Resolved Influenza A Virus Infection Has Extended Effects on Lung Homeostasis and Attenuates Allergic Airway Inflammation in a Mouse Model

 ,
, {kind=link}
{kind=link}
{kind=link}
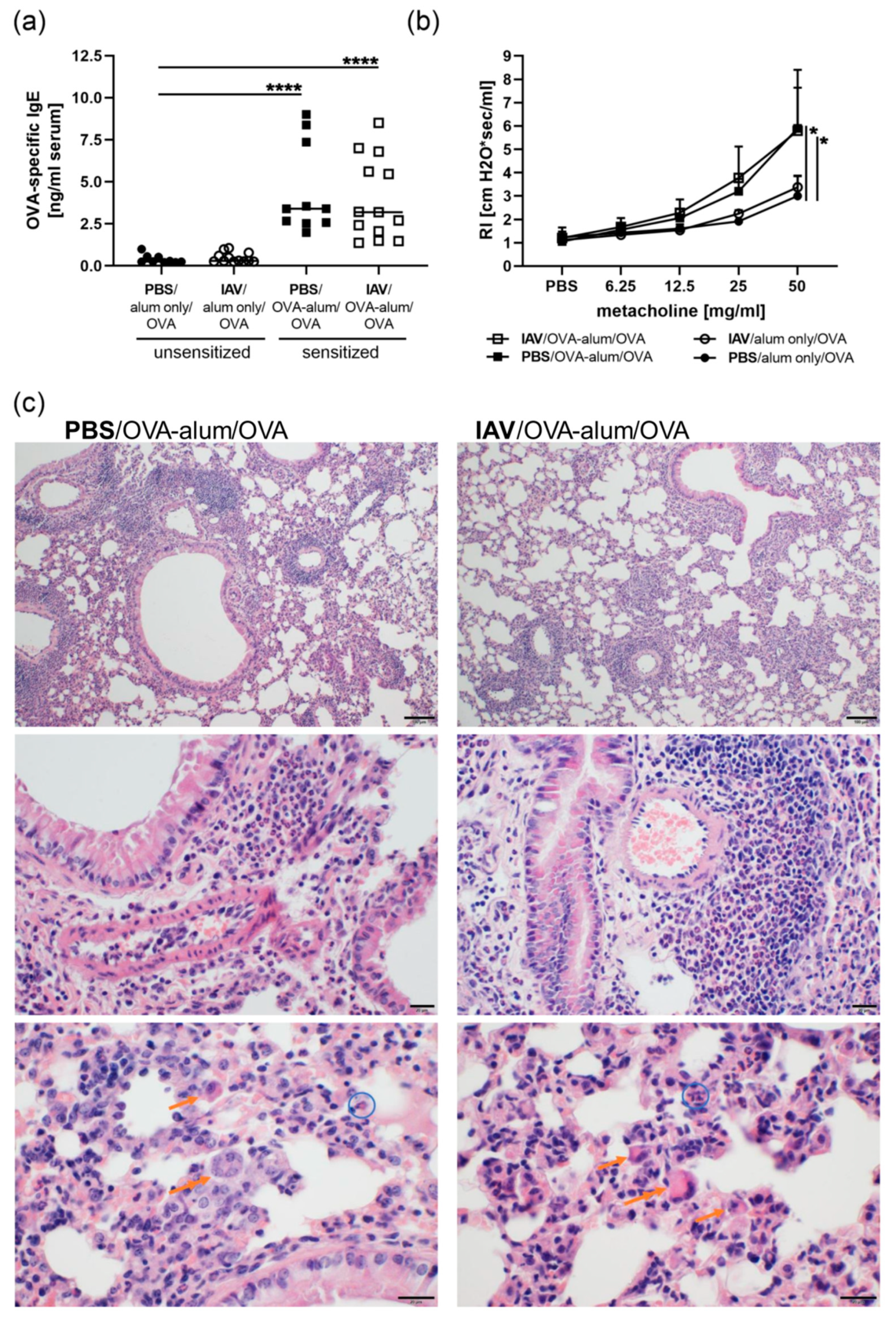{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Influenza A Virus Infection
2.3. Induction of Allergic Airway Inflammation
2.4. Serum
2.5. Isolation of Leukocytes
2.6. Flow Cytometry
2.7. ELISA
2.8. Quantification of Cytokines in BAL
2.9. Assessment of Airway Hyperreactivity
2.10. Histopathological Analysis
2.11. Statistical Analysis
3. Results
3.1. Influenza A Virus Infection Results in Persisting Changes in the Pulmonary Immune Cell Composition
3.2. Resolved IAV Infection Affects the Histological Presentation of AAI
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holt, P.G.; Strickland, D.H.; Wikstrom, M.E.; Jahnsen, F.L. Regulation of immunological homeostasis in the respiratory tract. Nat. Rev. Immunol. 2008, 8, 142–152. [Google Scholar] [CrossRef]
- Farne, H.A.; Johnston, S.L. Immune mechanisms of respiratory viral infections in asthma. Curr. Opin. Immunol. 2017, 48, 31–37. [Google Scholar] [CrossRef]
- Lloyd, C.M.; Marsland, B.J. Lung homeostasis: Influence of age, microbes, and the immune system. Immunity 2017, 46, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Braciale, T.J.; Sun, J.; Kim, T.S. Regulating the adaptive immune response to respiratory virus infection. Nat. Rev. Immunol. 2012, 12, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.L. Host immune response to influenza a virus infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.K.; Kim, T.S.; Hufford, M.M.; Braciale, T.J. Viral infection of the lung: Host response and sequelae. J. Allergy Clin. Immunol. 2013, 132, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Klomp, M.; Ghosh, S.; Mohammed, S.; Nadeem Khan, M. From virus to inflammation, how influenza promotes lung damage. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef]
- Major, J.; Crotta, S.; Llorian, M.; McCabe, T.M.; Gad, H.H.; Priestnall, S.L.; Hartmann, R.; Wack, A. Type i and iii interferons disrupt lung epithelial repair during recovery from viral infection. Science 2020, 369, 712–717. [Google Scholar] [CrossRef]
- Sharma-Chawla, N.; Stegemann-Koniszewski, S.; Christen, H.; Boehme, J.D.; Kershaw, O.; Schreiber, J.; Guzmán, C.A.; Bruder, D.; Hernandez-Vargas, E.A. In vivo neutralization of pro-inflammatory cytokines during secondary streptococcus pneumoniae infection post influenza a virus infection. Front. Immunol. 2019, 10, 1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma-Chawla, N.; Sender, V.; Kershaw, O.; Gruber, A.D.; Volckmar, J.; Henriques-Normark, B.; Stegemann-Koniszewski, S.; Bruder, D. Influenza a virus infection predisposes hosts to secondary infection with different streptococcus pneumoniae serotypes with similar outcome but serotype-specific manifestation. Infect. Immun. 2016, 84, 3445–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rynda-Apple, A.; Robinson, K.M.; Alcorn, J.F. Influenza and bacterial superinfection: Illuminating the immunologic mechanisms of disease. Infect. Immun. 2015, 83, 3764–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvigneau, S.; Sharma-Chawla, N.; Boianelli, A.; Stegemann-Koniszewski, S.; Nguyen, V.K.; Bruder, D.; Hernandez-Vargas, E.A. Hierarchical effects of pro-inflammatory cytokines on the post-influenza susceptibility to pneumococcal coinfection. Sci. Rep. 2016, 6, 37045. [Google Scholar] [CrossRef] [Green Version]
