic Pathway. Microorganisms. 2019; 7(11):553. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7110553
Chicago/Turabian StyleDemko, Martin, Lukáš Chrást, Pavel Dvořák, Jiří Damborský, and David Šafránek. 2019. "Computational Modelling of Metabolic Burden and Substrate Toxicity in Escherichia coli Carrying a Synthetic Metabolic Pathway" Microorganisms 7, no. 11: 553. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7110553