- Metzger, D.W.; Sun, K. Immune dysfunction and bacterial coinfections following influenza. J. Immunol. 2013, 191, 2047–2052. [Google Scholar] [CrossRef] [Green Version]
- Aegerter, H.; Kulikauskaite, J.; Crotta, S.; Patel, H.; Kelly, G.; Hessel, E.M.; Mack, M.; Beinke, S.; Wack, A. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat. Immunol. 2020, 21, 145–157. [Google Scholar] [CrossRef]
- Didierlaurent, A.; Goulding, J.; Patel, S.; Snelgrove, R.; Low, L.; Bebien, M.; Lawrence, T.; van Rijt, L.S.; Lambrecht, B.N.; Sirard, J.C.; et al. Sustained desensitization to bacterial toll-like receptor ligands after resolution of respiratory influenza infection. J. Exp. Med. 2008, 205, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Kanegai, C.M.; Xi, Y.; Donne, M.L.; Gotts, J.E.; Driver, I.H.; Amidzic, G.; Lechner, A.J.; Jones, K.D.; Vaughan, A.E.; Chapman, H.A.; et al. Persistent pathology in influenza-infected mouse lungs. Am. J. Respir. Cell Mol. Biol. 2016, 55, 613–615. [Google Scholar] [CrossRef] [Green Version]
- Keeler, S.P.; Agapov, E.V.; Hinojosa, M.E.; Letvin, A.N.; Wu, K.; Holtzman, M.J. Influenza a virus infection causes chronic lung disease linked to sites of active viral RNA remnants. J. Immunol. 2018, 201, 2354–2368. [Google Scholar] [CrossRef] [Green Version]
- Stegemann-Koniszewski, S.; Gereke, M.; Orrskog, S.; Lienenklaus, S.; Pasche, B.; Bader, S.R.; Gruber, A.D.; Akira, S.; Weiss, S.; Henriques-Normark, B.; et al. Tlr7 contributes to the rapid progression but not to the overall fatal outcome of secondary pneumococcal disease following influenza a virus infection. J. Innate Immun. 2013, 5, 84–96. [Google Scholar] [CrossRef]
- Zarogoulidis, P.; Kouliatsis, G.; Papanas, N.; Spyratos, D.; Constantinidis, T.C.; Kouroumichakis, I.; Steiropoulos, P.; Mabroudi, M.; Matthaios, D.; Kerenidi, T.; et al. Long-term respiratory follow-up of h1n1 infection. Virol. J. 2011, 8, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toufen, C., Jr.; Costa, E.L.; Hirota, A.S.; Li, H.Y.; Amato, M.B.; Carvalho, C.R. Follow-up after acute respiratory distress syndrome caused by influenza a (h1n1) virus infection. Clinics 2011, 66, 933–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Peng, L.; Liu, H.; Hua, S. Pulmonary function and clinical manifestations of patients infected with mild influenza a virus subtype h1n1: A one-year follow-up. PLoS ONE 2015, 10, e0133698. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, J.; Hao, S.; Yang, M.; Lu, X.; Chen, X.; Li, L. Long term outcomes in survivors of epidemic influenza a (h7n9) virus infection. Sci. Rep. 2017, 7, 17275. [Google Scholar] [CrossRef]
- Al-Garawi, A.; Fattouh, R.; Botelho, F.; Walker, T.D.; Goncharova, S.; Moore, C.L.; Mori, M.; Erjefalt, J.S.; Chu, D.K.; Humbles, A.A.; et al. Influenza a facilitates sensitization to house dust mite in infant mice leading to an asthma phenotype in adulthood. Mucosal. Immunol. 2011, 4, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Al-Garawi, A.A.; Fattouh, R.; Walker, T.D.; Jamula, E.B.; Botelho, F.; Goncharova, S.; Reed, J.; Stampfli, M.R.; O’Byrne, P.M.; Coyle, A.J.; et al. Acute, but not resolved, influenza a infection enhances susceptibility to house dust mite-induced allergic disease. J. Immunol. 2009, 182, 3095–3104. [Google Scholar] [CrossRef] [Green Version]
- Dahl, M.E.; Dabbagh, K.; Liggitt, D.; Kim, S.; Lewis, D.B. Viral-induced t helper type 1 responses enhance allergic disease by effects on lung dendritic cells. Nat. Immunol. 2004, 5, 337–343. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Global Inititative for Asthma. Global Strategy for Asthma Management and Prevention, 2020. Available online: https://ginasthma.org/reports/ (accessed on 9 November 2020).
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P.D. Asthma. Nat. Rev. Dis. Primers 2015, 1, 15025. [Google Scholar] [CrossRef]
- Fuhlbrigge, A.L.; Castro, M. Precision medicine in asthma-using phenotypes to understand endotypes that lead us to new therapeutic options. J. Allergy Clin. Immunol. Pr. 2020, 8, 496–497. [Google Scholar] [CrossRef]
- von Mutius, E.; Smits, H.H. Primary prevention of asthma: From risk and protective factors to targeted strategies for prevention. Lancet 2020, 396, 854–866. [Google Scholar] [CrossRef]
- Busse, W.W.; Lemanske, R.F., Jr.; Gern, J.E. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet 2010, 376, 826–834. [Google Scholar] [CrossRef]
- Bonnelykke, K.; Vissing, N.H.; Sevelsted, A.; Johnston, S.L.; Bisgaard, H. Association between respiratory infections in early life and later asthma is independent of virus type. J. Allergy Clin. Immunol. 2015, 136, 81–86 e84. [Google Scholar] [CrossRef] [PubMed]
- Rantala, A.; Jaakkola, J.J.; Jaakkola, M.S. Respiratory infections precede adult-onset asthma. PLoS ONE 2011, 6, e27912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegemann, S.; Dahlberg, S.; Kroger, A.; Gereke, M.; Bruder, D.; Henriques-Normark, B.; Gunzer, M. Increased susceptibility for superinfection with streptococcus pneumoniae during influenza virus infection is not caused by tlr7-mediated lymphopenia. PLoS ONE 2009, 4, e4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorde, I.; Hildebrand, C.B.; Kershaw, O.; Lücke, E.; Stegemann-Koniszewski, S.; Schreiber, J. Modulation of allergic sensitization and allergic inflammation by staphylococcus aureus enterotoxin b in an ovalbumin mouse model. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Europe. Influenza—Estimating Burden of Disease. Available online: https://www.euro.who.int/en/health-topics/communicable-diseases/influenza/seasonal-influenza/burden-of-influenza (accessed on 9 November 2020).
- Buchweitz, J.P.; Harkema, J.R.; Kaminski, N.E. Time-dependent airway epithelial and inflammatory cell responses induced by influenza virus a/pr/8/34 in c57bl/6 mice. Toxicol. Pathol. 2007, 35, 424–435. [Google Scholar] [CrossRef]
- Wohlleben, G.; Muller, J.; Tatsch, U.; Hambrecht, C.; Herz, U.; Renz, H.; Schmitt, E.; Moll, H.; Erb, K.J. Influenza a virus infection inhibits the efficient recruitment of th2 cells into the airways and the development of airway eosinophilia. J. Immunol. 2003, 170, 4601–4611. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G.L.; Valenzuela, A.; Manzoni, T.; Vaughan, A.E.; Lopez, C.B. Distinct chronic post-viral lung diseases upon infection with influenza or parainfluenza viruses differentially impact superinfection outcome. Am. J. Pathol. 2020, 190, 543–553. [Google Scholar] [CrossRef]
- Ural, B.B.; Yeung, S.T.; Damani-Yokota, P.; Devlin, J.C.; de Vries, M.; Vera-Licona, P.; Samji, T.; Sawai, C.M.; Jang, G.; Perez, O.A.; et al. Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Rowe, R.K.; Gill, M.A. Effects of allergic sensitization on antiviral immunity: Allergen, virus, and host cell mechanisms. Curr. Allergy Asthma Rep. 2017, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Gourbal, B.; Pinaud, S.; Beckers, G.J.M.; Van Der Meer, J.W.M.; Conrath, U.; Netea, M.G. Innate immune memory: An evolutionary perspective. Immunol. Rev. 2018, 283, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Wanka, L.; Jappe, U. Trained immunity and allergy: State of the art and future perspectives. Allergy 2020. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, J.M.; Gillespie, V.L.; Srivastava, K.; Li, X.M.; Busse, P.J. Influenza a infection enhances antigen-induced airway inflammation and hyperresponsiveness in young but not aged mice. Clin. Exp. Allergy 2014, 44, 1188–1199. [Google Scholar] [CrossRef]
- Skevaki, C.; Hudemann, C.; Matrosovich, M.; Mobs, C.; Paul, S.; Wachtendorf, A.; Alashkar Alhamwe, B.; Potaczek, D.P.; Hagner, S.; Gemsa, D.; et al. Influenza-derived peptides cross-react with allergens and provide asthma protection. J. Allergy Clin. Immunol. 2018, 142, 804–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veerapandian, R.; Snyder, J.D.; Samarasinghe, A.E. Influenza in asthmatics: For better or for worse? Front. Immunol. 2018, 9, 1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, M.R.; Strong, K.; Cameron, A.; Walton, R.P.; Jackson, D.J.; Johnston, S.L. Viral infections in allergy and immunology: How allergic inflammation influences viral infections and illness. J. Allergy Clin. Immunol. 2017, 140, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, J.; Openshaw, P.; Jha, A.; Del Giacco, S.R.; Firinu, D.; Tsilochristou, O.; Roberts, G.; Selby, A.; Akdis, C.; Agache, I.; et al. Influenza burden, prevention, and treatment in asthma-a scoping review by the eaaci influenza in asthma task force. Allergy 2018, 73, 1151–1181. [Google Scholar] [CrossRef]
- Eberl, G. Immunity by equilibrium. Nat. Rev. Immunol. 2016, 16, 524–532. [Google Scholar] [CrossRef]
- Minne, A.; Jaworska, J.; Gerhold, K.; Ahrens, B.; Avagyan, A.; Vanbever, R.; Matricardi, P.M.; Schmidt, A.C.; Hamelmann, E. Intranasal delivery of whole influenza vaccine prevents subsequent allergen-induced sensitization and airway hyper-reactivity in mice. Clin. Exp. Allergy 2007, 37, 1250–1258. [Google Scholar] [CrossRef]
- Xirakia, C.; Koltsida, O.; Stavropoulos, A.; Thanassopoulou, A.; Aidinis, V.; Sideras, P.; Andreakos, E. Toll-like receptor 7-triggered immune response in the lung mediates acute and long-lasting suppression of experimental asthma. Am. J. Respir. Crit. Care Med. 2010, 181, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Gorski, S.A.; Hahn, Y.S.; Braciale, T.J. Group 2 innate lymphoid cell production of il-5 is regulated by nkt cells during influenza virus infection. PLoS Pathog. 2013, 9, e1003615. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.; Mendez-Enriquez, E.; Westin, A.; Soderberg, C.; Dahlin, J.S.; Gronvik, K.O.; Hallgren, J. Influenza infection in mice induces accumulation of lung mast cells through the recruitment and maturation of mast cell progenitors. Front. Immunol. 2017, 8, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsland, B.J.; Harris, N.L.; Camberis, M.; Kopf, M.; Hook, S.M.; Le Gros, G. Bystander suppression of allergic airway inflammation by lung resident memory cd8+ t cells. Proc. Natl. Acad. Sci. USA 2004, 101, 6116–6121. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Jeyanathan, M.; Haddadi, S.; Barra, N.G.; Vaseghi-Shanjani, M.; Damjanovic, D.; Lai, R.; Afkhami, S.; Chen, Y.; Dvorkin-Gheva, A.; et al. Induction of autonomous memory alveolar macrophages requires t cell help and is critical to trained immunity. Cell 2018, 175, 1634–1650.e17. [Google Scholar] [CrossRef] [Green Version]
- Machiels, B.; Dourcy, M.; Xiao, X.; Javaux, J.; Mesnil, C.; Sabatel, C.; Desmecht, D.; Lallemand, F.; Martinive, P.; Hammad, H.; et al. A gammaherpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat. Immunol. 2017, 18, 1310–1320. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Quell, K.M.; Dutta, K.; Korkmaz, U.R.; Nogueira de Almeida, L.; Vollbrandt, T.; Konig, P.; Lewkowich, I.; Deepe, G.S., Jr.; Vershoor, A.; Kohl, J.; et al. Gm-csf and il-33 orchestrate polynucleation and polyploidy of resident murine alveolar macrophages in a murine model of allergic asthma. Int. J. Mol. Sci. 2020, 21, 7487. [Google Scholar] [CrossRef]
- Stegemann-Koniszewski, S.; Jeron, A.; Gereke, M.; Geffers, R.; Kroger, A.; Gunzer, M.; Bruder, D. Alveolar type ii epithelial cells contribute to the anti-influenza a virus response in the lung by integrating pathogen- and microenvironment-derived signals. MBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Taubenberger, J.K.; Morens, D.M. The pathology of influenza virus infections. Annu. Rev. Pathol. 2008, 3, 499–522. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.; Lunding, L.P.; Ehlers, J.C.; Weckmann, M.; Zissler, U.M.; Wegmann, M. More than just a barrier: The immune functions of the airway epithelium in asthma pathogenesis. Front. Immunol. 2020, 11, 761. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Jorde, I.; Kershaw, O.; Jeron, A.; Bruder, D.; Schreiber, J.; Stegemann-Koniszewski, S. Resolved Influenza A Virus Infection Has Extended Effects on Lung Homeostasis and Attenuates Allergic Airway Inflammation in a Mouse Model. Microorganisms 2020, 8, 1878. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121878
Wu Q, Jorde I, Kershaw O, Jeron A, Bruder D, Schreiber J, Stegemann-Koniszewski S. Resolved Influenza A Virus Infection Has Extended Effects on Lung Homeostasis and Attenuates Allergic Airway Inflammation in a Mouse Model. Microorganisms. 2020; 8(12):1878. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121878
Chicago/Turabian StyleWu, Qingyu, Ilka Jorde, Olivia Kershaw, Andreas Jeron, Dunja Bruder, Jens Schreiber, and Sabine Stegemann-Koniszewski. 2020. "Resolved Influenza A Virus Infection Has Extended Effects on Lung Homeostasis and Attenuates Allergic Airway Inflammation in a Mouse Model" Microorganisms 8, no. 12: 1878. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121878